

Abstract
Asymmetric aldol reactions are a powerful method for the construction of carbon-carbon bonds in an enantioselective fashion. Historically this reaction has been performed in a stoichiometric fashion to control the various aspects of chemo-, diastereo-, regio- and enantioselectivity, however, a more atom economical approach would unite high selectivity with the use of only a catalytic amount of a chiral promoter. This critical review documents the development of direct catalytic asymmetric aldol methodologies, including organocatalytic and metal-based strategies. New methods have improved the reactivity, selectivity and substrate scope of the direct aldol reaction and enabled the synthesis of complex molecular targets
1. Introduction
The aldol reaction, first discovered by Wurtz in 1872,1 is one of the most powerful transformations in organic chemistry. The process unites two carbonyl partners to give β-hydroxyketones with up to two new stereocenters (Scheme 1). The aldol reaction presents numerous challenges, including issues of chemo-, regio-, diastereo-, and enantioselectivity to the synthetic chemist, which has spurred the development of many powerful stoichiometric processes to address these issues.2 Development of catalytic methods that avoid the production of stoichiometric by-products while maintaining the high levels of control available from such processes provides an atom-economical alternative for these important transformations.3 Indeed, numerous catalytic methods have been reported in recent years, including enzymes,4 catalytic antibodies5 and small molecules.6–8 The focus of this review will be on small molecule catalysts, including organocatalysts and metal complexes, with special emphasis on selectivity, substrate scope and current limitations.
Scheme 1.
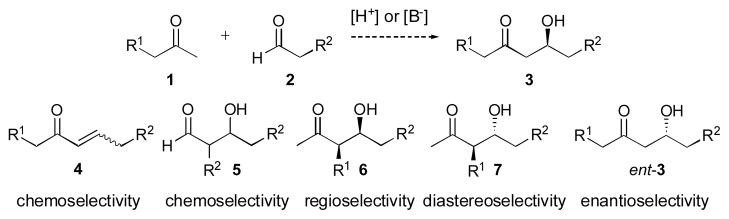
The direct catalytic asymmetric aldol and issues of selectivity.
In biological systems, the aldol reaction is accomplished by two types of aldolases, classified by their different mechanisms (Scheme 2).9 Type I aldolases function via an enamine mechanism, in which an enzyme lysine residue reacts with the donor component 8 to generate an enamine (B) in the active site. This enamine (C) then attacks the acceptor electrophile (9) to give iminium adduct D. Hydrolysis frees the substrate from the enzyme and releases the aldol adduct 10.
Scheme 2.

Mechanism of type I (RAMA FDP) and type II (fuculose-1-phosphate) aldolases.9
Type II aldolases catalyze the aldol reaction by activation of the donor substrate 8 with an active site histidine-bound zinc ion. This acidifies the α-proton, allowing for facile generation of zinc enolate F. Activation of the carbonyl of the acceptor 11 through hydrogen-bonding is followed by attack of the zinc enolate to provide aldol adduct G. Protonation and decomplexation yields aldol adduct 10.
These biological processes have provided a template for the development of small molecule catalysts. Indeed, most of the reported catalytic systems fall into these two mechanistic classes. Nature’s aldolases inspire chemists to create small molecule mimics capable of both imitating the power of enzymes in terms of selectivity and efficiency, but also to develop systems capable of catalyzing aldol reactions for a wide array of substrates, which nature’s enzymes will not tolerate.
2. Enamine-Catalyzed Aldol Reactions
Small molecule catalysts which take advantage of an enamine mechanism analogous to the type I aldolases comprise the vast majority of reported methods for carrying out direct catalytic asymmetric aldol reactions. As these approaches have been reviewed previously,10–24 the goal of this review will be to highlight and compare the reactivity, scope and selectivity of a wide range of catalysts, rather than catalogue every example of their use. This review will not focus on design parameters such as catalyst immobilization,25–35 attaching hydrophobic groups to the catalyst,30, 36–38 reactions in micelles,39, 40 host-guest catalytic systems,41, 42 or the use of ionic liquids43–54 or surfactants,55–60 which can be found elsewhere.12, 13 Instead, this review will highlight what is currently possible synthetically and what challenges remain unmet for enamine-based catalysts.
2.1 Proline-Catalyzed Aldol Reactions
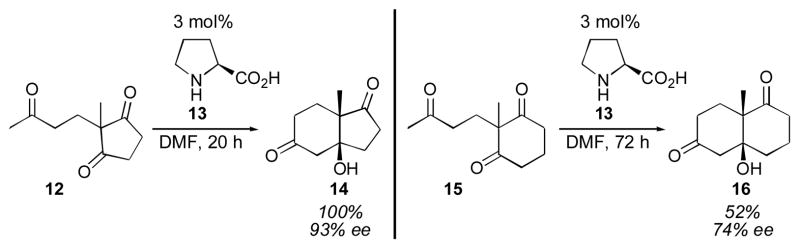
The first report of a direct asymmetric aldol reaction catalyzed by a small molecule was the Hajos-Parrish-Eder-Sauer-Wiechert cyclization, disclosed in 1971 (Scheme 3).61, 62 This intramolecular aldol cyclization proceeded with only 3 mol% proline (13) to give the cyclized product 14 in excellent yield and enantioselectivity. Unfortunately, both the yield and the enantioselectivity dropped for the 6-membered ring substrate 15. Despite this reaction’s utility, particularly for the synthesis of steroids, its mechanism was not determined, nor was proline appreciated as a broadly applicable catalyst, for many years. Amino acids such as proline are particularly appealing catalysts, due to their natural abundance and low cost.
Scheme 3.
The Hajos-Parrish-Eder-Sauer-Wiechert reaction is an enol-endo aldolization. Enol-exo cyclizations are also possible using proline catalysis. In 2003 List and coworkers reported the enol-exo cyclization of a variety of dialdehydes, such as 17, yielding 6-membered rings (18) in high yields, diastereo-and enantioselectivities. Five-membered rings (20) were formed with reduced diastereo- and enantioselectivities (Scheme 4).63, 64 List has also demonstrated that transannular intramolecular cyclizations are possible using catalytic proline, though the enantioselectivity was not high. Using modified catalyst 22 the enantioselectivity was greatly improved. The utility of this transannular cyclization was demonstrated in List’s synthesis of (+)-hirsutene (24).
Scheme 4.

Proline-catalyzed enol-exo and transannular cyclizations.63, 64
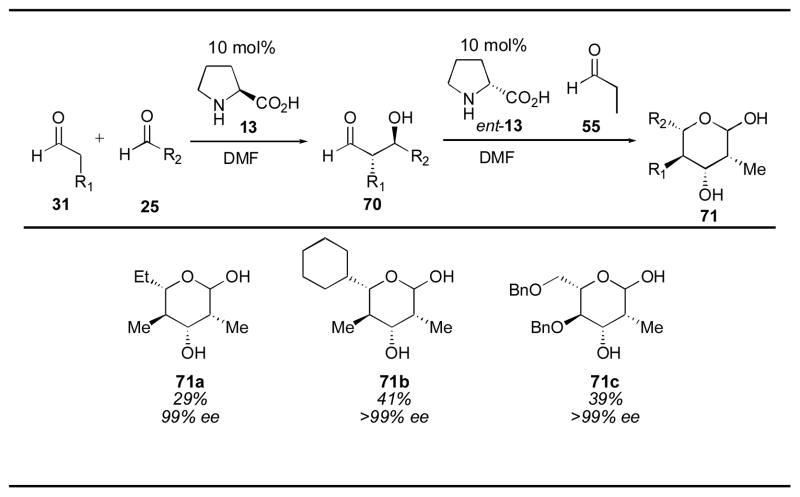
The first intermolecular proline-catalyzed direct aldol reaction was described by List and coworkers in 2000 (Table 1).65 Using 20–30 mol% L-proline and a 4:1 solution of DMSO:acetone, the desired aldol adducts 27 were obtained. Aryl aldehydes were good substrates for this reaction, as were branched aliphatic substrates, which gave the highest yields and enantioselectivities. α-Unbranched aldehydes were less successful substrates, giving low yields and moderate enantioselectivity after tuning of the reaction conditions to prevent aldehyde self-condensation.66 Unfortunately, even under these optimized conditions (employing neat acetone or chloroform as cosolvent for 3–7 days), the cross-aldol condensation product 4 (Scheme 1) was a significant byproduct, resulting in low overall yield. Despite the low yield and modest enantioselection of this process, the simplicity and mildness of these reaction conditions is exceptional. This methodology was used by List and coworkers to complete the synthesis of (S)-ipsenol (28), a sex pheromone of the bark beetle. The acetone aldol has also been used for the syntheses of 4-hydroxypipecolic acid derivative 2967 and carboxylic acid 30, a building block for the synthesis of epothilone.68
Table 1.
Proline-catalyzed acetone aldol.
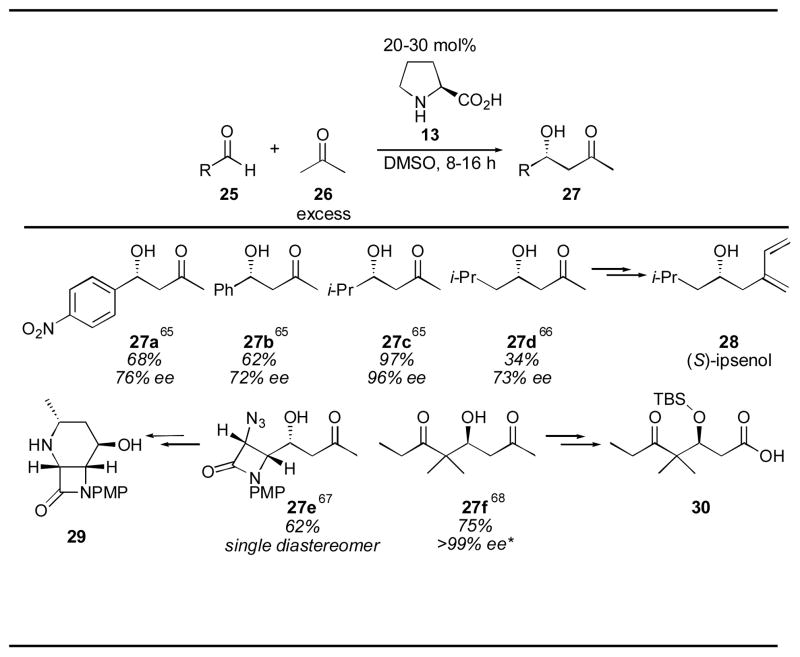
|
D-Proline was used. References are given for each product as a superscript.
A great deal of research has clarified the mechanism of the proline-catalyzed aldol reaction, which is essentially that of a class I aldolase (Scheme 5).24, 61, 69–105 The accepted mechanism for the intermolecular process begins with rate-limiting enamine formation (A), followed by carbonyl addition, activated by the carboxylic acid of proline (B), followed by hydrolysis of iminium ion C to give aldol adduct 27. The addition step has a similar energy barrier as the enamine formation, indicating that under different conditions or with different substrates, the rate-determining step may be the addition step. In fact, recent kinetic evidence obtained by Armstrong and Blackmond indicates that under the reaction conditions studied, the addition step is rate-determining.106
Scheme 5.
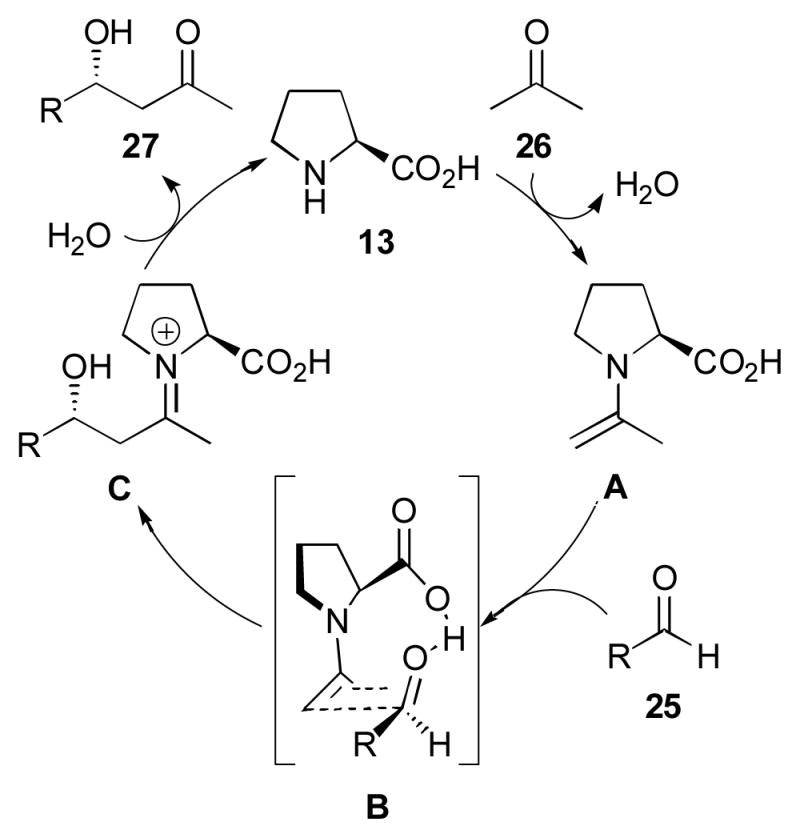
Mechanism of the intermolecular proline-catalyzed aldol.76
Several by-products have been observed in the proline-catalyzed acetone aldol reaction that lead to reduced yields, including aldol condensation (yielding enone 33), and the aldol reaction and condensation of the aldehyde component, which generates adducts 34 and 35, respectively (Scheme 6). Additionally, oxazolidinone 36 derived from the aldehyde component 31 has been observed. To achieve high yields of the desired adducts 32, acetone is used in a large excess to prevent homodimerization of the aldehyde and catalyst kill events, such as the formation of oxazolidinone 36.
Scheme 6.
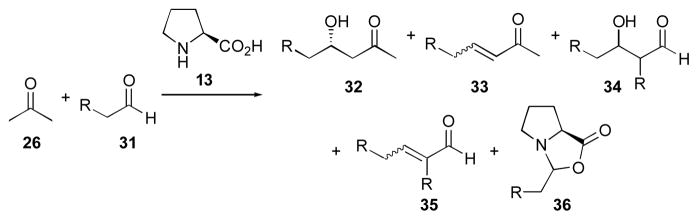
Side products obtained in the proline-catalyzed aldol reaction.
A beneficial effect of water in small amounts was first reported by Pihko and coworkers.107, 108 Armstrong and Blackmond have recently clarified the role of water in the reaction (Scheme 7).109 They demonstrated that water in fact decreases the rate of the reaction, as it must be extruded before the rate-limiting step (Scheme 5), but off cycle processes are shut down significantly in the presence of water, thereby giving an overall positive effect on the yield of the desired aldol product 27. By shifting the equilibrium between aldehyde 25 and iminium ion A toward aldehyde 25 with added water, the formation of byproducts can be avoided. Formation of oxazolidinone 37 and oxazole 38 (derived from decarboxylation of iminium ion A, followed by addition of ammonium ylide B to another equivalent of aldehyde 25) is largely prevented in this way.
Scheme 7.

Off-cycle processes that are ameliorated by the addition of water.106
An alternative mechanism for proline catalyzed reactions was proposed in 2007 by Seebach and Eschenmoser (Scheme 8).110 The authors propose that oxazolidinones, which are observable by NMR, are key players in the catalytic cycle, rather than mere catalytic sinks. Their proposed mechanism begins by formation of oxazolidinone A by condensation of proline with the donor componant 39. Regioselective formation of an enamine, either by E2 elimination or through a two-step iminium formation and intramolecular proton transfer sequence, then undergoes a trans addition to an electrophile (approach from the re face) to generate oxazolidinone C. Hydrolysis then gives the desired product 40. This mechanistic proposal differs greatly from the List-Houk model in that the key step is triggered by a η-lactonization process and does not involve activation of the acceptor componant by the catalyst. It is difficult to see how this model would predict high levels of enantioselectivity for the aldol reaction catalyzed by proline since the approach of the aldehyde to the enamine is not controlled through an H-bonding interaction. Also, this model can only be applied to catalysts bearing a free carboxylic acid group, which can participate in oxazolidinone formation.
Scheme 8.

Alternative oxazolidinone mechanism proposed by Seebach and Eschenmoser.110
The proline-catalyzed aldol reaction was extended to ketones other than acetone in 2001 (Table 2).22, 66 Since the ketone was required in near solvent quantities, the reaction was limited to simple ketone substrates, such as cyclohexanone and cyclopentanone, for practical reasons. The anti aldol adducts 42 were favored over the syn configuration in most cases. For cyclic ketones, only one enamine geometry is possible, the E enamine A. The orientation of the aldehyde when approaching this enamine, therefore, gives rise to the diastereoselectivity. The orientation depicted in A is generally favored, as it avoids steric clashes with the alkyl group on the other side of the enamine, as long as RS is smaller than this alkyl group. Cyclohexanone tends to give higher diastereoselectivities than cyclopentanone, as do aliphatic aldehydes when compared to aromatic aldehydes. These reactions are typically run for 3 days with 10–30 mol% catalyst, indicating that the reaction rate is rather slow. The original conditions reported by Barbas111 for the synthesis of aldol adduct 42 were improved upon by using the solvent-free conditions reported by Hayashi to give the cyclohexanone adduct 42a in 73% yield, 9:1 dr and in >99% ee favoring the anti isomer.112 The cyclopentanone aldol catalyzed by proline has been used for the synthesis of adduct 42f, which was used for the synthesis of the antimalarial (+)-(11R, 12S)-mefloquine hydrochloride.113
Table 2.
Substituted ketone donors in the proline-catalyzed aldol reaction.
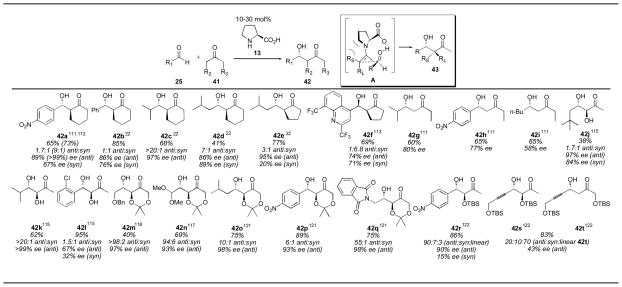
|
References are given for each product as a superscript.
In 2001 Barbas and coworkers described the use of proline with 2-butanone, an unsymmetrical ketone.111 Under the reaction conditions (20 mol% catalyst, 0.1M substrate in 1:4 ketone:DMSO, ambient temperature for 1–2 days) only the linear product was observed. The authors suggest that the formation of the kinetic enamine is rate-limiting, resulting in observation of only the linear product. Interestingly, employing N-ethyl-N-methylimadazolium trifluoromethanesulfonate ([emim][OTf]), an ionic liquid, rather than using DMSO as solvent led to isolation of the branched product, with a strong preference for the anti diastereomer.46, 114
Hydroxyacetone has also been used successfully as a donor for the proline-catalyzed direct aldol reaction, using similar reaction conditions (20–30 mol% catalyst, 0.1M substrate in 1:4 ketone:DMSO, ambient temperature for 1–3 days).115 Formation of the branched products is observed, with high diastereoselectivity for α-branched aldehydes, and lower diastereoselectivity observed for aryl aldehydes and α-unbranched aldehydes. Low yields were observed for α-unbranched aldehydes, similar to the acetone aldol. This reaction has been used for a formal synthesis of the steroidal plant-growth inhibitor brassinolide (Scheme 9).116
Scheme 9.
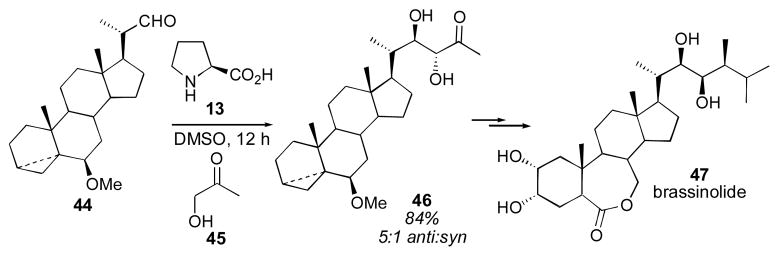
Use of the proline-catalyzed hydroxyacetone aldol reaction for the synthesis of brassinolide.116
The dihydroxyacetone derivative 2,2-dimethyl-1,3-diox-5- one has also been used successfully as a donor in the proline-catalyzed aldol reaction.117–119 In 2005 Enders and Grondal reported a highly anti selective aldol reaction that can be used to generate protected sugar derivatives in high enantioselectivity by selecting the appropriate aldehyde substrate. Aldol adduct 42m, for example, is a protected L-ribulose derivative. Gratifyingly, only one equivalent of the ketone donor was needed to obtain the desired adducts in good yields, employing 30 mol% proline as catalyst. Previous work by Barbas and coworkers demonstrated that unprotected dihydroxyacetone could be used and gave good diastereoselectivity, however, the aldol adducts obtained were racemic in this case.120 Later in the same year as the Enders and Grondal report, Barbas and coworkers disclosed similar reaction conditions for the use of 2,2-dimethyl-1,3-diox-5- one, demonstrating that the reaction proceeded well with aliphatic, aromatic, and even protected phthalimide substrates using 20 mol% proline.121 Aliphatic aldehydes gave higher diastereoselectivity than aromatic derivatives. It is notable that the α-unbranched derivative 42o was obtained in high yield, in contrast to the adduct derived from reaction of this aldehyde with acetone or cyclohexanone. The discovery of these simple reaction conditions for the use of a protected form of dihydroxyacetone in a direct catalytic aldol reaction has opened up new synthetic routes for the synthesis of various sugar derivatives.
A protected variant of hydroxyacetone have also been used to generate diols in which one of the alcohols is differentiated from the other with a protecting group. TBS-protected hydroxyacetone undergoes anti-selective aldol addition with aromatic substrates.122 Aldol adduct 42r is obtained with 90% ee, however, the ee was lower for other aromatic aldehydes (for example, 40% ee was obtained when benzaldehyde was used). α,β-Unsaturated aldehydes gave either no yield or gave predominantly the linear isomer, such as adduct 42t. The ee in this case was also quite low.
Proline catalysis has also been used for reaction with highly activated ketone electrophiles 48 (Table 3). The self condensation of 2,2-dimethyl-1,3-diox-5-one was observed by Enders and Grondal, producing adduct 49a in 94% ee.118 Zhang an coworkers reported the use of α-trifluoromethyl ketones for the synthesis of aldol adducts such as 49b with only 10 mol% proline with excellent yields, though the best ee observed was 64%.123 Maruoka demonstrated the feasibility of using α-keto esters as electrophiles with cyclohexanone as donor in 2005.124 Aldol adduct 49c was obtained in excellent enantio- and diastereoselectivity, although 50 mol% catalyst was needed to achieve these excellent results. Adduct 49c was taken on to complete the synthesis of a key intermediate for the synthesis of (S)-oxybutynin 50. The research groups of Zhang and Shao reported the use of α-keto esters as electrophiles for the acetone aldol catalyzed by proline in 2006 using 60 and 50 mol% proline, respectively.125, 126 α-Keto phosphonates have also been used as substrates for the proline-catalyzed acetone aldol, yielding the desired aldol adducts 49 in high yield and enantioselectivity when electron-deficient aldehydes are employed as the acceptors, using a catalyst loading of 20 mol%.127 Electron-rich aromatic substrates, however, gave lower yields due to their lower reactivity. Adduct 49g was obtained in only 32% yield using 50 mol% proline.
Table 3.
Ketone electrophiles in the proline-catalyzed aldol reaction.
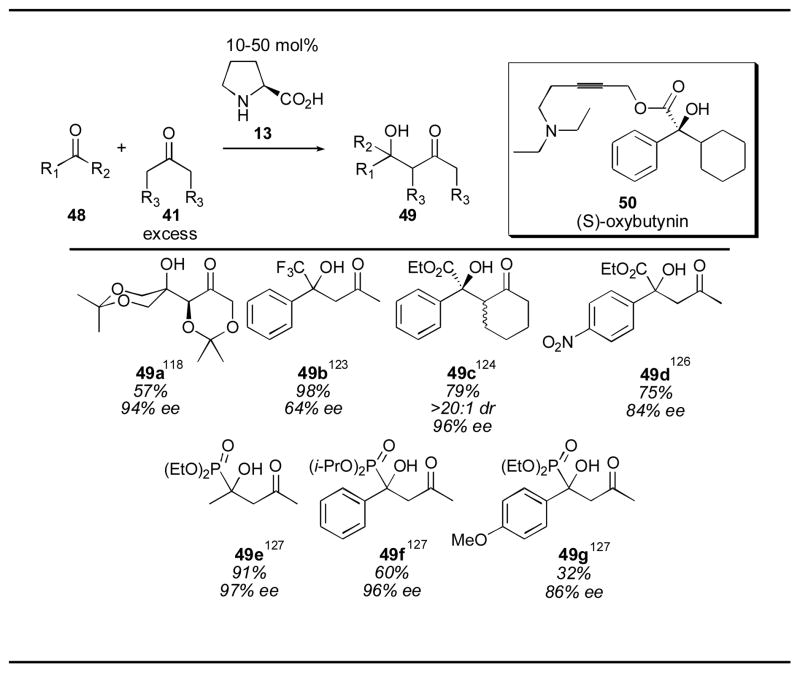
|
References are given for each product as a superscript.
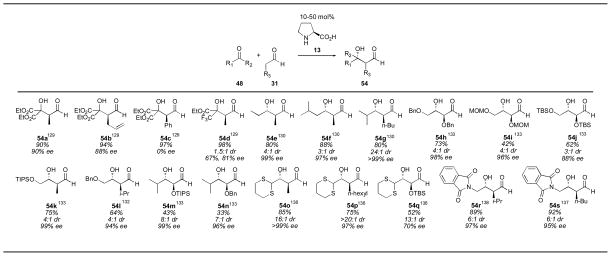
Aldehydes can also be used as donors for the proline-catalyzed aldol reaction. Barbas first noted the trimerization of acetaldehyde 51 in 10% yield and 90% ee (Scheme 10).128 Dienal 53 was isolated together with the interesting trimer 52. The first proline-catalyzed cross-aldol reaction of aldehyde donors was reported by Jørgensen (Table 4).129 Using 50 mol% proline, aldehydes were coupled with non-enolizable α-keto esters to give α-chiral aldehydes 54. While adducts 54a and 54b were isolated in good enantioselectivity, the phenyl substituted adduct 54c was found to be racemic. The trifluoromethyl derivative 54d was isolated in poor diastereoselectivity with moderate enantioselectivity.
Scheme 10.
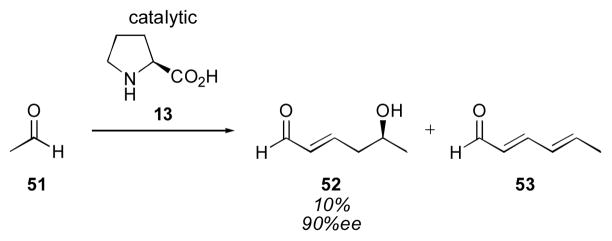
Trimerization of acetaldehyde.128
Table 4.
Aldehyde donors in the proline-catalyzed aldol reaction.
|
References are given for each product as a superscript.
MacMillan and coworkers reported a remarkable breakthrough for the cross-aldol reaction of aldehydes in 2002.130 Using slow addition of the donor aldehyde (2 equivalents) to the desired acceptor aldehyde the desired cross-aldol products 54 could be obtained in excellent yields and enantioselectivities with only 10 mol% proline. The diastereoselectivity was moderate with α-unbranched or aromatic acceptor aldehydes and excellent with α-branched acceptor aldehydes. Even remarkably similar aldehydes gave exclusively the desired cross-aldol adduct, such as adduct 54f. The acceptor substrate (in the case where the aldehydes are different) in each case is either non-enolizable or generates a less-reactive enamine. In this way the dimerization of the acceptor aldehyde is suppressed. Pihko and coworkers used this methodology for a short synthesis of prelactone B (Scheme 11).131 MacMillan and coworkers also used this method for the synthesis of callipeltoside C.132
Scheme 11.

Application of the aldehyde aldol reaction to the syntheses of prelactone B and callipeltoside C.131, 132
In 2004 MacMillan and coworkers extended this methodology to the dimerization of protected α-hydroxy aldehydes to give highly functionalized aldehydes.133 These substrates could themselves be used in a subsequent Mukaiyama aldol to give protected sugar derivatives.134 MacMillan and Mangion used this methodology to complete the synthesis of (−)-littoralisone (Scheme 12).135 Cross-aldol reactions with aliphatic aldehyde donors could also be used to give the desired aldol adducts, even with sterically-hindered donor aldehydes (adduct 54l for example). When more hindered aldehydes, such as isobutyraldehyde, that do not readily participate in enamine formation, are employed, both triisopropylsilyl- and benzyl-protected hydroxyacetaldehyde can be used as the donor component in the cross aldol reaction, albeit in rather low yields.
Scheme 12.
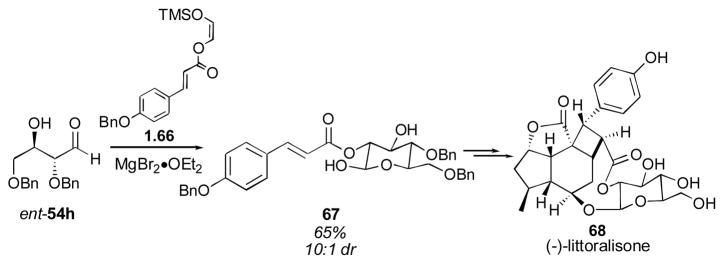
MacMillan’s direct aldol/Mukaiyama aldol sequence: application to the synthesis of (−)-littoralisone.135
MacMillan reported the use of α-thioacetal aldehydes as acceptors for the cross-aldol reaction of aldehydes in 2004.136 The highly functionalized products 54o-q were formed in moderate to excellent yields, and in excellent diastereo- and enantioselectivities with a variety of aldehyde donors. Tanaka and Barbas reported the use of a protected nitrogen-containing aldehyde for the production of adduct 54r,s in 2004.137 The nitrogen-containing aldehyde could also be used as the donor.
Aldehyde cross-trimerizations have also been reported for the synthesis of complex products in only one step from simple substrates. Barbas and coworkers reported the trimerization of alkyl aldehydes in 2002 using syringe-pump addition of two equivalents of propionaldehyde to one equivalent of the acceptor aldehyde (Table 5).138 The trimers were isolated after oxidation to the corresponding lactones 69 (The proposed relative configuration was later corrected by Córdova139 after a crystal structure was obtained, the corrected relative configuration is illustrated). The trimers were isolated in rather low yield (together with the C4 epimer) and disappointingly low ee, but given the simplicity of the reaction conditions and the amazing complexity generated in a single reaction, the transformation is exceptional. The authors note that the ee of the reaction degrades with time, for example 69a was obtained in 47% ee after 10 h at 4 °C.
Table 5.
Pyranose formation with proline-catalysis.135
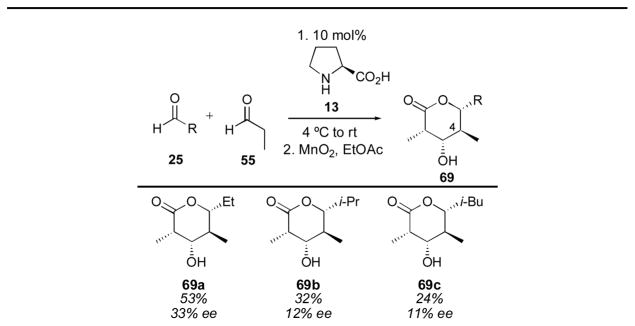
|
Relative stereochemistry between C3, C4 and C4, C5 corrected.136
Córdova and coworkers reported a similar reaction with considerably higher enantioselectivity in 2005 (Table 6).139 By altering the reaction conditions they were able to increase the enantioselectivity considerably, likely by shutting down the racemization observed under the conditions described by Barbas. Córdova observed that the ee was highly dependent on the reaction conditions and was best controlled by portion-wise addition of propionaldehyde. By separating the two sequential aldol steps, and using the opposite enantiomer for the second step, they were able to effectively shut down the retro-aldol for the first step, and thereby increase the enantioselectivity. This also allowed them to vary the three component aldehydes. The yields are rather low, but given the rapid assembly of complexity, the modest yield is easily compensated for.
Table 6.
Two-step polyketide synthesis.139
|
Remarkably, this trimerization was expanded to the synthesis of sugars by Córdova and coworkers in 2005 (Scheme 14).140, 141 For this class of aldehyde it appears that the retro-aldol is not problematic. Using only 10 mol% L-proline, 41% of protected hexose 73 was generated in excellent enantioselectivity, together with 51% of the dimer 54h. A large nonlinear effect was observed for this reaction, with proline of only 40% ee generating the product hexose 73 in >99% ee. This observation led the authors to propose a model for the evolution of homochirality, in which simple amino acids with low levels of enantiomeric excess amplify this small imbalance of one enantiomer to homochirality through prebiotic gluconeogenesis.
Scheme 14.

Improved reactivity and enantioselectivity with primary amine catalysts for sterically-hindered substrates.214, 215
The ability of such a simple molecule to generate such high levels of complexity is truly remarkable. Given the low cost and abundance of proline, as well as its wide scope, it is certainly the first choice for any enamine-catalyzed reaction, including the direct catalytic aldol reaction.
2.2 Catalyst Development
Despite the utility of proline for carrying out a wide array of aldol reactions, a great deal of effort has been exerted for the development of new catalysts. The long reaction times and poor results with certain substrates, such as α-unbranched aldehydes, have led numerous researchers to develop more reactive catalysts, as well as design catalysts that will not undergo oxazolidinone formation, which is the major catalyst deactivation pathway (Scheme 6 and 7). The need for high catalyst loadings, large excesses of ketone and long reaction times when using proline as catalyst has at times been imputed to its low solubility in the reaction media, and therefore many catalyst designs incorporate increased lipophilicity. New catalyst designs have also provided access to isomers not favored using reported proline-catalyzed conditions.
The initial screen of catalysts reported by List and coworkers in 2000 for the asymmetric aldol of acetone with para-nitrobenzaldehyde 74 illustrates the effect of a number of different modifications of the proline structure (Table 7).65, 66 Primary amino acids histidine (75), tyrosine (76), phenylalanine (77), and valine (78) provide less than 10% yield of the desired adduct 27a. Methylation of valine to produce an acyclic secondary amine (79) did not improve the yield. Both the azetidine (80) and piperidine (81) were inferior to the pyrrolidine structure of proline (13). Changing the carboxylic acid to an amide (82) also gave little reactivity. The thiazolidium carboxylate (83) gave similar, though slightly inferior results. Interestingly, proline derivative 84 gave a higher yield and enantioselectivity than proline itself. The tert-butyl ether derivative 85 gave lower yield and enantioselectivity, as did the acylated derivative 86, though to a lesser extent. Diastereomer 87 provided the opposite enantiomer, with reduced yield and selectivity.
Table 7.
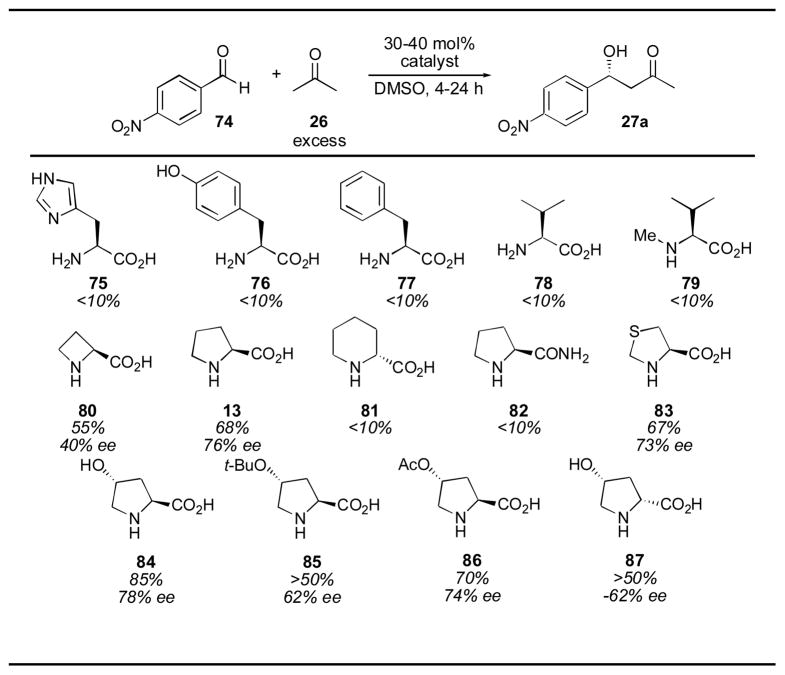
|
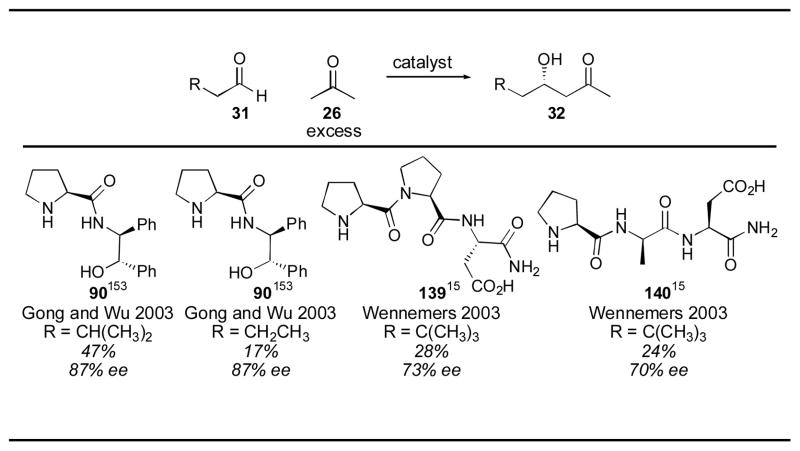
A number of other amino acids and small peptides have been evaluated as alternate catalysts for the direct catalytic asymmetric aldol reaction (Table 8).15, 142–150 The reaction between acetone and para-nitrobenzaldehyde has been used in this field as a model system to evaluate catalyst activity and is a convenient way to compare the performance of the multitude of available catalysts. Among the amino acids and peptides screened, L-Pro-L-Pro-L-Asp-NH2, reported by Wennemers and coworkers, gave the best results.148 The catalyst loading could be significantly decreased to 1 mol% to give the desired adduct ent-27a in 99% yield and 80% ee after 4 hour, using acetone as solvent. Compared to the other reported amino acids and small peptides, this is a significant increase in catalytic activity. Interestingly, this catalyst gives the opposite enantiomer than that obtained via proline- catalysis, an observation the authors trace to the secondary structure of this catalyst.
Table 8.
Screen of amino acids and small peptides for the acetone aldol reaction.
| |||
|---|---|---|---|
| Catalyst | catalyst loading (mol%) | Yield (%) | ee (%) |
| L-Pro65 | 20 | 68 | 76 |
| L-Asp142 | 20 | 35 | 40 |
| L-Glu142 | 20 | 36 | 18 |
| L-Thr142 | 20 | 37 | 42 |
| L-Pro-L-Ala-NPh, HOAc146 | 20 | 88 | 38 |
| 4-NHBoc-L-Pro-L-Pro144 | 15 | 83 | 73 |
| L-Pro-L-Ser143 | 30 | 87 | 77 |
| L-Pro-L-ThrOTBS147 | 20 | 91 | 82 |
| L-Leu-L-His145 | 30 | 87 | 71 |
| 4-NHBoc-L-Pro-L-Pro-L-Pro-L-Pro144 | 5 | 62 | 75 |
| L-Pro-D-Ala-D-Asp-NH2148 | 10 | 73 | 70 |
| L-Pro-L-Pro-L-Asp-NH2148 | 1 | 99 | −80 |
References are given for each product as a superscript.
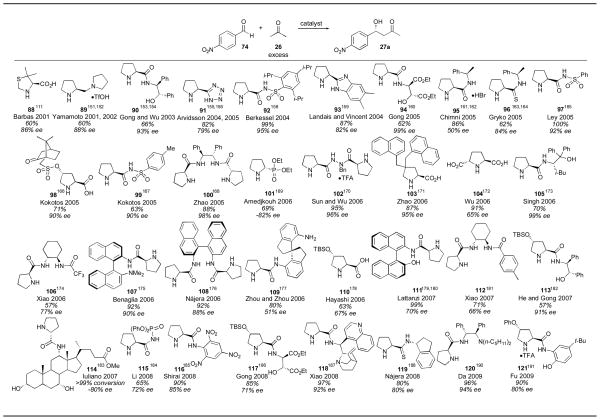
Many catalysts have been developed based on the structure of proline (Table 9). Some of the common modifications to the proline structure include increasing the hydrophobicity to improve solubility in organic solvents and modification of the carboxylic acid to a variety of other hydrogen-bonding groups. Other trends include adding steric bulk and stereocenters to enhance the enantioselectivity. Many of these catalysts require numerous steps for their synthesis, and this should be considered when choosing a catalyst. Proline, on the other hand, is abundant and inexpensive, and therefore new catalyst designs must provide significant improvements to surpass the utility of proline, both in terms of selectivity and reactivity.
Table 9.
Proline-based catalysts for the acetone aldol reaction
|
References are given for each product as a superscript.
One of the earliest designed catalysts was reported by Barbas in 2001.111 Using 20 mol% of catalyst 88 over the course of 1–2 days, adduct 27a was isolated in improved enantioselectivity when compared to proline, but the reactivity of this catalyst was rather low, reflected in the high catalyst loading, long reaction times and the need to use acetone as a co-solvent. Yamamoto reported the use of catalyst 89 for the direct catalytic aldol reaction in 2001.151, 152 With only 3 mol% catalyst 89, adduct 27a was obtained in moderate yield and ee after 2 hours. Despite this improved reactivity, acetone was still used as solvent under the optimized conditions. In 2003, Gong and Wu reported the use of 20 mol% of catalyst 90 over the course of 1–2 days.153, 154 The enantioselectivity obtained was quite good, though the reactivity was not high, and acetone was again needed as solvent. Arvidsson reported the use of tetrazole 91 for the direct catalytic aldol of acetone with aldehyde 74,155, 156 a catalyst that was reported in the same year by Saito and Yamamoto for reaction with chloral and trifluoroacetaldehyde (vide infra).157 A catalyst loading of 5 mol% and reaction time of 40 hours with acetone as a cosolvent gave the product 27a in 82% yield and 79% ee. Sulfonylcarboxamide catalyst 92 was reported in 2004 by Berkessel and coworkers.158 The catalyst was used in 30 mol% loading with acetone as a cosolvent over the course of 1 day to yield the desired adduct 27a in excellent yield and enantioselectivity. Benzoimidazole catalyst 93, reported by Landais and Vincent,159 could be used with a catalyst loading as low as 2 mol%, together with trifluoroacetic acid, in neat acetone over the course of 1 day to give the desired adduct 27a in 87% yield and 82% ee. In 2005 Gong reported an extremely enantioselective catalyst (94) possessing a chiral diester sidechain.160 Using only 2 mol% 94 the aldol reaction yielded adduct 27a in 62% yield and in 99% ee. The reaction was carried out in neat acetone over the course of 1–2 days, indicating that the reactivity was still rather low. Chimni and coworkers reported the use of proline derivative 95 in 2005.161, 162 The reaction conditions required the use of 20 mol% 95 in wet acetone over the course of 40 hours. Unfortunately the enantioselectivity was lower than that obtained using proline alone. Thioamide 96, reported by Gryko, delivered adduct 27a in 62% yield and 84% ee using acetone as solvent and 20 mol% catalyst loading after 68 hours.163, 164 Ley and coworkers reported sulfonylcarboxamide catalyst 97, similar to catalyst 92, which gave adduct 27a in 100% yield and 92% ee, using acetone as solvent and 20 mol% catalyst loading over 1–2 days.165
Camphorsulfonyl-derivative 98 was disclosed by Kokotos and coworkers in 2005.166 The catalyst could be used in 10 mol% loading with an equal molar amount of triethylamine with acetone as a cosolvent over 18–24 hours yielding adduct 27a in 71% yield and 90% ee. Kokotos also reported sulfonylcarboxamide catalyst 99 in the same year, similar to catalysts 92 and 97, which gave adduct 27a in 63% yield and in 90% ee using 20 mol% catalyst and an equivalent amount of triethylamine, with acetone as a cosolvent over the course of 18–24 hours.167 C2-Symmetric stilbene derivative 100 was disclosed by Zhao and coworkers in 2005.168 The catalyst was used in 10 mol% loading in acetone over 12–24 hours, delivering adduct 27a in 88% yield and 98% ee. Phosphonate catalyst 101 was reported in 2006 by Dinér and Amedjkouh to give the opposite enantiomer of adduct 27a in 69% yield and 82% ee.169 The catalyst was used in 20 mol% loading with acetone as cosolvent over the course of 1 day. Sun and Wu disclosed hydrazide catalyst 102 for the direct catalytic aldol in 2006.170 With 20 mol% of catalyst 102 and an equal amount of trifluoroacetic acid, employing acetone as a cosolvent over 7 hours the desired adduct 27a was obtained in 95% yield and in 96% ee. Zhao reported 4-disubstituted proline derivative 103 in 2006 for the direct catalytic aldol reaction.171 Using 10 mol% of this catalyst with acetone as cosolvent over 2 days the adduct 27a was obtained in 87% yield and 95% ee. C2-Symmetric proline derivative 104, together with an equal amount of triethylamine, was employed by Wu and coworkers in 2006.172 The catalyst was used in 30 mol% loading and the reaction was run in acetone as solvent over 30 hours to deliver the desired adduct 27a in 91% yield but in only 65% ee. Singh reported the highly selective catalyst 105 in 2006.173 Using 10 mol% catalyst in acetone for 1–2 days the product 27a was obtained in 70% yield and 99% ee.
Catalyst 106 was reported for the asymmetric aldol reaction by Xiao and coworkers.174 The catalyst was used in 20 mol% loading with 40 mol% acetic acid over 18 hours to give the adduct 27a in 57% yield and 77% ee. Binaphthyl-derivative 107, disclosed by Benaglia and coworkers, can be used in 10 mol% with acetone as solvent over 88 hours to deliver adduct 27a in 92% yield and in 90% ee.175 A C2-symmetric binaphthyl derivative 108 was disclosed by Nájera and coworkers, used in 10 mol% loading along with 20 mol% benzoic acid, with acetone as cosolvent over 36 hours. These conditions delivered the adduct 23a in 92% yield and in 88% ee.176 Zhou and Zhou reported the use of spiro-derivative 109, employing only 1 mol% catalyst over 4 hours in acetone.177 This increased reactivity resulted in an 80% yield of adduct 27a, but unfortunately only in 51% ee. Hayashi reported the utility of silylprotected 4-hydroxyproline 110 in 2006 for the direct catalytic asymmetric aldol reaction.178 The catalyst loading used was 10 mol% with 5 equivalents of acetone in water over 18 hours. The adduct 27a was obtained in 63% yield and 67% ee. Binaphthyl derivative 111 was reported in 2007 by Lattanzi.179, 180 The catalyst was used in 5 mol% loading using only 3 equivalents of acetone over 10 hours to deliver the adduct in 99% yield and in 70% ee. Catalyst 112, reported by Xiao in 2007, was used in 20 mol% loading with an equal amount of acetic acid.181 The reaction was run in brine with 10 equivalents of acetone over 39 hours to yield adduct 27a in 71% yield and 66% ee. He and Gong disclosed the use of 5 mol% catalyst 113 using acetone as solvent over 60 hours.182 The adduct 27a was obtained in 57% yield with 91% ee.
Iuliano and coworkers reported the synthesis and use of steroid-derived catalyst 114 containing a D-proline tethered to the 6-membered ring.183 The catalyst was tested for reactivity in the direct catalytic aldol reaction with para-nitrobenzaldehyde 74. The catalyst was used in 5 mol% loading with acetone as a cosolvent over the course of 2 days. The reaction yield was not given, but proceeded in complete conversion to give adduct 27a in 80% ee. Phosphoramide catalyst 115 was reported in 2008 by Li and coworkers.184 With 10 mol% catalyst, an equal amount of N-methylmorpholine and 6 equivalents of acetone the product 27a was obtained in 65% yield and 72% yield. Trinitroanilide catalyst 116 was reported by Shirai and coworkers in 2008.185 The reaction was performed using HMPA as solvent with 30 equivalents of water and 20 equivalents of acetone to give the adduct 27a in 90% yield and 85% ee after 4 days. Catalyst 117, which is similar to catalyst 94, was reported by Gong in 2008.186 Using only 1 mol% catalyst loading and 2 equivalents of acetone the adduct was obtained in 85% yield and 71% ee after 14 hours. Cinchona alkaloid derivative 118 was reported by Xiao in 2008.187 Similar conditions were reported by Liu and coworkers in 2009.188 The reaction was carried out using 10 mol% catalyst 118 and 20 mol% acetic acid in acetone over the course of 1–3 days to deliver adduct 27a in excellent yield and in 92% ee. Nájera and coworkers reported the use of prolinethioamide 119 for the direct catalytic asymmetric aldol reaction in 2008.189 The reaction conditions consisted of using 5 mol% of catalyst 119 for 2 days, yielding adduct 27a in 80% yield and 80% ee. In 2009, Da and coworkers reported the use of stilbene catalyst 120.190 Using only 1 mol% catalyst, together with an equal amount of 2,4-dinitrophenol, and 10 equivalents of acetone a 96% yield of adduct 27a was obtained in 94% ee after 20 hours. Fu and coworkers reported the use of catalyst 121 in 2009. Adduct 27a was obtained in 90% yield in 80% ee using 10 mol% catalyst loading and only two equivalents of acetone.191
Although many of these catalysts have successfully improved the enantioselectivity of the aldol reaction between acetone and para-nitrobenzaldehyde, reactivity remains a problem. Da’s catalyst 120 gives perhaps the best combination of low catalyst loadings, good yield, and good enantioselectivity amongst the reported catalysts. The catalyst can be made in three steps with 76% overall yield. There is still a great deal of room for improvement in terms of reactivity, as the reaction times are almost uniformly very long (days). The need for huge excesses of the ketone component is also a huge drawback in cases in which the ketone is of more value than acetone.
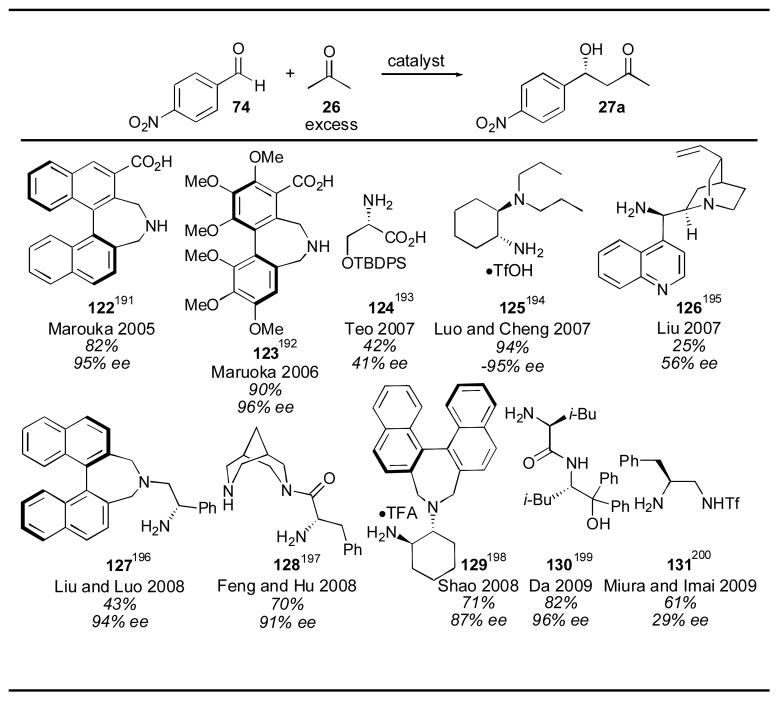
A variety of catalysts have been reported that are not based on the structure of proline (Table 10). Maruoka reported binaphthyl catalyst 122 in 2005, which catalyzes the aldol reaction between para-nitrobenzaldehyde 74 and acetone using 5 mol% 122 with 27 equivalents of acetone in 82% yield and with 95% ee over the course of 1 day.192 Later the same group reported the use of the highly substituted biphenyl catalyst 123 which could be used in as little as 0.5 mol% to give the desired adduct 27a in 90% yield and 96% ee, although the reaction took 2–3 days to complete and required the use of acetone as solvent to achieve these results.193 In 2007 Teo reported the use of siloxy serine derivative 124 for the direct catalytic aldol reaction with cyclic ketones.194 Unfortunately, the yield and selectivity were low when applied to acetone. Luo and coworkers reported the use of a primary amine catalyst 125 for the asymmetric aldol.195 Using 10 mol% catalyst with 20 equivalents of acetone, adduct 27a was obtained in 94% yield and 95% ee after 20–72 hours. Liu reported the use of cinchona alkaloid derivative 126 for use with cyclic ketones in 2007.196 When applied to acetone, the catalyst 126 gave disappointingly low yield and enantioselectivity.
Table 10.
Non-proline based catalysts for the acetone aldol reaction
|
References are given for each product as a superscript.
Binaphthyl catalyst 127 was reported by Liu and Luo in 2008.197 The catalyst 127 was used in a 10 mol% loading with 20 mol% trifluoroacetic acid using acetone as solvent to deliver adduct 27a in 43% yield and 94% ee. The rest of the material was almost entirely the aldol condensation product, rather than unreacted starting aldehyde 74. Bispidine catalyst 128 was reported by Feng and Hu in 2008 and applied to the acetone aldol with para-nitrobenzaldehyde 74.198 Using 30 mol% catalyst 128 in acetone with 30 mol% 3,3′,5,5′-tetrabromobiphenol as an additive adduct 27a was obtained in 70% yield with 91% ee after 2 days. Binaphthyl derivative 129, disclosed by Shao in 2008, was used in only 3.5 mol% catalyst loading with 20 equivalents of acetone.199 The aldol adduct 27a was obtained in 71% yield and 87% ee after 2 days. In 2009, Da reported the use of catalyst 130 for the asymmetric aldol.200 When applied to para-nitrobenzaldehyde 74 and acetone, the reaction proceeded with 82% yield and 96% ee. The reaction conditions included the use of 20 mol% catalyst 130 and 20 mol% 2,4-dinitrophenol in acetone as solvent over the course of 26 hours. Triflamide catalyst 131 was reported in 2009 for use with cyclic ketones.201 Unfortunately, when the reaction conditions were applied to acetone, both the yield and enantioselectivity were disappointingly low. Among the non-proline derived catalysts, the biphenyl catalyst 123 of Maruoka and the cyclohexanediamine catalyst 125 of Luo and Cheng stand out. Although the Maruoka catalyst 123 is the more active, the simplicity of Luo and Cheng’s catalyst 125, which can be synthesized in far fewer steps, make it an appealing choice.
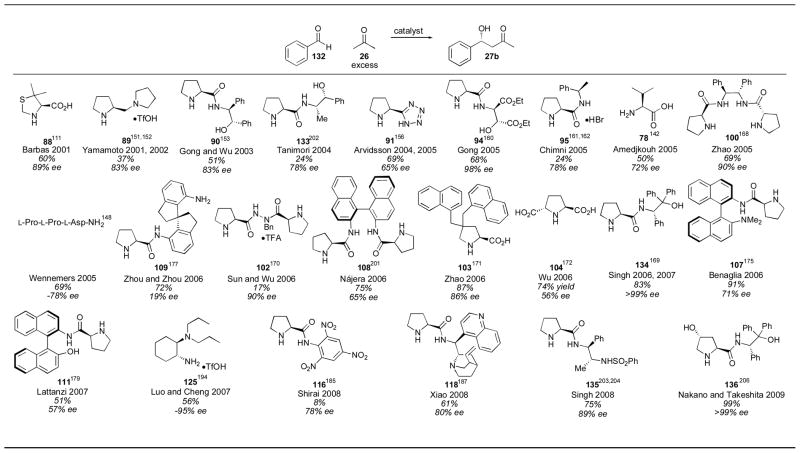
Given the high reactivity of para-nitrobenzaldehyde 74 relative to typical aldehydes, it is an excellent substrate for finding reactivity initially, but it is less useful as a measure of the general applicability of a catalyst for less reactive (and more typical) substrates. For this reason the reactivity of various catalysts using benzaldehyde (132) as an acceptor have been compared (Table 11). Proline was found to catalyze this reaction in 62% yield and in 72% ee using 30–40 mol% catalyst loading with acetone as cosolvent for 2–8 hours (Table 1). Among the catalysts described, many do not surpass this level of reactivity and enantioselectivity.142, 151, 156, 161, 162, 170, 172, 177, 179, 185, 202, 203 Among those that do are thiazole 88,111 stilbene derivative 90,153 diester 94,160 C2-symmetric catalyst 100,163 4-disubstituted proline derivative 103,171 prolinamide 134,173, 204 binaphthyl derivative 107,175 diamine 125,195 cinchona alkaloid derivative 118,187 sulfonamide 135,205 and 4-hydroxy proline derivative 136.206 The reaction conditions for most of these catalysts have already been described (vide supra) and were not changed when using benzaldehyde 132 in place of para-nitrobenzaldehyde 74. Singh’s catalyst 134 was used in an analogous fashion to derivative 105 (Table 9). Sulfamide catalyst 135, also developed by Singh,205 was used in 5 mol% loading using 4 equivalents of acetone to give adduct 27b in 75% yield with 89% ee after 20–52 hours. Among the catalysts reported with benzaldehyde 132, the 4-hydroxyproline catalyst 136 of Nakano and Takeshita stands out for its high yield and enantioselectivity.206 With only 5 mol% catalyst (using acetone as solvent) adduct 27b was obtained in 99% yield and >99% ee in 36 hours. Interestingly, although the enantioselectivity of the catalytic system remained excellent, the yield of the reaction dropped to 48% when cyclohexanecarboxaldehyde was used. Notably, although Wennemers catalyst L-Pro-L-Pro-L-Asp-NH2 gave decreased enantioselectivity, the catalyst could be used in only 1 mol% loading.
Table 11.
Use of benzaldehyde for the acetone aldol reaction.
|
References are given for each product as a superscript.
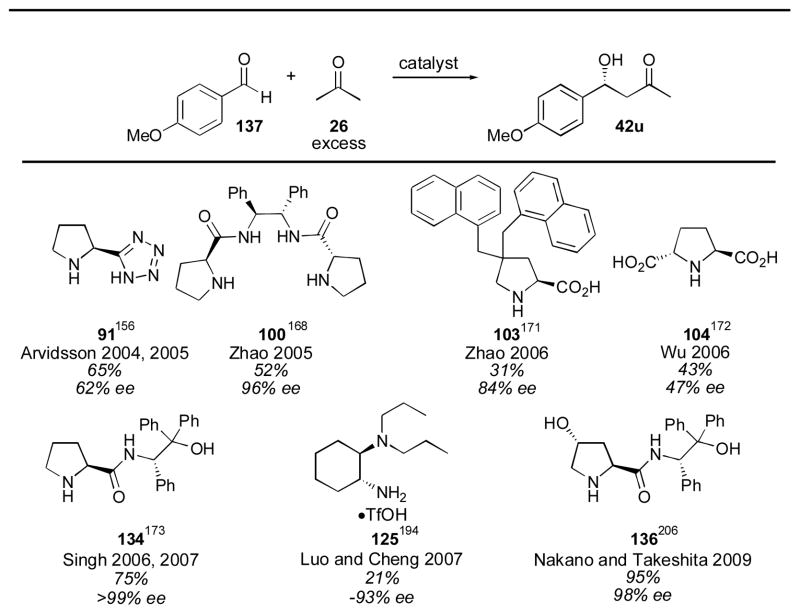
When the even more electron-rich aromatic aldehyde para-methoxybenzaldehyde 137 was used, reactivity dropped off significantly for many of the catalysts (Table 12). Singh’s prolinamide catalyst 134 and Nakano and Takeshita’s catalyst 136, using identical conditions as described above, retained their high activity and excellent enantioselectivity. These catalysts should make excellent choices for use with aromatic aldehydes when the enantioselectivity afforded by proline is deemed to be insufficient for one’s purposes.
Table 12.
Use of para-methoxybenzaldehyde for the acetone aldol.
|
References are given for each product as a superscript.
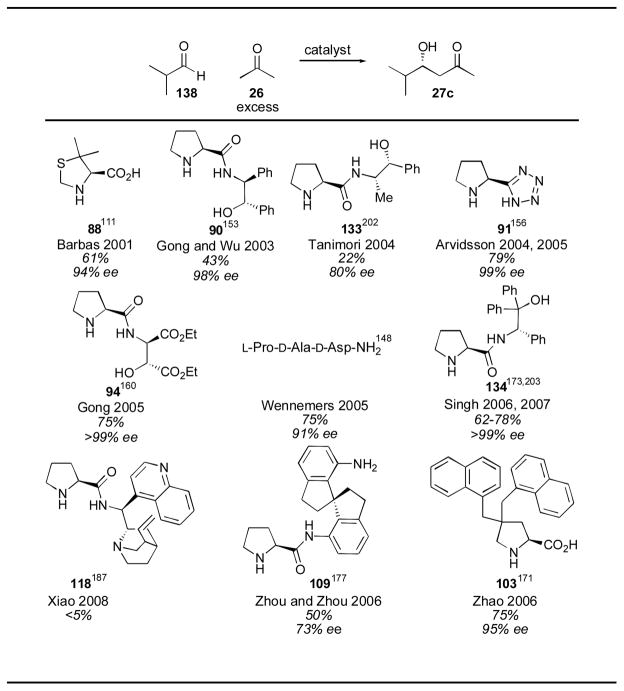
As noted in the case of catalyst 136, the results obtained can vary widely with the substrate employed. α-Branched aldehydes are among the best substrates for proline-catalysis, delivering adduct 27c in 97% yield and 96% ee (Table 1). Although many alternate catalysts give good results, they cannot improve upon the high yield and enantioselectivity, coupled with the ready availability, of proline (Table 13). α-Unbranched aldehydes are problematic in the proline-catalyzed aldol reaction. When isovaleraldehyde is used as a substrate, adduct 27d is obtained in 34% yield and 73% ee (Table 1). Very few reports of α-unbranched aldehyde substrates have been disclosed with alternate catalysts, and these show no significant improvement (Table 14).
Table 13.
Catalysts examined for the acetone aldol with isobutyraldehyde.
|
References are given for each product as a superscript.
Table 14.
Aldol reactions with α-unbranched aldehydes.
|
References are given for each product as a superscript.
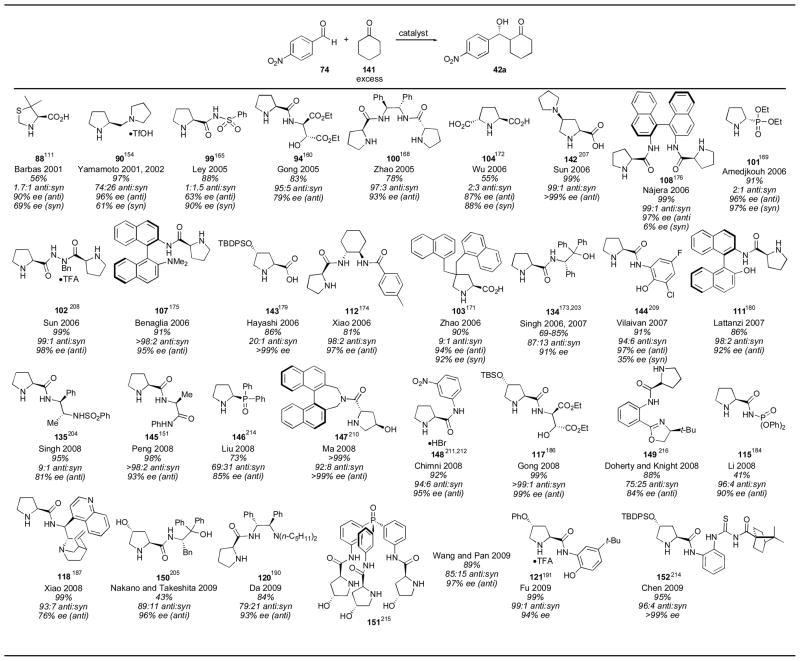
Many catalysts have been evaluated for the reaction between cyclohexanone and para-nitrobenzaldehyde (Table 15). This reaction is highly successful using proline as catalyst delivering adduct 42a in 73% yield, with a 9:1 anti:syn ratio and >99% ee (Table 2). Diastereoselectivity with cyclic ketones is not always high when employing proline, however, particularly with other aromatic aldehydes. Among the reported catalysts, stilbene derivative 100,168 pyrrolidine 142,207 binaphthyl catalyst 108,176 hydrazide 102,208 binaphthyl catalyst 107,175 silylated proline derivative 143,178 cyclohexanediamine 112,174 4-disubstituted proline catalyst 103,171 aryl prolinamide 144,209 binaphthyl derivative 111,180 prolinamide 145,146 binaphthyl catalyst 147,210 prolinamide 148,211, 212 diester 117,186 phosphine oxide 151,213 prolinamide 121191 and thiourea 152214 gave particularly good results. Of these, pyrrolidine 145, binaphthyl catalyst 147, prolinamide 148, diester 117 and thiourea 152 gave the best combination of high yield, diastereoselectivity and enantioselectivity. Phosphine oxide catalyst 151215 and oxazoline 149,216 two novel catalyst designs, did not give improved results.
Table 15.
Catalysts for the aldol reaction of cyclohexanone with para-nitrobenzaldehyde.
|
References are given for each product as a superscript.
The reaction conditions used by Zhao employing stilbene catalyst 100 were the same as used previously (10 mol% catalyst loading, vide supra).168 Sun’s pyrrolidine catalyst 117 was used in 20 mol% loading, using cyclohexanone as a cosolvent over the course of 4 hours.207 Sun also reported the use of hydrazide catalyst 77 for use with cyclohexanone as the donor. These conditions employed 20 mol% catalyst with cyclohexanone as cosolvent for 1 hour.208 Binaphthyl catalyst 82 was used in 10 mol% loading using cyclohexanone as solvent over 88 hours.175 The silylated catalyst 118 was used in 10 mol% with 5 equivalents of cyclohexanone over 18 hours.178 Xiao reported the use of 20 mol% of catalyst 112, along with 20% acetic acid, with cyclohexanone as cosolvent over the course of 6–24 hours.174 The 4-disubstituted proline catalyst 103 was used in 10 mol% catalyst loading, as before.171 Aryl prolinamide catalyst 144, reported by Sathaporvajana and Vilaivan in 2007, was used in 10 mol% loading with 2 equivalents of cyclohexanone and a reaction time of 1 day. Lattanzi reported in 2007 that 10 mol% of binaphthyl catalyst 111 with 3 equivalents of cyclohexanone yielded adduct 42a in 65 hours.180 Prolinamide catalyst 145 was used in 20 mol% with 20 mol% acetic acid using cyclohexanone as a cosolvent with a reaction time of 24–72 hours.146 Binaphthyl catalyst 147 was used by Ma and coworkers in 10 mol% loading with 5 equivalents of cyclohexanone in 72 hours.210 Chimni’s catalyst 148 was used in 20 mol% loading with 5 equivalents of cyclohexanone with a reaction time of 1 day.211, 212 Gong’s catalyst 117 was used in only 1 mol% loading with 2 equivalents of cyclohexanone, over the course of 5 hours.186 The phosphine oxide catalyst 151, containing three proline units, was used in 2 mol% loading.213 Despite this low catalyst loading, the reaction took 72–120 hours to complete. Thiourea catalyst 152 was used in 20 mol% loading with 2 equivalents of cyclohexanone over the course of 1.5 days.214 Of these catalysts, Gong’s diester catalyst 117 appears to be not only highly selective, but also the most reactive.
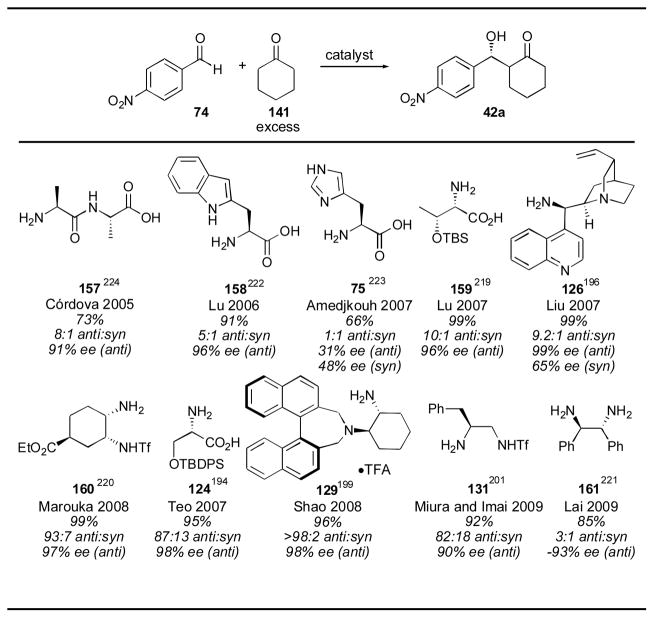
Although primary amino acids give poorer selectivity than proline for the aldol reaction in many cases, in certain instances they show improved results. In the Hajos-Parrish- Eder-Sauer-Wiechert of substrate 153 (S)-phenylalanine gave superior results for the aldol condensation (Scheme 14).217 Ethyl ketone 155 also gave superior results with (S)- phenylalanine as catalyst.218 These results suggest that with more hindered ketones, primary amines may have an advantage over secondary amines. This has led many investigators to evaluate primary amine catalysts for the reaction of cyclohexanone 141 with para-nitrobenzaldehyde 74 (Table 16). Among the catalysts examined, protected threonine catalyst 159,219 cinchona derivative 125,196 cyclohexyl catalyst 160,220 protected serine derivative 124,194 binaphthyl catalyst 129,199 triflamide catalyst 131201 and stilbene catalyst 161221 gave good results. Unprotected amino acids tryptophan 158222 and histidine 75,223 and dipeptide 157,224 gave inferior results. The conditions used for the threonine catalyst 159, as reported by Lu and coworkers, required only 2 mol% catalyst loading and 2 equivalents of cyclohexanone for 20 hours.219 The cinchona derivative 126 was used in 10 mol% loading with 15 mol% triflic acid, using cyclohexanone as solvent over the course of 9 hours.196 Maruoka’s catalyst 160 was used in 5 mol%, though the reaction took 3–4 days.220 Shao’s catalyst 129 could be used in only 3.5 mol% with 3 equivalents of cyclohexanone over 10 hours.199 Triflamide catalyst 131 was used in 10 mol% loading with 10 equivalents of cyclohexanone over the course of 2 days.201 Lai’s catalyst 161 was used in 10 mol% with 10 equivalents of cyclohexanone over the course of 12 hours.221 Among these catalysts, the silylated threonine catalyst 159 gives perhaps the best combination of good reactivity, good selectivity, and ease of synthesis.
Table 16.
Non-proline derived catalysts for the aldol reaction of cyclohexanone with para-nitrobenzaldehyde.
|
References are given for each product as a superscript.
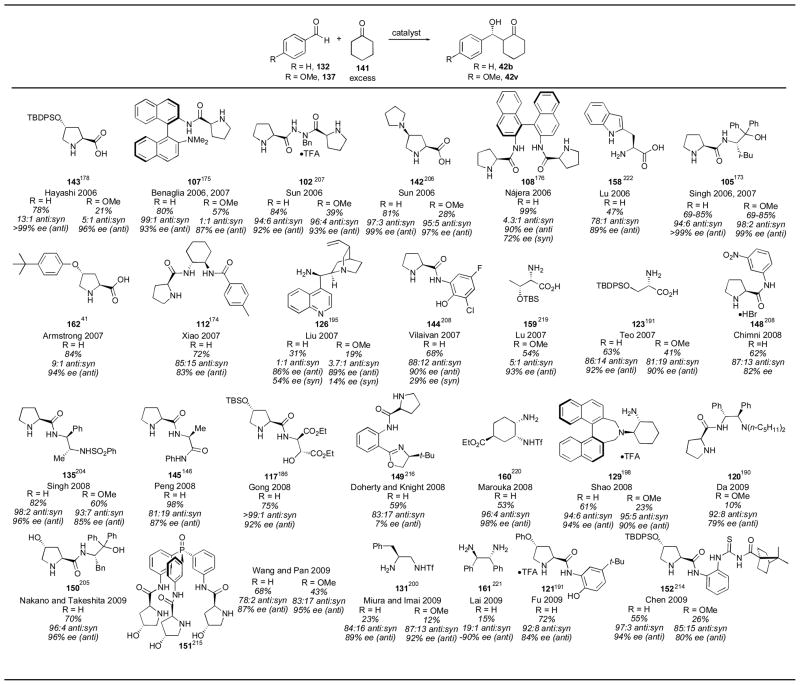
With less reactive aldehydes such as benzaldehyde 132 or para-methoxybenzaldehyde 137, much poorer results were obtained with most of the catalysts (Table 17). Singh’s catalyst 105 retained good reactivity, however, even with para-methoxybenzaldehyde 137.173 Armstrong’s aryl ether catalyst 162 also showed good reactivity, and was needed in only 2 mol% loading with only one equivalent of cyclohexanone, though the reaction still took 2 days to complete.41 The cyclohexyldiamine catalyst 112,174 anilide catalyst 144,209 protected threonine derivative 159,219 sulfamide 134,205 dipeptide catalyst 145,146 diester catalyst 117,186 4-hydroxyproline derivative 150,206 and Fu’s catalyst 121191 all gave fair results as well. The reaction conditions for these catalysts have been given above.
Table 17.
Catalysts for less reactive aldehydes with cyclohexanone.
|
References are given for each product as a superscript.
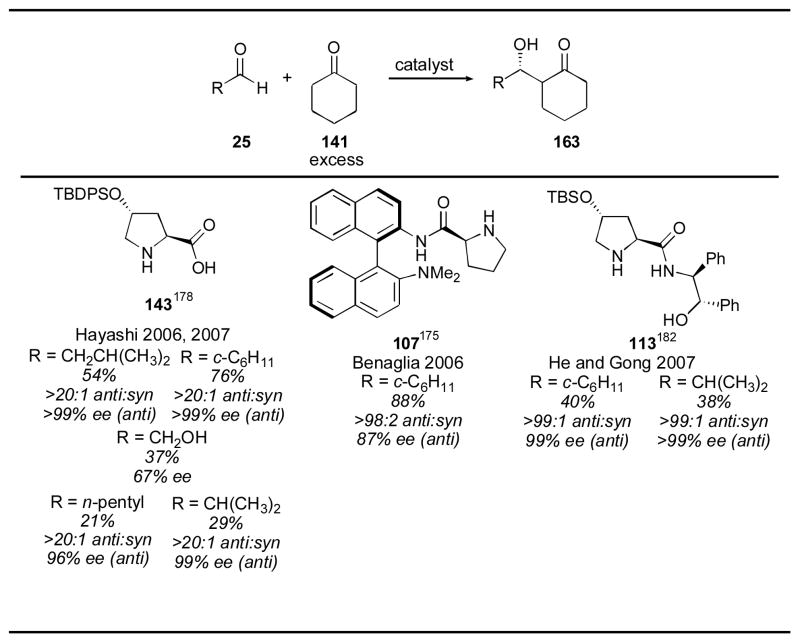
Proline is an excellent catalyst for aldol reactions of cyclohexanone with alkyl aldehydes, especially α-branched aldehydes (Table 2). Use of more elaborate catalysts has been reported, but with generally worse results than using proline itself (Table 18). Silyl derivative 143 and binaphthyl catalyst 107 give high yields for cyclohexanecarboxaldehyde, but other aldehydes give poor yields, though with excellent selectivities. Proline also gives high diastereo- and enantioselectivity, however, and remains the preferred catalyst for such substrates, due to its ready availability.
Table 18.
Aldol reactions with alkyl aldehydes.
|
References are given for each product as a superscript.
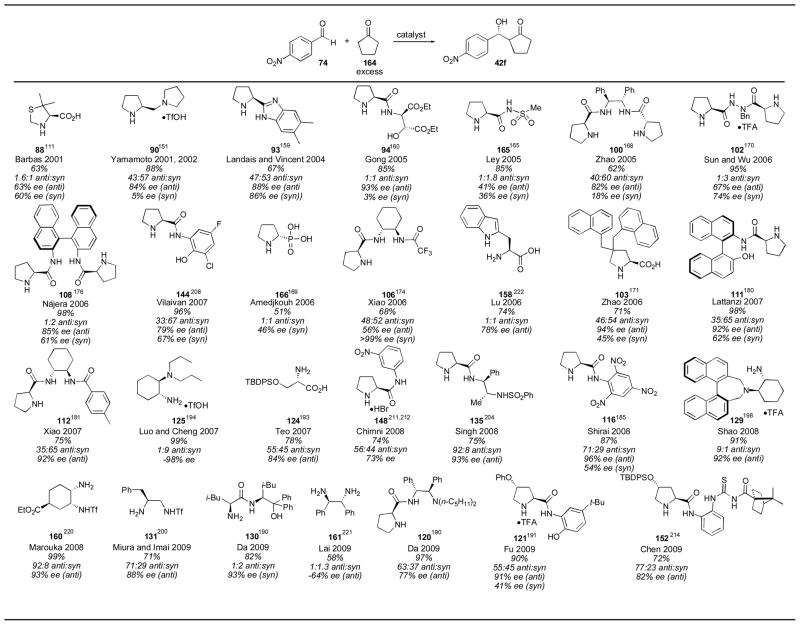
When cyclopentanone is used as a donor in the proline-catalyzed asymmetric aldol reaction poor diastereoselectivity is observed. Many of the developed catalysts have been applied to this substrate, and fortunately, several catalysts were found with improved diastereoselectivity for the anti diastereomer (Table 19). Singh’s sulfamide catalyst 135,205 Shirai’s trinitrocatalyst 116,185 Shao’s binaphthyl catalyst 129,199 Maruoka’s cyclohexyl catalyst 160,220 Miura and Imai’s triflamide catalyst 131201 and Chen’s thiourea catalyst 152214 all showed improved diastereoselectivity over the 1:1 selectivity observed for proline. Of these catalysts, Maruoka’s gives the best balance of selectivity and yield. Interestingly, several catalysts showed selectivity for the syn adduct 42f, including hydrazide 102,170 binaphthyl catalyst 108,176 anilide catalyst 144,209 cyclohexanediamine catalyst 125195 and Da’s stilbene catalyst 130.190 Luo and Cheng’s catalyst 125 is remarkable in its selectivity, and should prove quite useful for the synthesis of syn products.
Table 19.
Catalysts used in the aldol reaction between cyclopentanone and para-nitrobenzaldehyde.
|
References are given for each product as a superscript.
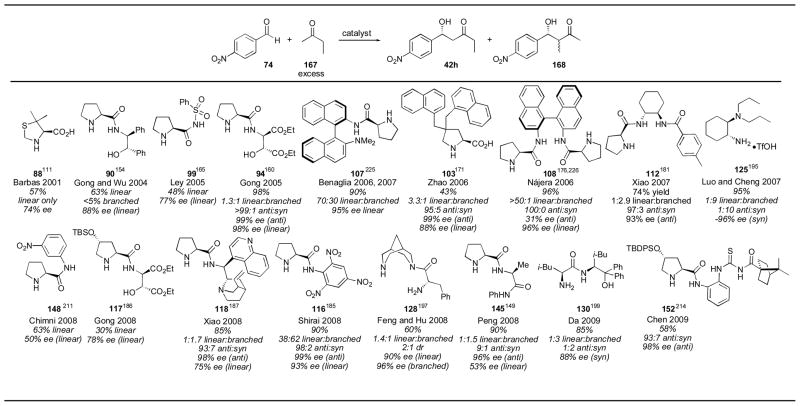
When proline was used with 2-butanone 167 and para-nitrobenzaldehyde 74, the linear adduct 42h was obtained in 65% yield and 77% ee (Table 2). Many catalysts have been applied to this reaction enhancing both the yield and selectivity of the linear adduct 42h, but also provided access to both diastereomers of the branched adduct 168 (Table 20). The linear product was favored by catalysts 88,111 90,154 99,165 94,160 107,225 103,171 108,176, 226 148,211 117,186 and 128.198 Among these catalysts, Nájera’s binaphthyl amine 108 provided the linear adduct 42h with the best yield and selectivity. Xiao’s cyclohexanediamine catalyst 112, on the other hand, gave the branched product 168 with excellent selectivity for the anti diastereomer, though with only moderate selectivity for the branched product over the linear.181 The enantioselectivity for catalyst 112 was also high. Trinitroanilide catalyst 116 also favors the anti adduct, but the reaction conditions are less appealing, as HMPA is used as solvent.185 Thiourea catalyst 152 also provide the anti adduct in good selectivity.214 Luo and Cheng’s cyclohexanediamine catalyst 125 gave the syn adduct in excellent selectivity and yield.195 With these developments, each aldol adduct isomer can be synthesized with good selectivity.
Table 20.
Catalysts used in the aldol reaction between 2-butanone and para-nitrobenzaldehyde.
|
References are given for each product as a superscript.
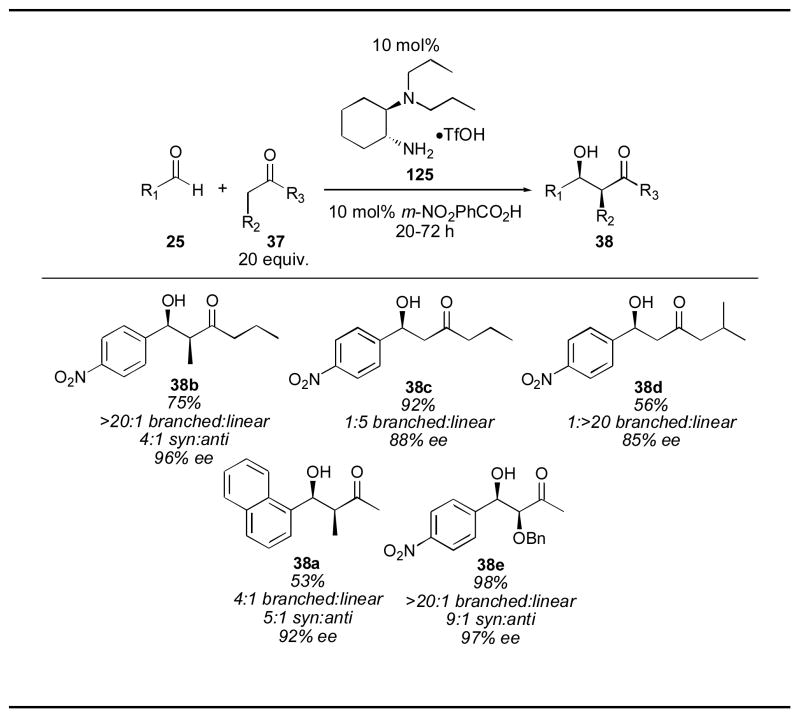
Luo and Cheng’s diamine catalyst 125 displays very interesting selectivity (Table 21). When unsymmetrical ketones are employed, such as 2-butanone 167, the adduct (38a for example) resulting from reaction through the more substituted enamine is observed, producing the syn branched isomer 38. When 3-hexanone is used, the adduct resulting from the least sterically-hindered enamine (38b) was formed almost exclusively. When 2-pentanone was used, a competition between reaction through the more reactive highly-substituted enamine and the less sterically-hindered enamine. The major product resulted from reaction through the less hindered enamine, yielding adduct 38c in 5:1 selectivity. When the ketone was made even more hindered, the selectivity increased dramatically, giving adduct 38d. Benzyl-protected hydroxyacetone also gave excellent selectively for the syn branched product 38e. Luo and Cheng’s catalyst 125 provides a very useful selectivity unavailable from proline.
Table 21.
Lou and Cheng’s syn selective aldol.195
|
When hydroxyacetone is used as a donor under proline-catalysis, good diastereoselectivity is observed with α-branched aldehydes, favoring the anti branched isomer, but some aryl (particularly ortho-chlorobenzaldehyde) and α-unbranched aldehydes result in poor diastereoselectivity. Silyl protected hydroxyacetone, as well as methylated derivatives,227 also gave the anti branched isomer selectively. A variety of catalysts have been tested with hydroxyacetone and protected derivatives (Table 22). When hydroxyacetone was used as substrate, diester catalyst 95 gave excellent selectivity for the linear isomer, with excellent enantioselectivity.228 Other catalysts were not very selective with unprotected hydroxyacetone.111, 168, 224, 229 Wu and Zhao’s primary amine catalyst 175 gave the syn isomer in 4:1 selectivity with 80% ee.230 Luo and Cheng’s catalyst 176 gave even higher selectivity for the syn isomer.231 Silyl protected hydroxyacetone was also used as a substrate with threonine derivatives 173219 and 174,232 both of which favored the syn branched isomer in excellent enantioselectivity. Methyl and benzyl protected hydroxyacetone were also used, and depending on the chosen catalyst, could be obtained in either the anti or syn configuration. Binaphthyl catalysts 108,227, 233 172,175 and 111,180 as well as thioamide 119189 all gave the anti isomer selectively. The syn isomer was obtained when threonine derivative 174 or primary-tertiary amine catalyst 176 was used. All three isomers are now available through judicious choice of protecting group and catalyst.
Table 22.
Hydroxyacetone aldol reactions and protected derivatives.
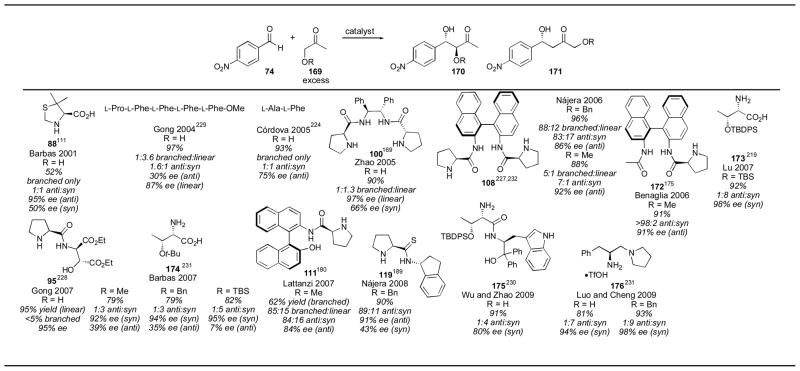
|
References are given for each product as a superscript.
Dihydroxyacetone and its protected derivatives have also been used with many different catalysts. Proline gives the anti derivative with excellent selectivity with a variety of aldehydes. When untethered dihydroxyacetone derivatives are used only binaphthyl catalyst 108 gave the anti isomer with moderate selectivity (Table 23).227 The syn isomer, on the other hand, is favored by a variety of primary amine catalysts including threonine derivative 174,232 cyclohexyl catalyst 125,195, 234 primary-tertiary amine catalyst 176,231 and amino alcohols 179,235 180,60 and 130.200 When tethered dihydroxyacetone derivative 2,2-dimethyl-1,3-dioxan-5-one (177, R = -CMe2-) was used, only the anti isomer was observed with all of the catalysts.236 When the anti isomer is desired, proline should be the catalyst of choice, as it gives good selectivity as is inexpensive. When the syn isomer is desired, a number of primary amine catalysts can be used. Diamine catalyst 125 is particularly useful, as it can be used in the lowest catalyst loading (10 mol%).
Table 23.
Dihydroxyacetone aldol reactions.
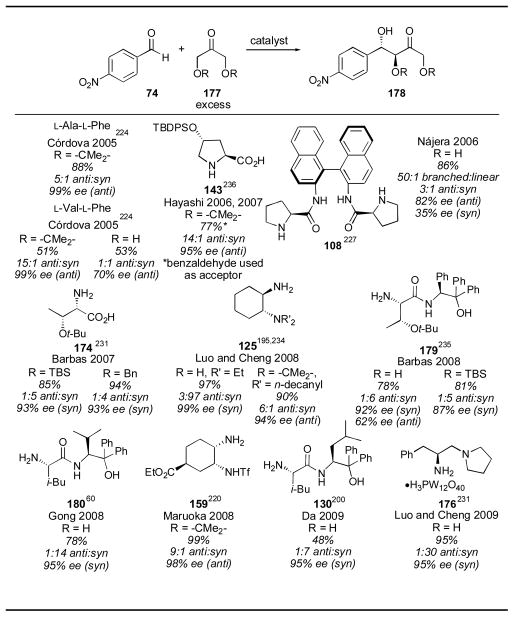
|
References are given for each product as a superscript.
Thioether and halogenated ketone derivatives have also been examined (Table 24). When α-chloroacetone was used as a substrate, the anti branched product was obtained in excellent selectivity with a variety of catalysts,180, 237 though only good yield was obtained with the binaphthyl catalyst 108180, 238 and the trinitroanilide catalyst 116.185 α-Fluoroacetone was used as a substrate with Gong’s diester catalyst 94,228, 239 and was capable of yielding either the branched product in 2:1 dr or the linear product, depending on the reaction conditions. When water was added, the linear adduct 183 was favored, while running the reaction in dry THF gave the branched adduct 182. A thioether-substituted ketone was also used as a substrate, both with the diester catalyst 116 and with binaphthyl catalyst 108. Both catalysts favor the linear product 183, with diester catalyst 116 giving slightly better selectivity.
Table 24.
Halogenated and sulfur-containing substrates.
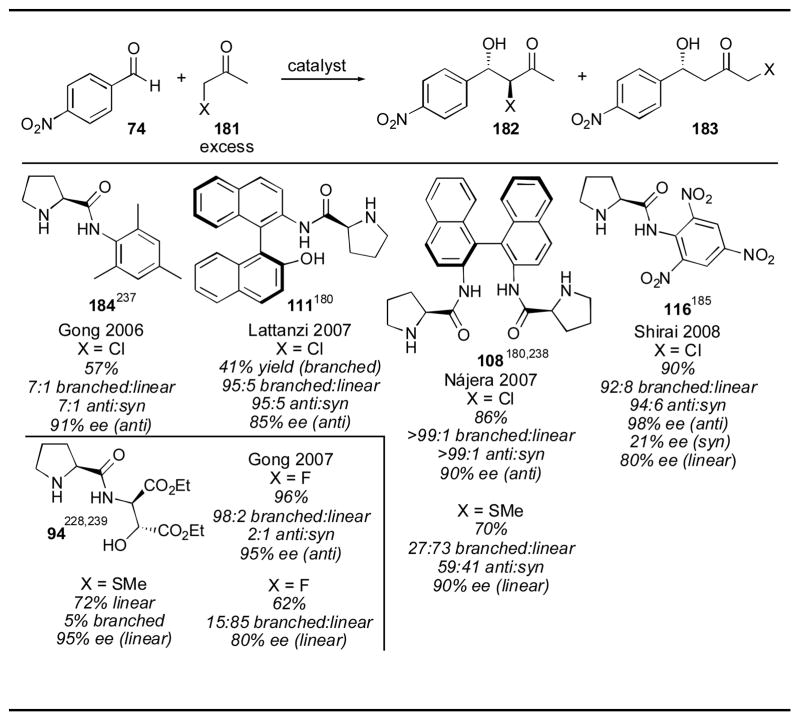
|
References are given for each product as a superscript.
A number of interesting substrates other than aryl and alkyl aldehydes have been employed for the direct catalytic aldol reaction (Scheme 15). The use of enals 185 and 186 was reported by Maruoka using binaphthyl amino acid 123 in the aldol reaction with acetone.192 The adducts were obtained in 73% yield with 90% ee and 81% yield with 96% ee, respectively. Saito and Yamamoto demonstrated the use of chloral 187 and trifluoroacetaldehyde ethyl hemiacetal 188 as acceptors using tetrazole catalyst 91 in 2004. The authors used a variety of ketones with these acceptors, including α-ketoester 189 and methyl aryl ketones 190 with yields between 55–93% and with 82–97% ee.157 Zhang and Wang have used methyl aryl ketones 190 as well, employing triflamide catalyst 191.240 Trifluoroacetaldehyde ethyl hemiacetal 188 was also used by Gong as an acceptor using cyclohexanone as the donor. The authors found that prolinamide catalyst 82 gave the best results.241 Acetal derivatives 192 were employed by Luo and Cheng using cyclohexanediamine catalyst 125, giving the desired adducts in 30–98% yield and with 49–99% ee.242 Ynone donor 193 was used by Gouverneur and coworkers with aromatic aldehydes as acceptors employing sulfonamide catalyst 99. The adducts were obtained in 26–90% yield, 3:1–19:1 dr, and 77–95% ee.243 The reaction appears to be limited to electron-poor aryl aldehydes. This collection of interesting substrates should prove useful for the synthesis of more functionalized products.
Scheme 15.
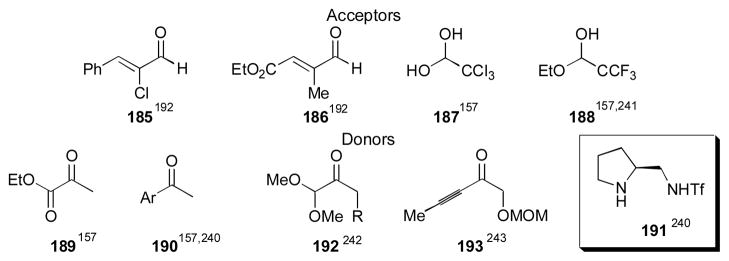
Additional acceptors and donors.
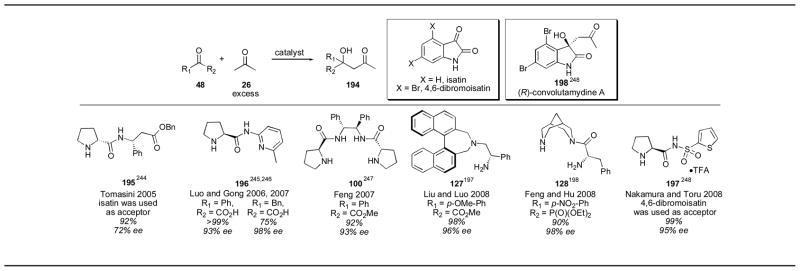
There have been a few reports of ketone electrophiles in the direct catalytic asymmetric aldol reaction (Table 25). Tomasini used catalyst 195 with isatin, a highly reactive and non-enolizable ketone to give the aldol adduct 194 in 92% yield and 72% ee.244 Luo and Gong developed pyridine catalyst 196 for the aldol reaction of acetone with α-keto acids.245, 246 The aldol adducts were obtained in excellent yield and enantioselectivity, even when an enolizable substrate was used. Feng reported the use of stilbene catalyst 100 with α-keto esters.247 When R1 = Ph and R2 = CO2Me, the aldol adduct was obtained in 92% yield and in 93% ee. Binaphthyl catalyst 127 also was used with α-keto esters, for example, when R1 = pMeO-Ph and R2 = CO2Me, the aldol adduct was obtained in 98% yield and 96% ee.197 Bispidine catalyst 128 was used with benzoyl phosphonates.198 When R1 = p-NO2-Ph and R2 = P(O)(OEt)2, the α-hydroxyphosphonate was obtained in 90% yield and in 98% ee. Thiophene catalyst 195 was used for substituted isatins, such as 4,6-dibromoisatin, which yielded the desired adduct 194 in 99% yield and 95% ee.248 Isatin itself gave the aldol adduct in 99% yield, however, the product was only obtained in only 3% ee. Nakamura and Toru used this methodology for the synthesis of (R)-convolutamydine A. Non-enolizable and highly reactive ketone derivatives have been used successfully with proline as well, which should be the first choice of catalyst for most substrates (Table 3).
Table 25.
Ketone electrophiles in the acetone aldol reaction.
|
References are given for each product as a superscript.
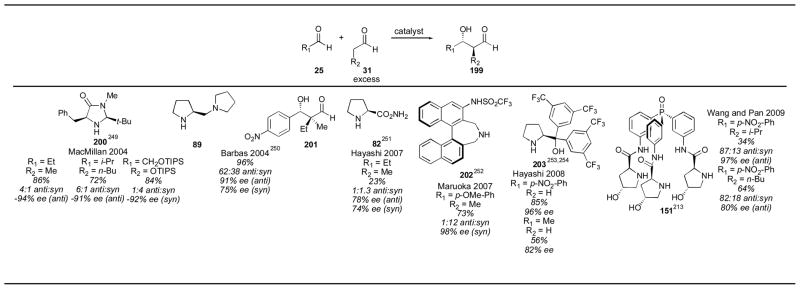
Aldehyde cross-aldol reactions have been achieved with a number of different catalysts (Table 26). MacMillan first described the cross-aldol reaction with proline,130 and later reported the use of catalyst 200 for the reaction.249 The reaction proceeds with good yield and enantioselectivity for a wide array of substrates, favoring the anti isomer. Proline, however, is also a good catalyst for this reaction, and is a simpler catalyst (Table 4). Barbas reported the use of diamine 89, together with an equal amount of trifluoroacetic acid, for the creation of quaternary stereocenters adjacent to an aldehyde (201).250 The reaction works well with electron poor aryl aldehydes, but the yields are lower for benzaldehyde and other more electron rich aryl aldehydes. The diastereoselectivity is only moderate with this catalyst, and future developments should improve this highly useful reaction. Hayashi reported the use of prolinamide 82 for the self-aldol reaction of propanal in an aqueous environment, which was followed by reduction of the resulting aldehyde with sodium borohydride to give the diol.251 Unfortunately low diastereoselectivity and yield was obtained under these conditions. Maruoka’s binaphthyl catalyst 202 gave very impressive results when applied to the cross-aldol reaction.252 Unlike proline, catalyst 202 produces the syn diastereomer in good diastereoselectivity, yield and enantioselectivity. This provides a complement to the proline catalyst, making the selective production of either diastereomer possible. Prolinol 203, reported by Hayashi in 2008, has been reported for the use of acetaldehyde in self-aldol and cross-aldol reactions.253, 254 Hayashi demonstrated that proline itself was an inefficient catalyst for this reaction (which was followed by sodium borohydride reduction of the resulting aldehyde) giving very little of the desired adducts. The use of catalyst 203 allows the substrate scope of enamine-catalyzed cross-aldol reactions to be extended to the use of acetaldehyde. Wang and Pan have also used their phosphine oxide catalyst 151 for the cross-aldol reaction, but this catalyst appears to have no advantage over use of the far simpler catalyst proline.213 The aldehyde self- and cross-aldol reaction may now be used for a variety of aldehydes, including α-branched and α-unbranched aldehydes, and even acetaldehyde. The syn diastereomeric products are also now available, providing access to an ever- growing family of products.
Table 26.
Aldehyde donors in self-aldol and cross-aldol reactions.
|
References are given for each product as a superscript.
Enamine-catalysis is a burgeoning field, and its impact on the direct aldol reaction is far-reaching. The scope of this reaction has been extended from the original report of acetone as a donor to include more substituted ketones, cyclic ketones, unsymmetrical ketones, β-keto esters, aryl ketones, ynones, and even aldehydes. The acceptor component can be a variety of different aldehydes and highly reactive ketone derivatives. Importantly, the selectivity of the reaction has been extended to include production of isomers not observed when proline is used, through the development of alternate catalysts. Selective formation of linear and branched products is possible, as well as control over the diastereoselectivity. One of the remaining challenges is the development of more reactive catalysts, as the need for excess donor component, high catalyst loadings, and long reaction times remains a problem for most of the described methods.
3. Non-Enamine Organocatalysts for the Aldol Reaction
Several reports of tertiary amines and quaternary ammonium salts for use aldol reactions have been reported. Some of the catalytic systems reported for the aldol reaction reported in this section use stoichiometric or even larger amounts of base, and are therefore not direct aldol reactions. This includes phase-transfer catalysts that use hydroxide-based aqueous phases. The tertiary amine catalysts can act through either of two mechanisms, formation of a chiral enolate (Scheme 16, equation 1), or formation of a tight ion pair (equation 2), followed by aldehyde addition. The quaternary ammonium salt catalyzed processes proceed through an ion pair mechanism (equation 3). Formation of the enolate requires the counterion to act as a base, limiting the substrate scope to highly acidic substrates, or requiring the use of stoichiometric base. The substrate scope for these types of processes is typically much smaller than that available to enamine- catalyzed processes due to mechanistic requirements.
Scheme 16.

Potential mechanisms for the direct aldol catalyzed by tertiary amines and quaternary ammonium salts.
Romo and coworkers reported an intramolecular aldol-lactonization protocol in 2001 for the generation of bicyclic β-lactones (Scheme 17).255–257 The mechanism of this process is proposed to go through an iminium enolate as shown in equation 1 (Scheme 16). This reaction uses super- stoichiometric amounts of Hünig’s base, as well as Mukaiyama’s reagent (2-chloro-1-methyl-pyridinium iodide)258 in order to activate the carboxylic acid and therefore is not a direct aldol process. The authors note that Hünig’s base was sufficiently hindered so as not to cause a significant background reaction. The adducts 211 are obtained in 70–82% yield with 91–98% ee using syringe-pump addition of the substrate 209. Related intermolecular reactions have been reported by Wynberg259, 260 and Romo.261, 262
Scheme 17.

One of the most popular substrate classes for phase-transfer catalysis are the glycine Schiff bases, such as tert-butyl ester 212. In 1991 Gasparski and Miller reported the first aldol reaction of a glycine Schiff base using cinchona alkaloids as catalysts (Scheme 18).263 The process favored the syn adduct 214 with about 2:1 selectivity, but with almost no enantioinduction. Since that time several new catalysts have been reported for this reaction, but the enantioselectivity of these processes remains low (Scheme 19).264, 265 It is noteworthy that either diastereomer of adduct 214 can be favored (though in a modest excess) by choosing the appropriate catalyst.
Scheme 18.

Cinchona alkaloid-catalyzed aldol reactions of glycine Schiff bases.263
Scheme 19.

A significant improvement to the enantioselectivity of the aldol reactions employing glycine Schiff bases was reported by Maruoka in 2002 (Scheme 20).266, 267 The chiral C2- symmetric quaternary salts designed by Maruoka are not based on the cinchona alkaloid family. Using only 2 mol% of catalyst 217 delivers the desired aldol adducts 214 with 40–78% yield. The anti adduct is favored and the enantioselectivity is better than the previous reports employing cinchona-derived catalysts.
Scheme 20.

Maruoka reported several further modifications of the catalyst, one of which, catalyst 218, favors the syn adduct, providing a complement to the original catalyst (Scheme 21).268, 269 These catalysts take numerous steps to synthesize and have a rather large molecular weight. Fortunately, they can be used in only 1 mol% catalyst loading. These catalysts provide significantly better results than the more easily prepared cinchona-derived catalysts.
Scheme 21.
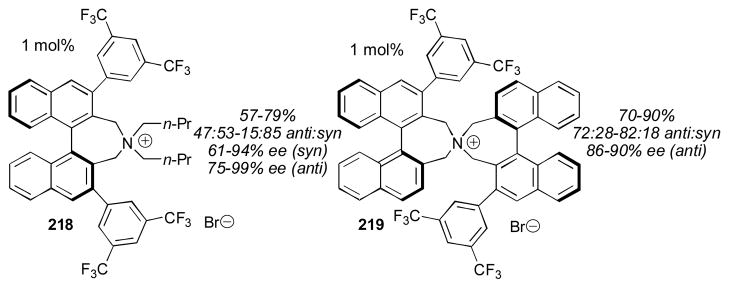
Additional non-cinchona derived quaternary ammonium salts.268,269
In 2009 Mlynarski and coworkers reported the use of quinidine for the direct catalytic aldol reaction of hydroxyacetone 220 (only 2 equivalents of the donor were needed) with a variety of aldehydes 25 (Scheme 22).270 The aldol adducts 222 were isolated in 11–96% yield (with aliphatic aldehydes giving the lowest yields) with the syn diastereomer favored in up to 9:1 selectivity. A syn selective direct aldol employing hydroxyacetone as the donor is not currently available using enamine catalysis, and would be valuable. Unfortunately, the enantioselectivity of the quinidine-catalyzed (221) process was rather low.
Scheme 22.
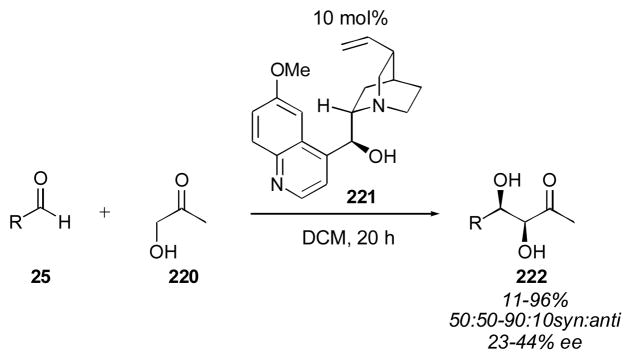
Quinidine-catalyzed direct aldol of hydroxyacetone.270
Arai and Nishida reported the use of cinchona alkaloid-derivatives for the aldol reaction of diazo ester 223 in 2004 (Scheme 23).271, 272 The reaction requires long reaction times and the enantioselectivity is highly dependent on the aldehyde used. para-Methoxybenzaldehyde, for example, gave racemic product, while 1-naphthaldehyde gave the product in 79% ee. This reaction also employs rubidium hydroxide as the basic aqueous phase, which is fairly expensive. The authors demonstrated that these diazo products 225 could be reduced to give amino acids.
Scheme 23.
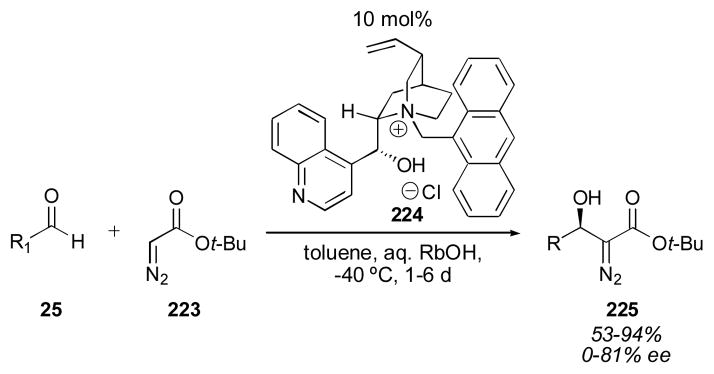
Diazo ester aldol catalyzed by cinchona alkaloid derivative 224.271,272
The use of highly activated ketone electrophiles has also been demonstrated. Calter and coworkers reported an aldol-cyclization reaction in 2005 catalyzed by cinchona dimer 228 (Scheme 24).273 The authors propose a mechanism in which the α-keto ester 226 is activated by hydrogen-bonding to protonated catalyst 228, followed by rate-determining attack of the enol form of diketone 227. This reaction could also proceed via an ion-pair mechanism (equation 3, Scheme 16). Proton sponge was used to neutralize the equivalent of hydrogen bromide that is generated in this reaction. Tetrabutylammonium bromide was found to enhance, rather than deteriorate the enantioselectivity, suggesting that an ion-pair mechanism is not likely, as achiral ammonium salts would be expected to give a background reaction. The reaction produced bicyclic compounds 229 in excellent yields and enantioselectivities, favoring the cis diastereomer.
Scheme 24.
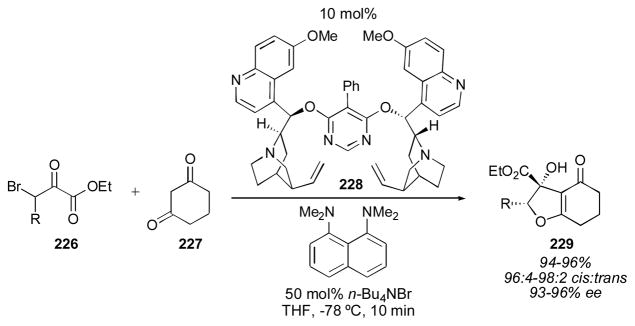
Cinchona alkaloid-catalyzed aldol-cyclization.273
In 2007 Shibata and Toru reported the use of oxindoles 231 as donors with ethyl trifluoropyruvate 230 as the acceptor (Scheme 25).274 This reaction is a direct catalytic aldol reaction, as there is no need for stoichiometric reagents. A variety of cinchona alkaloids were screened for the reaction, and (DHQD)2PHAL 232 was found to give high yield, diastereo- and enantioselectivity. (DHQ)2PHAL was used to provide access to the enantiomer of adduct 233 with similar levels of selectivity. The authors note the importance of the trifluoromethyl group, both for reactivity and enantioselectivity. An ion-pairing mechanism is proposed for this transformation (equation 3, Scheme 16).
Scheme 25.
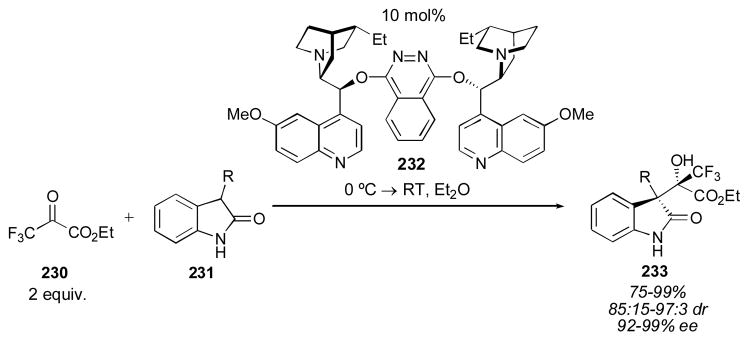
Oxindole aldol reaction with ethyl trifluoropyruvate.274
Tertiary amines and quaternary ammonium salts have been used successfully for a number of aldol reactions, although most of these processes require stoichiometric or superstoichiometric amounts of base. The syn selective hydroxyacetone direct aldol reaction described by Mlynarski is among those reactions that does not require stoichiometric base. This reaction could prove useful if the enantioselectivity were improved as a complement to the reactivity observed with proline-catalysis. The oxindole chemistry is also noteworthy, as these motifs are frequently found in biologically-active compounds.
4. Metal-Catalyzed Aldol Reactions
A variety of metal-catalyzed processes have been reported for the direct catalytic asymmetric aldol reaction. These can be compared mechanistically to the type II aldolases, which employ a zinc ion to acidify the α-proton of the donor component to form an enolate. Many of the catalysts described in this section function by dual activation of the donor and the acceptor, and are therefore classified as bifunctional catalysts. Numerous reviews of this type of activation mode have appeared, particularly focusing on BINOL-based systems.275–278 These catalysts are frequently milder than enamine-based catalysts as they do not employ nucleophilic amines that may undergo side reactions, allowing for the use of sensitive substrates such as methyl vinyl ketone and ynones.
4.1 Precious Metals as Catalysts for the Aldol Reaction
One of the first direct catalytic aldol reactions was reported by Ito, Sawamura and Hayashi (Scheme 26).279–281 This gold-catalyzed reaction of α-isocyanocarboxylate 234 with aldehydes 25 produces trans oxazolines 236 in good yield, diastereo- and enantioselectivity. The gold catalyst is believed to bind both the enolate and the aldehyde, with the ligand acting as a base to generate the enolate (A). This reaction has been developed extensively, expanding the scope of the donor to include more substituted derivatives as well as substrates in which the ester group is replaced with sulphonic and phosphonic esters.8, 282–303
Scheme 26.
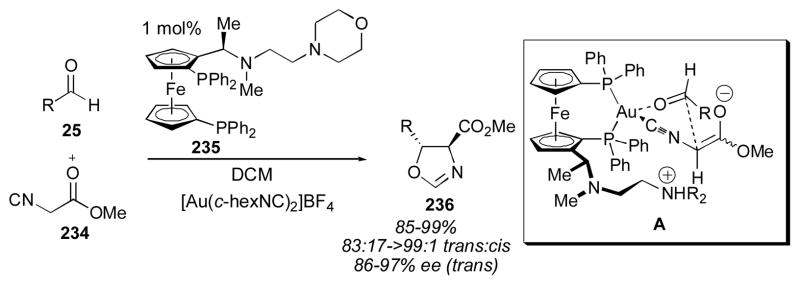
Silver has also been used with ferrocenyl ligand 235 to catalyze the direct aldol reaction (Scheme 27).304, 305 Slow addition of the donor is required to maintain high enantioselectivity when α-isocyanocarboxylates 234 were used. Unlike the corresponding gold catalyst, the silver complex resting state is tetracoordinate when a high concentration of the α-isocyanocarboxylate 234 is present, and this complex leads to the production of adduct 236 in decreased selectivity. Palladium catalysts have also been used for this transformation (Scheme 28).306–314 The reported enantioselectivities are much lower than those obtained with gold. These complexes also favor the trans isomer.
Scheme 27.
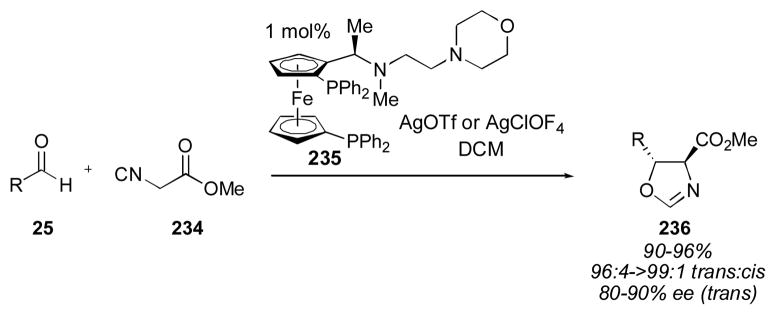
Scheme 28.
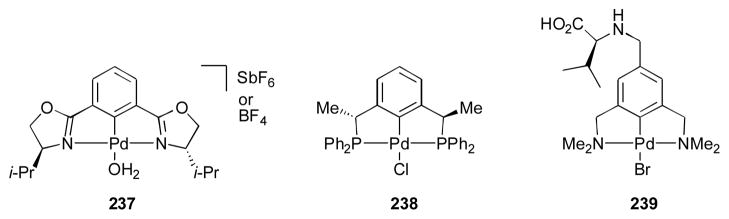
Palladium catalysts for the direct aldol of α-isocyanocarboxylates and aldehydes.306–314
Rhodium has also been used to catalyze direct aldol reactions. Substituted α-cyanocarboxylates have been employed with 1 mol% of a ferrocenyl rhodium complex to deliver adducts 242 bearing a quaternary carbon center in up to 91% ee (Scheme 29).315 The reaction was not very general, for example, no reaction was observed with benzaldehyde. The bulky ester was necessary to achieve these levels of enantioselectivity. Kuwano and Ito propose coordination of the acceptor through the cyanide nitrogen, followed by attack on the aldehyde through an open transition state.
Scheme 29.
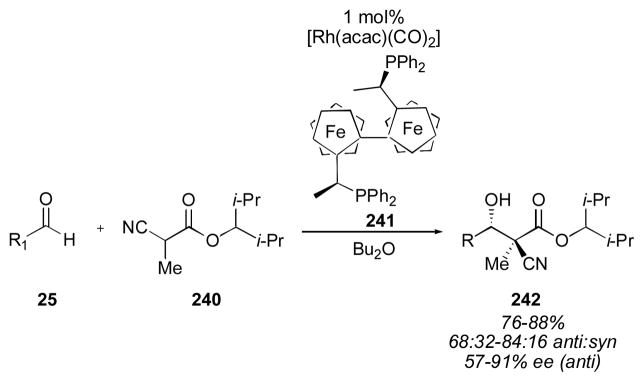
Rhodium-catalyzed direct aldol reaction of α-cyanocarboxylates.315
In 2007 Nishiyama reported the use of a rhodium-Phebox complex for the direct catalytic asymmetric aldol reaction of cyclohexanone, cyclopentanone, and acetone with aromatic aldehydes 243 (Scheme 30).316 Recently, the same catalyst has been reported for the use of cyclohexenones and cyclopentenones as donors, though with generally low yields.317 When acetone was used with para-nitrobenzaldehyde the adduct 245 was obtained in 63% yield with 74% ee. Cyclohexanone gave 57% yield in a 11:89 syn:anti ratio with 86% ee for the anti product 245 when para-trifluoromethylbenzaldehyde was used. With cyclopentanone, the adduct 245 was obtained in 75% yield in a 20:80 syn:anti ratio with 91% ee for the anti isomer. The authors propose structure A as the transition state to account for the observed selectivity. Though these results are fairly good, particularly the ability to use α,β-unsaturated donors, use of an expensive metal catalyst is unlikely to supplant the use of an inexpensive catalyst such as proline. The reaction was not applied to electron-rich aromatic substrates for which proline is a poor catalyst, but given that the reactions with cyclohexanone and cyclopentanone took 72 hours with electron-poor substrates at 60 °C, this system likely is not reactive enough for such substrates.
Scheme 30.
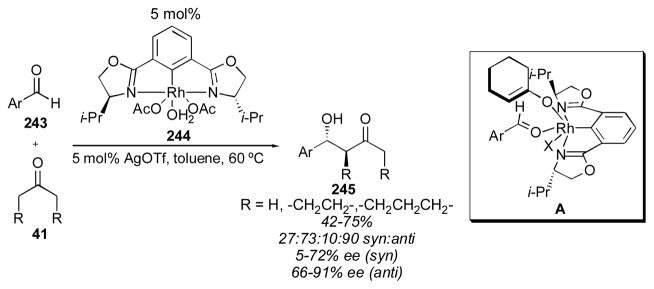
Rhodium-catalyzed aldol reaction of cyclohexanone and cyclopentanone.316
4.2 BINOL-Based Catalysts
Numerous catalysts have been developed that are based on the structure of BINOL, most notably, those designed and developed by Shibasaki. Shibasaki’s first report on catalysis for the direct aldol reaction, however, employed a TADDOL-based complex (Scheme 31). In 1992 Shibasaki reported the use of a catalyst generated from yttrium trichloride, a lithiated TADDOL ligand (246), and sodium tert-butoxide, for the Hajos-Parrish-Eder-Sauer-Wiechert reaction of triketone 12, the first report of a direct catalytic asymmetric intramolecular aldol other than proline.318 The adduct 247 was obtained in 60% yield with 52% ee after 6 days at −52 °C. The authors note that the ee of the isolated product 247 was degraded when exposed to the catalyst at −30 °C, suggesting a retro-aldol reaction.319 This provided a proof of concept that rare-earth alkoxides could be used catalytically for the direct aldol reaction.
Scheme 31.
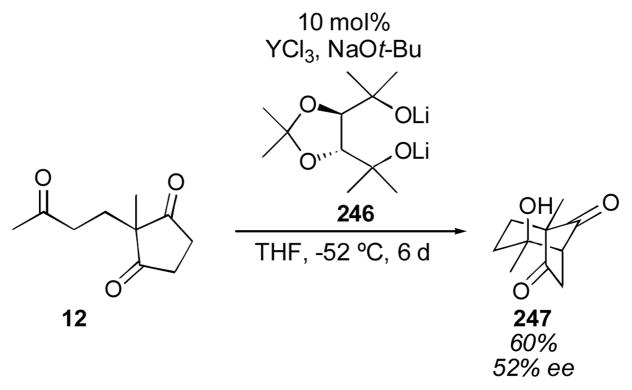
Rare-earth alkoxide catalyzed aldol reaction.318
Shibasaki later reported the first intermolecular direct catalytic aldol of simple ketones using lanthanum-lithium-BINOL complex 248 (Table 26).320 With 20 mol% catalyst 248 a variety of ketones, including aryl ketones, acetone and 2-butanone (205) were coupled with alkyl aldehydes (25). Neopentyl aldehydes gave the best enantioselectivities, in moderate yield. α-Branched aldehydes gave decreased enantioselectivity, while α-Unbranched aldehydes gave both poor enantioselectivity and yield. Interestingly, 2-butanone gave the linear product selectively, with only a trace amount of the branched product. The reactivity of this catalyst was rather low, necessitating the use of 7.4–50 equivalents of the donor (205) relative to the aldehyde (25). The reaction times were very long as a result of this low activity. Adduct 206e, for example, was obtained in 59% yield in 54% ee after 11.5 days.
Table 26.
Lanthanum-lithium-BINOL catalyzed direct aldol reactions.320
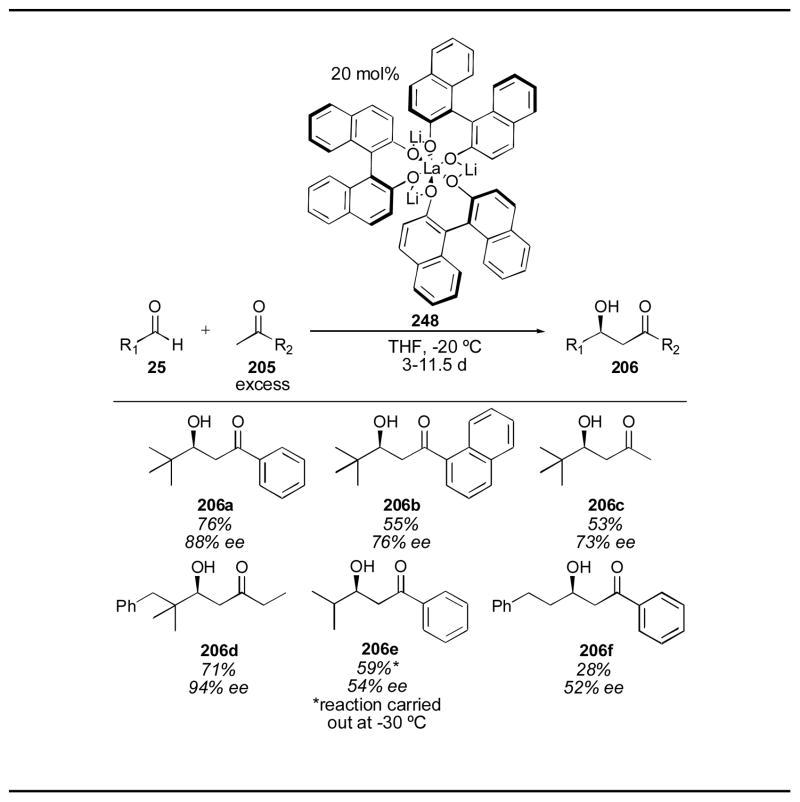
|
References are given for each product as a superscript.
Shibasaki reported an improved catalyst for the direct aldol reaction in 1999 (Table 27).321 The addition of an equimolar amount of KHMDS to complex 248 gave a more reactive catalyst, allowing the loading to be dropped to 8 mol% and the number of equivalents of the ketone to be decreased to 3–15 equivalents. The reaction time was also shorter, although still could take up to 4 days. The enantioselectivity was slightly lower than when catalyst 248 was employed without added KHMDS. Cyclopentanone was used as a donor, giving adduct 38b in good yield and syn selectivity with 76% ee. This provides a nice complement to the enamine-catalyzed reaction, which favors the anti adduct. Shibasaki demonstrated the utility of aryl ketones as ester surrogates through a Baeyer-Villiger oxidation, which was used to synthesize a key intermediate for the synthesis of bryostatin 7 and for the total syntheses of epothilones A (both arising from adduct 38a). This strategy was later used to complete the syntheses of epothilones A and B.322
Table 27.
Improved activity of potassium modified catalyst.321
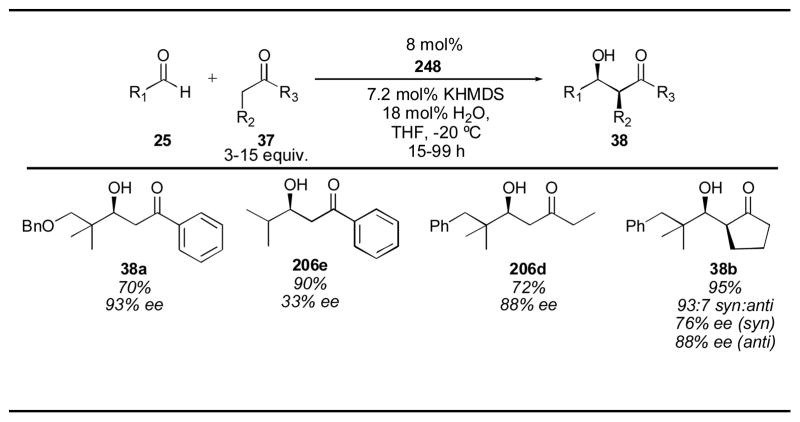
|
Shibasaki also reported a barium BINOL-derived catalyst for the direct aldol reaction of acetophenone (249) with alkyl aldehydes (25) (Scheme 32).323 The catalyst could be used in 5 mol% loading with only 2 equivalents of the donor (249). The reaction took between 18–48 hours to complete. Unfortunately, although the catalytic activity was improved, the enantioselectivity was not as high as the original lanthanum-lithium-BINOL catalyst 248. A similar catalyst (253) was recently used with β,η-unsaturated esters to generate Baylis-Hillman adducts 254 in a dynamic kinetic asymmetric transformation through isomerization of the initial aldol adduct (DYKAT).324
Scheme 32.

In 2001 Shibasaki extended the use of his BINOL-based catalysts to 2-hydroxy-1-phenylethanone 255 (Scheme 33).325, 326 With 10 mol% of catalyst 248, modified with KHMDS, the anti adduct 256 was obtained in 2:1–5:1 selectivity in 78–92% yield, and in 90–95% ee for the major diastereomer. The syn diastereomer epi-256 could be obtained by using 2 equivalents of diethylzinc together with BINOL-derived ligand 257 in 10 mol% loading, as well as with 20 mol% triphenylphosphine oxide. Although this catalyst was originally thought to be dinuclear in zinc, mechanistic studies later indicated the active catalyst likely contains 7 zinc atoms.327 Shibasaki demonstrated the applicability of both sets of conditions to α-unbranched aldehydes, a substrate class that gives poor results with enamine catalysis.
Scheme 33.

BINOL-based catalysts for the aldol reaction of 2-hydroxy-1-phenylethanone.325,326
Shibasaki reported the development of the lanthanum-lithium-BINOL-derived catalyst 259 for the asymmetric direct aldol in 2001 (Scheme 34).328 A variety of unsymmetrical ketones were examined with catalyst 259, including an enone substrate. These reaction conditions produced the desired adduct 260 in 62–71% yield with 40–67% ee. The results were generally poorer with this catalyst than the original BINOL- derived catalyst 248. 3-Pentanone was also examined as a substrate, but gave poor reactivity and enantioselectivity under a variety of conditions.
Scheme 34.
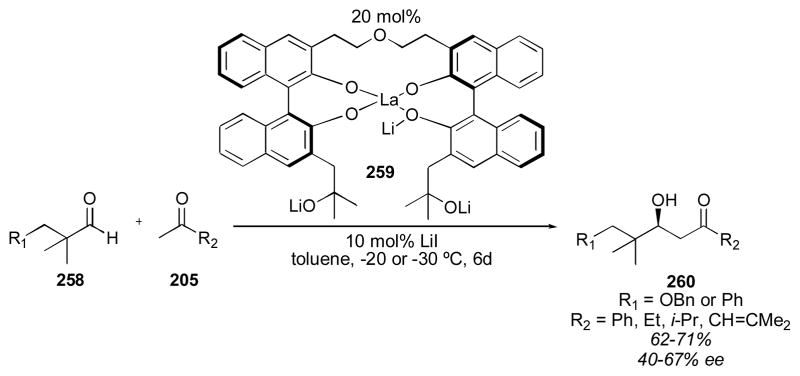
Linked BINOL catalyst for the direct aldol reaction.328
Mahrwald and Ziemer reported a titanium-based BINOL and (R)-mandelic acid catalyst 263 in 2002 for the direct aldol reaction of 3-pentanone 261 (Scheme 35).329 The syn isomer 262 was favored for a variety of aldehydes, including aryl, neopentyl, alkynyl, α-branched and α-unbranched aldehydes. Interestingly, the aldehyde (25) was used in excess under these conditions. These conditions provide a complement to the proline-catalyzed reaction, which favors the anti adduct.
Scheme 35.
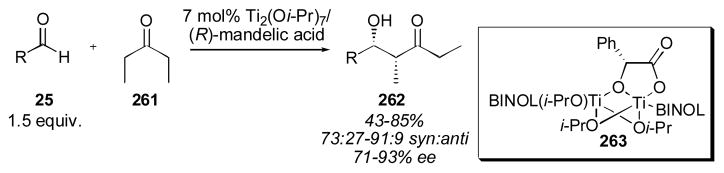
Titanium BINOL catalyst for the direct aldol reaction.329
Shibasaki reported the use of the previously developed BINOL-linked ligand 257 for reaction with α-hydroxy donors 264 using either 2 or 4 equivalents of diethylzinc (Scheme 36).327, 330 These donors provide syn adducts 265 in good yield and high diastereoselectivity (lower for R2 = Me). Changing the linker atom from an oxygen to a sulfur atom changes the diastereoselectivity to favor the anti adduct, though in generally poor ratios. Ligand 257 (X = O) could be used in as little as 1 mol% loading with substrate 264 (R2 = H). The ortho-methoxy group provides an additional binding site, likely responsible for the increased activity of this substrate. This aryl group was used as an auxiliary, and was removed with a Baeyer-Villiger oxidation.
Scheme 36.
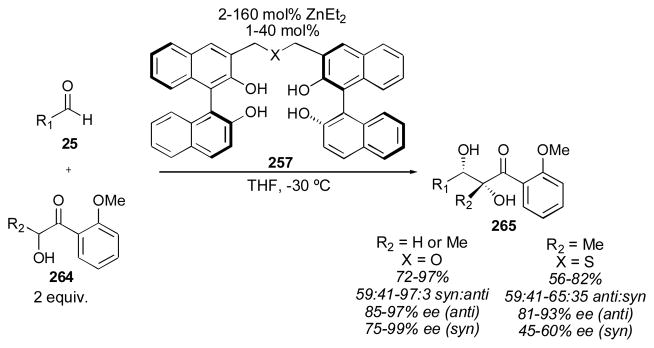
Linked-BINOL zinc-catalysts for the Synthesis of 1,2-diols.327, 330
The lanthanum-lithium-BINOL catalyst 248 was applied to glycine Schiff base substrate 266 in 2002 for the synthesis of serine derivatives 267 (Scheme 37).331 The anti adduct 267 was favored for α-branched and aryl aldehydes in modest selectivity. 1-Hexanal, however, favored the syn diastereomer in only 1% ee. 2-Furfural also gave poor enantioselectivity for both the syn and anti diastereomers. α-Branched aldehydes were the best substrates, delivering the desired products in 71–93% ee, in a 59:41–86:14 anti:syn ratio with 69–76% ee. The imine was hydrolyzed in a second step to give the free amino acids 267. The results obtained with catalyst 248 were inferior to that obtained with Maruoka’s quaternary ammonium salt 217, but given the synthetic ease for the preparation of the BINOL-based catalyst 248, this procedure may be preferred when high enantioselectivity is not required or when the product can be recrystallized to higher enantiomeric purity.
Scheme 37.
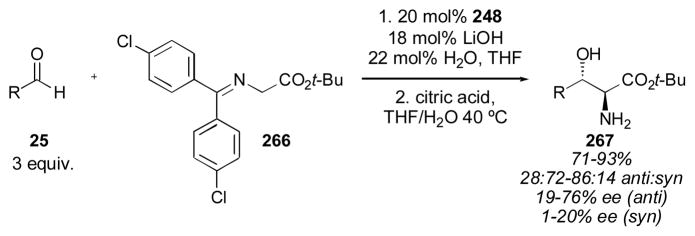
Application of catalyst 248 to glycine Schiff bases.322
In 2003 Yao and Wang reported a zirconium-BINOL catalyst for the direct aldol reaction of diazo ester 268 (Scheme 38).332 The reaction was applied to aryl, heteroaryl, styrenyl, and alkyl aldehydes. The adducts 270 were obtained in 47–82% yield and in 53–87% ee. These adducts could be acylated and then oxidized to transform the diazo esters 270 into α-keto esters 271. Use of diazo esters 268 allows for the extension of the substrate scope of BINOL-based catalysis to donors at the carboxylic acid oxidation state.
Scheme 38.
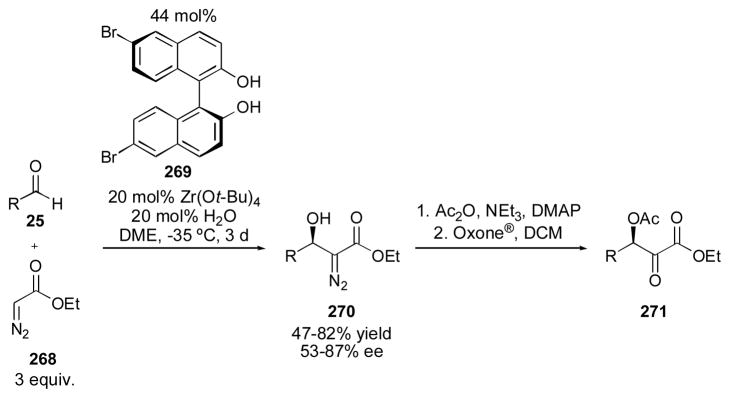
Zirconium-BINOL catalyzed diazo ester aldol.328
Shibasaki reported the use of ynones (273) as donors for the direct catalytic aldol reaction catalyzed by lanthanum-lithium-BINOL-derived complex 248 in 2005 (Scheme 39).333 This reaction demonstrates the mildness of these conditions. When unsubstituted ynones were used with enamine catalyst 99, very little of the desired adduct was observed.243, 334 Products resulting from aldol condensation and 1,4-addition were also obtained under these conditions. Shibasaki used this methodology for the synthesis of fostriecin 277 and a fostriecin epimer.
Scheme 39.
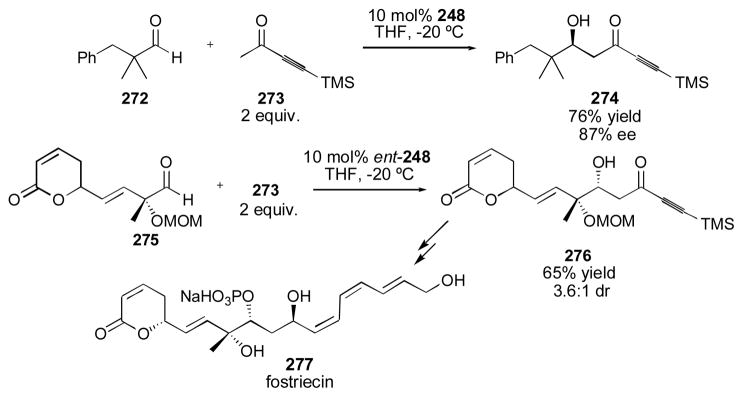
Lanthanum-lithium-BINOL-catalyzed ynone aldol and its application to the synthesis of fostriecin.333,
4.3 ProPhenol-Based Catalysts
In 2000 Trost reported dinuclear zinc catalyst 279, using the ligand known as ProPhenol, for the aldol reaction of aryl methyl ketones (278) with a wide variety of aldehydes (25) (Scheme 40).335 α-Unbranched aldehydes gave lower yields and enantioselectivities than α-branched aldehydes, which were excellent substrates, giving the adducts 280 in 93–99% ee. The reaction employed 5–10 equivalents of the donor 278 and 5 mol% of catalyst 279 over the course of 2–4 days.
Scheme 40.
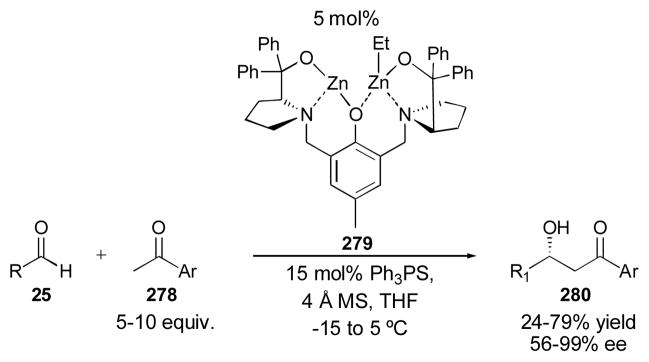
ProPhenol-catalyzed aldol reaction of aryl ketones.335
Modified catalyst 281 was reported in 2001 for the direct aldol reaction of acetone (Scheme 41).336 The best enantioselectivities were observed for α-branched aldehydes, as before, while α-unbranched and aryl aldehydes gave slightly lower enantioselectivities. The reactions proceeded with 5–10 mol% catalyst with 10–15 equivalents of acetone over 2 days. The success of the dinuclear zinc ProPhenol catalyst 281 with α-unbranched aldehydes is particularly noteworthy, as these are poor substrates for proline catalysis.
Scheme 41.
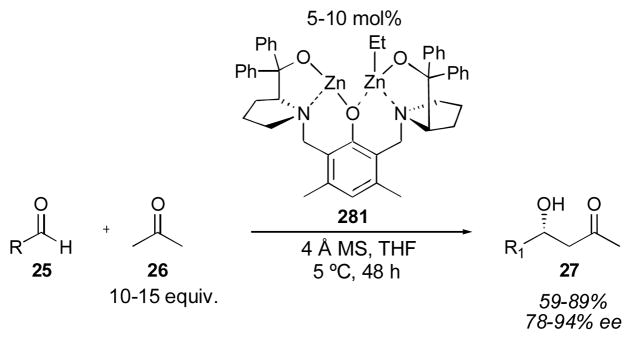
ProPhenol-catalyzed acetone aldol.336
Dinuclear zinc ProPhenol-catalysis has also been applied to the synthesis of 1,2-diols 283 (Scheme 42).337 The adducts 283 were obtained with syn selectivity (4:1–100:0) in 86–98% ee in 65–97% yield. The reaction only required 2.5–5 mol% catalyst and 1.5 equivalents of the donor. The reaction took less time than the ProPhenol-catalyzed methyl ketone aldol reaction as well, indicating the increased reactivity of α-hydroxy donors 282. The α-hydroxy ketone 282 was proposed to chelate one of the zinc atoms to give the α-stereocenter in excellent selectivity (A). Attack from the more open face gave the major syn isomer, while attack from the opposite face gave the anti isomer (with the opposite configuration at the β-stereocenter). The low catalyst loading, small excess of the donor, and commercial availability of the ProPhenol ligand together with the high selectivity and yields make this reaction a practical choice for the synthesis of syn diols, especially when sensitive functional groups are present. Indeed, this reaction has found use for the synthesis of spiroketals.338
Scheme 42.
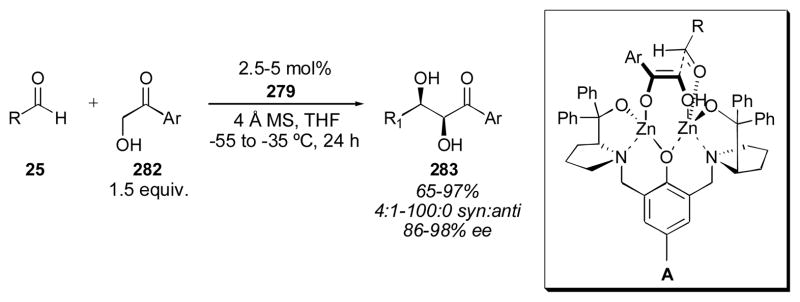
Dinuclear zinc ProPhenol-catalyzed aldol for the synthesis of syn diols.337
Dinuclear zinc ProPhenol catalysis has also been applied to sensitive ynone substrates 285 together with α-ketal aldehydes 284 (Scheme 43).339 The aldol adducts 286 were obtained in 61–84% yield with up to 98% ee using 5–10 mol% of catalyst 279 after 4–17.5 hours using only a slight excess of the ynone donor. The authors noted a large nonlinear effect and accounted for this observation by suggesting that the product 286 modifies the catalyst after the first catalyst turnover. Indeed, addition of several different chelating additives had a pronounced effect on the enantioselectivity of the reaction. The dinuclear zinc ProPhenol-catalyzed ynone aldol has found use for the synthesis of dephosphofostriecin (287).340
Scheme 43.

ProPhenol-catalyzed ynone aldol and its application to the synthesis of dephosphofostriecin.339, 340
In 2005 Trost reported the use of dinuclear zinc ProPhenol catalysis for the direct aldol reaction of methyl vinyl ketone (288) (Scheme 44).341 Given the propensity of methyl vinyl ketone (288) to undergo polymerization, the ability to use this donor under the ProPhenol catalysis conditions is a testament to the mildness of these conditions. With 10 mol% catalyst 279 the adducts 289 were obtained in moderate yield with generally high enantioselectivity. This reaction should prove quite useful as the aldol adducts (289) possess many functional groups for further modifications. Indeed, these adducts (289) were used as substrates for cycloadditions with nitrile oxides.
Scheme 44.
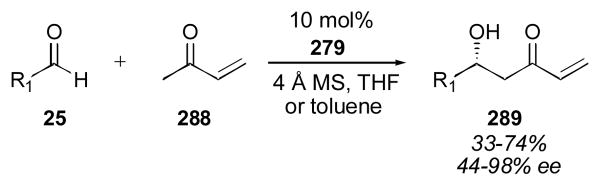
Dinuclear zinc ProPhenol-catalyzed aldol reaction of methyl vinyl ketone.341
In 2008 Da and coworkers reported the use of a catalyst resembling both the BINOL-catalysts of Shibasaki and the ProPhenol catalyst of Trost (Scheme 45).342 This catalyst was used for the aldol reaction of methyl aryl ketones 278 with aryl aldehydes 243. The adducts 291 were obtained in moderate yields and enantioselectivities using 20 mol% catalyst with 80 mol% triethylamine over 5 days.
Scheme 45.

BINOL-derived zincate catalyst for the aldol reaction of aryl ketones and aryl aldehydes.342
In 2009 Trost and coworkers reported the direct aldol of diazo esters 268 using dinuclear magnesium catalyst 292 (Scheme 46).343 With 5 mol% catalyst 292 and 5 mol% diol 293 the aldol adducts 270 were obtained in 50–92% yield with 87–98% ee. The reactants could be used in equimolar amounts, allowing for the minimization of waste. These adducts 270 can be modified in a number of ways to transform the diazo group into other functional groups. This allows for the use of substrates at the carboxylic acid oxidation state with the ProPhenol ligand. The results obtained with the ProPhenol ligand for diazo ester 268 are superior to those obtained with quaternary ammonium salt 217 and zirconium-BINOL catalyst generated from BINOL-derivative 269 (Scheme 23 and 38).
Scheme 46.

Magnesium-ProPhenol catalyzed diazo ester aldol.343
4.4 Miscellaneous Catalysts
Shibasaki reported the use of a calcium catalyst for the direct catalytic aldol reaction in 2001 (Scheme 47).344 α-Branched aldehydes gave adducts with 66–91% ee and in 70–88% yield, while α-unbranched aldehydes gave poor yield and enantioselectivity, favoring the opposite enantiomer of adduct 251. Only 3 mol% of the catalyst was needed with a reaction time of 10–24 hours. The donor was used in 10–100-fold excess under these conditions.
Scheme 47.
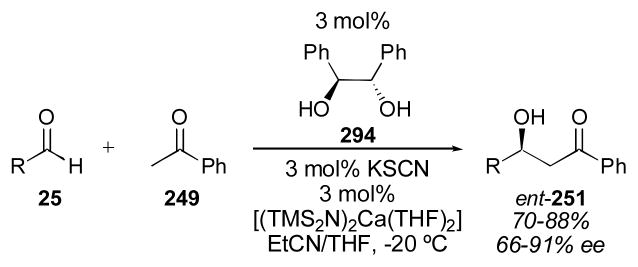
Calcium-catalyzed aldol of acetophenone.344
Calter and Orr reported the use of 20 mol% diamine 295 with 10 mol% zinc nitrate for the aldol reaction of acetone 26 with para-nitrobenzaldehyde 74 in 2003 (Scheme 48).345 Unfortunately, the adduct 27a was obtained in only 37% yield and 22% ee.
Scheme 48.
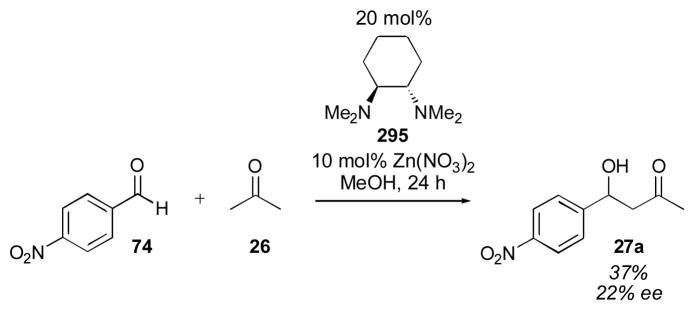
Zinc-diamine catalyzed acetone aldol.345
Darbre reported the use of a zinc proline catalyst for the asymmetric aldol of acetone with aromatic aldehydes in 2003 (Scheme 49).346, 347 The authors noted that under the reaction conditions proline itself gave only 6% yield of adduct 296 in 21% ee, favoring the opposite enantiomer as the one produced when proline and zinc were used together as the catalyst. These results indicate that the two reactions are mechanistically distinct and that zinc plays an important role in the reaction. A number of different mechanisms were suggested by the authors. One possibility is a pathway mirroring that of the type II aldolases, with the zinc activating the donor to generate a zinc enolate, or an enamine mechanism in which the zinc activates the aldehyde (243) and stabilizes the enamine. Unfortunately, the enantioselectivity of this process was rather low.
Scheme 49.
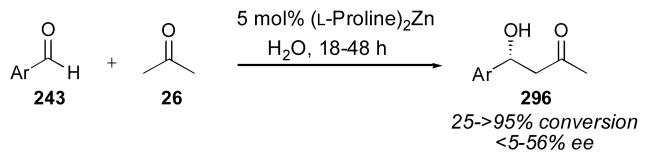
Gathergood and Jørgensen reported the use of copper-BOX catalyst 298 for the direct aldol of α-keto ester 297 with ethyl trifluoromethylpyruvate 230 (Scheme 50).348 Unfortunately the adduct 299 was obtained in low yield with poor diastereoselectivity, although the enantioselectivity was generally quite high.
Scheme 50.
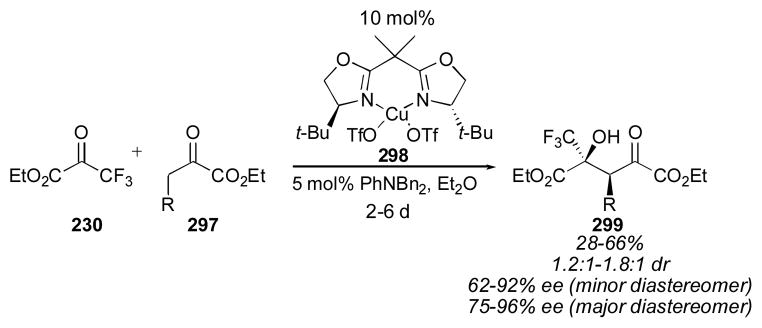
Copper-catalyzed aldol of trifluoropyruvate.348
Zhou and Shan reported the use of spiroboronate ester 300 for the direct acetone aldol reaction in 2006 (Scheme 51).349 The authors do not propose a mechanism for this unusual catalytic system. The adducts 296 were obtained in low to moderate yield with generally high enantioselectivity for most aryl aldehydes. The authors report the use of an α-unbranched aldehyde as well, which gives the aldol adduct 296 in 34% yield and in 96% ee.
Scheme 51.
Spiroborate ester-catalyzed acetone aldol.349
Stilbene ligand 100 together with an equimolar amount of zinc triflate was used for the direct catalytic aldol reaction by Mlynarski and coworkers in 2007 (Scheme 52).350 The reaction was applied to a variety of ketone donors, including acetone, cyclohexanone and cyclopentanone as well as unsymmetrical ketones. Unsymmetrical ketones gave the linear product with 21–54% yield and in 84–88% ee. Cyclohexanone gave the anti products in excellent diastereoselectivity and enantioselectivity. Use of cyclopentanone, however, produced the desired aldol with only 44:66 diastereoselectivity favoring the syn adduct. Electron-poor aryl aldehydes gave better yields than electron-rich aryl aldehydes, as observed with proline itself. The authors suggest an enamine mechanism, in which the zinc stabilizes the enolate as well as activates the aldehyde, as suggested by Dabre for the zinc proline catalyst.
Scheme 52.
Zinc-proline catalyzed aldol.350
Recently Shibasaki has reported a dinuclear nickel catalyst 302 for the direct catalytic aldol reaction of β-keto esters 301 with aqueous formaldehyde (also known as formalin) (Scheme 53).351 The reaction proceeds with only 0.1–1 mol% catalyst 302 to give the desired adducts in 22–94% yield and in 66–94% ee. The use of formaldehyde as the acceptor component in direct catalytic asymmetric aldol reactions is rare and should prove valuable. There have also been a few reports of the use of formaldehyde with enamine,157, 352, 353 rhodium,315, 354 and palladium catalysts.355
Scheme 53.

Dinuclear nickel Schiff base catalyzed aldol.351
In 2009, Feng and coworkers reported the use of cinchonine (304), together with BINOL and titanium for the direct catalytic aldol of diazo esters with a variety of aldehydes, including aryl, heteroaryl, and one example of an alkyl aldehyde (which gave the lowest enantioselectivity) (Scheme 54).356 The diazo ester is used in a significant excess under these conditions, and the reaction times are extremely long, even with 20 mol% catalyst loading, indicating that this catalytic system suffers from rather low activity.
Scheme 54.
Cinchonine-BINOL-titanium catalyzed diazo acetate aldol.356
Wang and coworkers reported the use of ligand 305 together with copper as a catalyst for the direct aldol reaction of aryl aldehydes in 2009 (Scheme 55).357 The authors propose an enamine mechanism, combined with activation of the aldehyde by the copper (II) bound to the ligand (A). Most of the reported aldehyde acceptors are electron poor species, and the donors include acetone, cyclopentanone, cyclohexanone and 2-butanone. The regioselectivity in the case of 2-butanone is rather poor, though the diastereoselectivity and enantioselectivity in such cases is high.
Scheme 55.
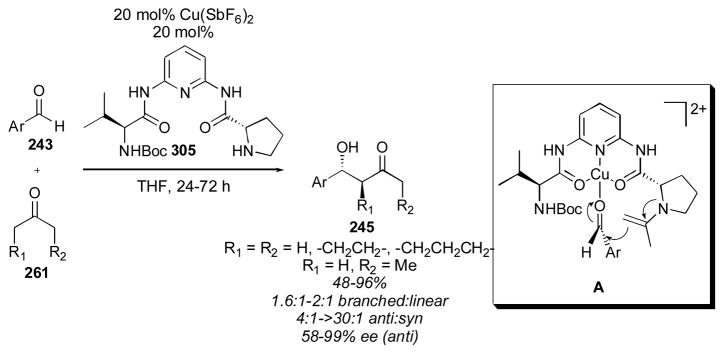
Copper-catalyzed aldol reaction of aryl aldehydes.357
5. Conclusions
A great deal of research has been dedicated to the development of the direct catalytic aldol reaction in recent years. The most practical of the described catalysts is proline, a simple and inexpensive amino acid that can be used for a wide array of substrates. The reactivity of proline, however, is rather low, and has spurred the development of a plethora of new catalysts. Despite the growing number of reported catalysts, however, very few increase the reactivity of the parent system dramatically, although the yield and enantioselectivity have been increased through use of alternate catalysts.
One of the most notable achievements in this area is the development of catalysts that provide access to isomers not favored by proline catalysis. The use of non-enamine catalysts is also noteworthy. Because these catalysts proceed through a different mechanism, they offer different drawbacks and benefits. One of these benefits is the exceptional mildness of some of the reported reaction conditions. The ability to use methyl vinyl ketone, for example, is extraordinary. The mildness of many of the metal-catalyzed conditions has allowed for a significant increase in the scope of the donor, and in some cases, the acceptor as well. The main area that remains to be improved is the low reactivity of the reported catalysts. Most of the reported conditions use a large excess of the donor, together with a high catalyst loading and long reaction time. This limits the utility of the aldol reaction to highly reactive substrates. A more reactive catalyst would allow the scope of the reaction to be extended to less reactive donors and acceptors. A number of catalysts have shown improved reactivity and this will undoubtedly continue to be an area of intense research in the future.
Scheme 13.
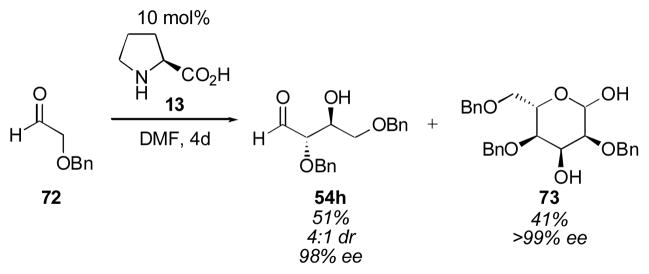
Proline-catalyzed sugar synthesis.140
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health (GM-13598 and GM-33049) for their generous support of our programs.
Notes and references
- 1.Wurtz A. Bull Soc Chim Fr. 1872;17:436. [Google Scholar]
- 2.Mahrwald R, editor. Modern Aldol Reactions. Wiley-VCH; Berlin: 2004. [Google Scholar]
- 3.Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 4.Fessner W-D. In: Modern Aldol Reactions. Mahrwald R, editor. Wiley-VCH; Berlin: 2004. [Google Scholar]
- 5.Tanaka A, Barbas CF., III . In: Modern Aldol Reactions. Mahrwald R, editor. Wiley-VCH; Berlin: 2004. [Google Scholar]
- 6.List B. In: Modern Aldol Reactions. Mahrwald R, editor. Wiley-VCH; Berlin: 2004. [Google Scholar]
- 7.Shibasaki M, Matsunaga S, Kumagai N. In: Modern Aldol Reactions. Mahrwald R, editor. Wiley-VCH; Berlin: 2004. [Google Scholar]
- 8.Yanagisawa A. In: Modern Aldol Reactions. Mahrwald R, editor. Wiley-VCH; Berlin: 2004. [Google Scholar]
- 9.Machajewski TD, Wong CH. Angew Chem, Int Ed. 2000;39:1352. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 10.Alcaide B, Almendros P. Eur J Org Chem. 2002:1595. [Google Scholar]
- 11.Alcaide B, Almendros P. Angew Chem, Int Ed. 2003;42:858. doi: 10.1002/anie.200390232. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 13.Guillena G, Najera C, Ramon DJ. Tetrahedron: Asymmetry. 2007;18:2249. [Google Scholar]
- 14.Notz W, Tanaka F, Barbas CF., III Acc Chem Res. 2004;37:580. doi: 10.1021/ar0300468. [DOI] [PubMed] [Google Scholar]
- 15.Wennemers H. Chimia. 2007;61:276. [Google Scholar]
- 16.Palomo C, Oiarbide M, Garcia JM. Chem Soc Rev. 2004;33:65. doi: 10.1039/b202901d. [DOI] [PubMed] [Google Scholar]
- 17.Seayad J, List B. Org Biomol Chem. 2005;3:719. doi: 10.1039/b415217b. [DOI] [PubMed] [Google Scholar]
- 18.MacMillan DWC. Nature. 2008;455:304. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- 19.Barbas CF., III Angew Chem Int Ed. 2007;46:2. [Google Scholar]
- 20.Enders D, Grondal C, Huttl MRM. Angew Chem, Int Ed. 2007;46:1570. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]
- 21.List B. Synlett. 2001:1675. [Google Scholar]
- 22.List B. Tetrahedron. 2002;58:5573. [Google Scholar]
- 23.List B. Chem Commun. 2006:819. doi: 10.1039/b514296m. [DOI] [PubMed] [Google Scholar]
- 24.List B. Acc Chem Res. 2004;37:548. doi: 10.1021/ar0300571. [DOI] [PubMed] [Google Scholar]
- 25.Benaglia M, Celentano G, Cozzi F. Adv Synth Catal. 2001;343:171. [Google Scholar]
- 26.Benaglia M, Cinquini M, Cozzi F, Puglisi A, Celentano G. Adv Synth Catal. 2002;344:533. [Google Scholar]
- 27.Dhar D, Beadham I, Chandrasekaran S. Proc Indian Acad Sci Chem Sci. 2003;115:365. [Google Scholar]
- 28.Font D, Jimeno C, Pericas MA. Org Lett. 2006;8:4653. doi: 10.1021/ol061964j. [DOI] [PubMed] [Google Scholar]
- 29.Font D, Sayalero S, Bastero A, Jimeno C, Pericas MA. Org Lett. 2008;10:337. doi: 10.1021/ol702901z. [DOI] [PubMed] [Google Scholar]
- 30.Gruttadauria M, Giacalone F, Marculescu AM, Lo Meo P, Riela S, Noto R. Eur J Org Chem. 2007:4688. [Google Scholar]
- 31.Kondo K, Yamano T, Takemoto K. Macromol Chem Phys. 1985;186:1781. [Google Scholar]
- 32.Luo SZ, Zheng XX, Cheng JP. Chem Commun. 2008:5719. doi: 10.1039/b812958d. [DOI] [PubMed] [Google Scholar]
- 33.Yan JC, Wang L. Synthesis. 2008:2065. [Google Scholar]
- 34.Yan J, Wang L. Chirality. 2009;21:413. doi: 10.1002/chir.20603. [DOI] [PubMed] [Google Scholar]
- 35.Luo SZ, Li JY, Zhang L, Xu H, Cheng JP. Chem Eur J. 2008;14:1273. doi: 10.1002/chem.200701129. [DOI] [PubMed] [Google Scholar]
- 36.Fu YQ, An YJ, Liu WM, Li ZC, Zhang G, Tao JC. Catal Lett. 2008;124:397. [Google Scholar]
- 37.Giacalone F, Gruttadauria M, Lo Meo P, Riela S, Noto R. Adv Synth Catal. 2008;350:2747. [Google Scholar]
- 38.Xu LW, Ju YD, Li L, Qiu HY, Jiang JX, Lai GQ. Tetrahedron Lett. 2008;49:7037. [Google Scholar]
- 39.Peng YY, Peng SJ, Ding QP, Wang Q, Cheng JP. Chin J Chem. 2007;25:356. [Google Scholar]
- 40.Deng DS, Cai JW. Helv Chim Acta. 2007;90:114. [Google Scholar]
- 41.Huang J, Zhang X, Armstrong DW. Angew Chem, Int Ed. 2007;46:9073. doi: 10.1002/anie.200703606. [DOI] [PubMed] [Google Scholar]
- 42.Reis O, Eymur S, Reis B, Demir AS. Chem Commun. 2009:1088. doi: 10.1039/b817474a. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Li Y, Xie H, Hu C-g, Dong X. Russ J Org Chem. 2008;44:1807. [Google Scholar]
- 44.Guo HM, Cun LF, Gong LZ, Mi AQ, Jiang YZ. Chem Commun. 2005:1450. doi: 10.1039/b417267a. [DOI] [PubMed] [Google Scholar]
- 45.Hu SQ, Jiang T, Zhang ZF, Zhu AL, Han BX, Song JL, Xie Y, Li WJ. Tetrahedron Lett. 2007;48:5613. [Google Scholar]
- 46.Kotrusz P, Kmentova I, Gotov B, Toma S, Solcaniova E. Chem Commun. 2002:2510. [Google Scholar]
- 47.Lombardo M, Easwar S, Pasi F, Trombini C. Adv Synth Catal. 2009;351:276. [Google Scholar]
- 48.Lombardo M, Easwar S, Pasi F, Trombini C, Dhavale DD. Tetrahedron. 2008;64:9203. [Google Scholar]
- 49.Lombardo M, Pasi F, Easwar S, Trombini C. Synlett. 2008:2471. doi: 10.1039/b812607k. [DOI] [PubMed] [Google Scholar]
- 50.Luo SZ, Mi XL, Zhang L, Liu S, Xu H, Cheng JP. Tetrahedron. 2007;63:1923. [Google Scholar]
- 51.Qian Y, Zheng X, Wang X, Xiao S, Wang Y. Chem Lett. 2009;38:576. [Google Scholar]
- 52.Reddy KR, Chakrapani L, Ramani T, Rajasekhar CV. Synth Commun. 2007;37:4301. [Google Scholar]
- 53.Shah J, Blumenthal H, Yacob Z, Liebscher J. Adv Synth Catal. 2008;350:1267. [Google Scholar]
- 54.Siyutkin DE, Kucherenko AS, Struchkova MI, Zlotin SG. Tetrahedron Lett. 2008;49:1212. [Google Scholar]
- 55.Chandrasekhar S, Narsihmulu C, Reddy NR, Sultana SS. Tetrahedron Lett. 2004;45:4581. [Google Scholar]
- 56.Hayashi Y, Aratake S, Okano T, Takahashi J, Sumiya T, Shoji M. Angew Chem, Int Ed. 2006;45:5527. doi: 10.1002/anie.200601156. [DOI] [PubMed] [Google Scholar]
- 57.Luo S, Xu H, Li J, Zhang L, Mi X, Zheng X, Cheng JP. Tetrahedron. 2007;63:11307. [Google Scholar]
- 58.Peng YY, Ding QP, Li ZC, Wang PG, Cheng JP. Tetrahedron Lett. 2003;44:3871. [Google Scholar]
- 59.Zhong L, Gao Q, Gao JB, Xiao JL, Li C. J Catal. 2007;250:360. [Google Scholar]
- 60.Zhu MK, Xu XY, Gong LZ. Adv Synth Catal. 2008;350:1390. [Google Scholar]
- 61.Hajos ZG, Parrish DR. German Patent DE 2102623. 1971
- 62.Eder U, Sauer GR, Wiechert R. German Patent DE 2014757. 1971
- 63.Pidathala C, Hoang L, Vignola N, List B. Angew Chem, Int Ed. 2003;42:2785. doi: 10.1002/anie.200351266. [DOI] [PubMed] [Google Scholar]
- 64.Chandler CL, List B. J Am Chem Soc. 2008;130:6737. doi: 10.1021/ja8024164. [DOI] [PubMed] [Google Scholar]
- 65.List B, Lerner RA, Barbas CF., III J Am Chem Soc. 2000;122:2395. [Google Scholar]
- 66.List B, Pojarliev P, Castello C. Org Lett. 2001;3:573. doi: 10.1021/ol006976y. [DOI] [PubMed] [Google Scholar]
- 67.Alcaide B, Almendros P, Luna A, del Campo TM. J Org Chem. 2008;73:1635. doi: 10.1021/jo702405h. [DOI] [PubMed] [Google Scholar]
- 68.Zheng YS, Avery MA. Tetrahedron. 2004;60:2091. [Google Scholar]
- 69.Agami C. Bull Soc Chim Fr. 1988:499. [Google Scholar]
- 70.Agami C, Levisalles J, Puchot C. J Chem Soc, Chem Commun. 1985:441. [Google Scholar]
- 71.Agami C, Levisalles J, Sevestre H. J Chem Soc, Chem Commun. 1984:418. [Google Scholar]
- 72.Agami C, Puchot C. J Mol Catal. 1986;38:341. [Google Scholar]
- 73.Agami C, Puchot C. Tetrahedron. 1986;42:2037. [Google Scholar]
- 74.Agami C, Puchot C, Sevestre H. Tetrahedron Lett. 1986;27:1501. [Google Scholar]
- 75.Allemann C, Gordillo R, Clemente FR, Cheong PHY, Houk KN. Acc Chem Res. 2004;37:558. doi: 10.1021/ar0300524. [DOI] [PubMed] [Google Scholar]
- 76.Arno M, Domingo LR. Theor Chem Acc. 2002;108:232. [Google Scholar]
- 77.Arno M, Zaragoza RJ, Domingo LR. Tetrahedron: Asymmetry. 2007;18:157. [Google Scholar]
- 78.Bahmanyar S, Houk KN. J Am Chem Soc. 2001;123:12911. doi: 10.1021/ja011714s. [DOI] [PubMed] [Google Scholar]
- 79.Bahmanyar S, Houk KN. J Am Chem Soc. 2001;123:11273. doi: 10.1021/ja011403h. [DOI] [PubMed] [Google Scholar]
- 80.Bassan A, Zou WB, Reyes E, Himo F, Córdova A. Angew Chem, Int Ed. 2005;44:7028. doi: 10.1002/anie.200502388. [DOI] [PubMed] [Google Scholar]
- 81.Cheong PHY, Houk KN. Synthesis. 2005:1533. [Google Scholar]
- 82.Clemente FR, Houk KN. Angew Chem Int Ed. 2004;43:5766. doi: 10.1002/anie.200460916. [DOI] [PubMed] [Google Scholar]
- 83.Clemente FR, Houk KN. J Am Chem Soc. 2005;127:11294. doi: 10.1021/ja0507620. [DOI] [PubMed] [Google Scholar]
- 84.Puchot C, Samuel O, Dunach E, Zhao S, Agami C, Kagan HB. J Am Chem Soc. 1986;108:2353. doi: 10.1021/ja00269a036. [DOI] [PubMed] [Google Scholar]
- 85.Rajagopal D, Moni MS, Subramanian S, Swaminathan S. Tetrahedron: Asymmetry. 1999;10:1631. [Google Scholar]
- 86.Zhu H, Clemente FR, Houk KN, Meyer MP. J Am Chem Soc. 2009;131:1632. doi: 10.1021/ja806672y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blackmond DG, Klussmann M. AlChE J. 2007;53:2. [Google Scholar]
- 88.Brown KL, Hobi R, Damm L, Dunitz JD, Eschenmoser A, Kratky C. Helv Chim Acta. 1978;61:3108. [Google Scholar]
- 89.Clemente FR, Houk KN. Angew Chem Int Ed. 2004;43:5765. doi: 10.1002/anie.200460916. [DOI] [PubMed] [Google Scholar]
- 90.Jung ME. Tetrahedron. 1976;32:3. [Google Scholar]
- 91.Kellogg RM. Angew Chem, Int Ed. 2007;46:494. doi: 10.1002/anie.200603028. [DOI] [PubMed] [Google Scholar]
- 92.Klussmann M, Iwamura H, Mathew SP, Wells DH, Pandya U, Armstrong A, Blackmond DG. Nature. 2006;441:621. doi: 10.1038/nature04780. [DOI] [PubMed] [Google Scholar]
- 93.Klussmann M, Mathew SR, Iwamura H, Wells DH, Armstrong A, Blackmond DG. Angew Chem, Int Ed. 2006;45:7989. doi: 10.1002/anie.200602521. [DOI] [PubMed] [Google Scholar]
- 94.Klussmann M, White AJR, Armstrong A, Blackmond DG. Angew Chem, Int Ed. 2006;45:7985. doi: 10.1002/anie.200602520. [DOI] [PubMed] [Google Scholar]
- 95.Marquez C, Metzger JO. Chem Commun. 2006:1539. doi: 10.1039/b518288c. [DOI] [PubMed] [Google Scholar]
- 96.Rankin KN, Gauld JW, Boyd RJ. J Phys Chem A. 2002;106:5155. [Google Scholar]
- 97.List B, Hoang L, Martin HJ. Proc Nat Acad Sci USA. 2004;101:5839. doi: 10.1073/pnas.0307979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hoang L, Bahmanyar S, Houk KN, List B. J Am Chem Soc. 2003;125:16. doi: 10.1021/ja028634o. [DOI] [PubMed] [Google Scholar]
- 99.Guillena G, Hita MD, Najera C, Viozquez SF. J Org Chem. 2008;73:5933. doi: 10.1021/jo800773q. [DOI] [PubMed] [Google Scholar]
- 100.Fu A, Li H, Tian F, Yuan S, Si H, Duan Y. Tetrahedron: Asymmetry. 2008;19:1288. [Google Scholar]
- 101.Gryko D, Zimnicka M, Lipinski R. J Org Chem. 2007;72:964. doi: 10.1021/jo062149k. [DOI] [PubMed] [Google Scholar]
- 102.Fan JF, He LJ, Sun YP. Chirality. 2008;20:54. doi: 10.1002/chir.20501. [DOI] [PubMed] [Google Scholar]
- 103.Fan JF, Wu LF, Sun YP. Chin J Chem. 2007;25:472. [Google Scholar]
- 104.Fan JF, Wu LF, Tao FM. Int J Quantum Chem. 2008;108:66. [Google Scholar]
- 105.Bahmanyar S, Houk KN, Martin HJ, List B. J Am Chem Soc. 2003;125:2475. doi: 10.1021/ja028812d. [DOI] [PubMed] [Google Scholar]
- 106.Zotova N, Broadbelt LJ, Armstrong A, Blackmond DG. Bioorg Med Chem Lett. 2009;19:3934. doi: 10.1016/j.bmcl.2009.03.112. [DOI] [PubMed] [Google Scholar]
- 107.Nyberg AI, Usano A, Pihko PM. Synlett. 2004;11:1891. [Google Scholar]
- 108.Pihko PM, Laurikainen KM, Usano A, Nyberg AI, Kaavi JA. Tetrahedron. 2006;62:317. [Google Scholar]
- 109.Zotova N, Franzke A, Armstrong A, Blackmond DG. J Am Chem Soc. 2007;129:15100. doi: 10.1021/ja0738881. [DOI] [PubMed] [Google Scholar]
- 110.Seebach D, Beck AK, Badine DM, Limbach M, Eschenmoser A, Treasurywala AM, Hobi R. Helv Chim Acta. 2007;90:425. [Google Scholar]
- 111.Sakthivel K, Notz W, Bui T, Barbas CF., III J Am Chem Soc. 2001;123:5260. doi: 10.1021/ja010037z. [DOI] [PubMed] [Google Scholar]
- 112.Hayashi Y, Aratake S, Itoh T, Okano T, Sumiya T, Shoji M. Chem Commun. 2007:957. doi: 10.1039/b613262f. [DOI] [PubMed] [Google Scholar]
- 113.Xie ZX, Zhang LZ, Ren XJ, Tang SY, Li Y. Chin J Chem. 2008;26:1272. [Google Scholar]
- 114.Kitazume T, Jiang Z, Kasai K, Mihara Y, Suzuki S. J Fluorine Chem. 2003;121:205. [Google Scholar]
- 115.Notz W, List B. J Am Chem Soc. 2000;122:7386–7387. [Google Scholar]
- 116.Peng L, Liu H, Zhang T, Zhang F, Mei T, Li Y. Tetrahedron Lett. 2003;44:5107. [Google Scholar]
- 117.Grondal C, Enders D. Adv Synth Catal. 2007;349:694. [Google Scholar]
- 118.Enders D, Grondal C. Angew Chem Int Ed. 2005;44:1210. doi: 10.1002/anie.200462428. [DOI] [PubMed] [Google Scholar]
- 119.Grondal C, Enders D. Tetrahedron. 2006;62:329. [Google Scholar]
- 120.Córdova A, Notz W, Barbas CF., III Chem Commun. 2002:3024. doi: 10.1039/b207664k. [DOI] [PubMed] [Google Scholar]
- 121.Suri JT, Ramachary DB, Barbas CF., III Org Lett. 2005;7:1383. doi: 10.1021/ol0502533. [DOI] [PubMed] [Google Scholar]
- 122.Liu HW, Peng LZ, Zhang T, Li YL. New J Chem. 2003;27:1159. [Google Scholar]
- 123.Qiu LH, Shen ZX, Shi CQ, Liu YH, Zhang YW. Chin J Chem. 2005;23:584. [Google Scholar]
- 124.Tokuda O, Kano T, Gao WG, Ikemoto T, Maruoka K. Org Lett. 2005;7:5103. doi: 10.1021/ol052164w. [DOI] [PubMed] [Google Scholar]
- 125.Wang YJ, Shen ZX, Li B, Zhang YW. Chin J Chem. 2006;24:1196. [Google Scholar]
- 126.Samanta S, Zhao CG. Tetrahedron Lett. 2006;47:3383. [Google Scholar]
- 127.Samanta S, Zhao CG. J Am Chem Soc. 2006;128:7442. doi: 10.1021/ja062091r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Córdova A, Notz W, Barbas CFI. J Org Chem. 2002;67:301. doi: 10.1021/jo015881m. [DOI] [PubMed] [Google Scholar]
- 129.Bogevig A, Kumaragurubaran N, Jorgensen KA. Chem Commun. 2002:620. doi: 10.1039/b200681b. [DOI] [PubMed] [Google Scholar]
- 130.Northrup AB, MacMillan DWC. J Am Chem Soc. 2002;124:6798. doi: 10.1021/ja0262378. [DOI] [PubMed] [Google Scholar]
- 131.Pihko PM, Erkkilä A. Tetrahedron Lett. 2003;44:7607. [Google Scholar]
- 132.Carpenter J, Northrup AB, Chung d, Wiener JJM, Kim SG, MacMillan DWC. Angew Chem Int Ed. 2008;47:3568. doi: 10.1002/anie.200800086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew Chem, Int Ed. 2004;43:2152. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]
- 134.Northrup AB, MacMillan DWC. Science. 2004;305:1752. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- 135.Mangion IK, MacMillan DWC. J Am Chem Soc. 2005;127:3696. doi: 10.1021/ja050064f. [DOI] [PubMed] [Google Scholar]
- 136.Storer RI, MacMillan DWC. Tetrahedron. 2004;60:7705. [Google Scholar]
- 137.Thayumanavan R, Tanaka F, Barbas CF., III Org Lett. 2004;6:3541. doi: 10.1021/ol0485417. [DOI] [PubMed] [Google Scholar]
- 138.Chowdari NS, Ramachary DB, Córdova A, Barbas CF., III Tetrahedron Lett. 2002;43:9591. [Google Scholar]
- 139.Casas J, Engqvist M, Ibrahem I, Kaynak B, Córdova A. Angew Chem, Int Ed. 2005;44:1343. doi: 10.1002/anie.200461400. [DOI] [PubMed] [Google Scholar]
- 140.Córdova A, Engqvist M, Ibrahem I, Casas J, Sunden H. Chem Commun. 2005:2047. doi: 10.1039/b500589b. [DOI] [PubMed] [Google Scholar]
- 141.Córdova A, Ibrahem I, Casas J, Sunden H, Engqvist M, Reyes E. Chem Eur J. 2005;11:4772. doi: 10.1002/chem.200500139. [DOI] [PubMed] [Google Scholar]
- 142.Amedjkouh M. Tetrahedron: Asymmetry. 2005;16:1411. [Google Scholar]
- 143.Martin HJ, List B. Synlett. 2003:1901. [Google Scholar]
- 144.Tsogoeva SB, Jagtap SB, Ardemasova ZA. Tetrahedron: Asymmetry. 2006;17:989. [Google Scholar]
- 145.Tsogoeva SB, Wei S. Tetrahedron: Asymmetry. 2005;16:1947. [Google Scholar]
- 146.Chen F, Huang S, Zhang H, Liu F, Peng Y. Tetrahedron. 2008;64:9585. [Google Scholar]
- 147.Chandrasekhar S, Johny K, Reddy CR. Tetrahedron: Asymmetry. 2009;20:1742. [Google Scholar]
- 148.Krattiger P, Kovasy R, Revell JD, Ivan S, Wennemers H. Org Lett. 2005;7:1101. doi: 10.1021/ol0500259. [DOI] [PubMed] [Google Scholar]
- 149.Revell JD, Gantenbein D, Krattiger P, Wennemers H. Biopolymers (Peptide Science) 2006;84:105. doi: 10.1002/bip.20393. [DOI] [PubMed] [Google Scholar]
- 150.Revell JD, Wennemers H. Adv Synth Catal. 2008;350:1046. [Google Scholar]
- 151.Nakadai M, Saito S, Yamamoto H. Tetrahedron. 2002;58:8167. [Google Scholar]
- 152.Saito S, Nakadai M, Yamamoto H. Synlett. 2001;8:1245. [Google Scholar]
- 153.Tang Z, Jiang F, Yu LT, Cui X, Gong LZ, Mi AQ, Jiang YZ, Wu YD. J Am Chem Soc. 2003;125:5262. doi: 10.1021/ja034528q. [DOI] [PubMed] [Google Scholar]
- 154.Tang Z, Jiang F, Cui X, Gong LZ, Mi AQ, Jiang YZ, Wu YD. Proc Nat Acad Sci USA. 2004;101:5755. doi: 10.1073/pnas.0307176101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hartikka A, Arvidsson PI. Tetrahedron: Asymmetry. 2004;15:1831. [Google Scholar]
- 156.Hartikka A, Arvidsson PI. Eur J Org Chem. 2005:4287. [Google Scholar]
- 157.Torii H, Nakadai M, Ishihara K, Saito S, Yamamoto H. Angew Chem, Int Ed. 2004;43:1983. doi: 10.1002/anie.200352724. [DOI] [PubMed] [Google Scholar]
- 158.Berkessel A, Koch B, Lex J. Adv Synth Catal. 2004;346:1141. [Google Scholar]
- 159.Lacoste E, Landais Y, Schenk K, Verlhac JB, Vincent JM. Tetrahedron Lett. 2004;45:8035. [Google Scholar]
- 160.Tang Z, Yang ZH, Chen XH, Cun LF, Mi AQ, Jiang YZ, Gong LZ. J Am Chem Soc. 2005;127:9285. doi: 10.1021/ja0510156. [DOI] [PubMed] [Google Scholar]
- 161.Chimni SS, Mahajan D. Tetrahedron: Asymmetry. 2006;17:2108. [Google Scholar]
- 162.Chimni SS, Mahajan D, Babu VVS. Tetrahedron Lett. 2005;46:5617. [Google Scholar]
- 163.Gryko D, Lipinski R. Adv Synth Catal. 2005;347:1948. [Google Scholar]
- 164.Gryko D, Lipinski R. Eur J Org Chem. 2006:3864. doi: 10.1021/jo062149k. [DOI] [PubMed] [Google Scholar]
- 165.Cobb AJA, Shaw DM, Longbottom DA, Gold JB, Ley SV. Org Biomol Chem. 2005;3:84. doi: 10.1039/b414742a. [DOI] [PubMed] [Google Scholar]
- 166.Bellis E, Kokotos G. Tetrahedron. 2005;61:8669. [Google Scholar]
- 167.Bellis E, Vasilatou K, Kokotos G. Synthesis. 2005;14:2407. [Google Scholar]
- 168.Samanta S, Liu JY, Dodda R, Zhao CG. Org Lett. 2005;7:5321. doi: 10.1021/ol052277f. [DOI] [PubMed] [Google Scholar]
- 169.Diner P, Amedjkouh M. Org Biomol Chem. 2006;4:2091. doi: 10.1039/b605091c. [DOI] [PubMed] [Google Scholar]
- 170.Cheng CL, Sun J, Wang C, Zhang Y, Wei SY, Jiang F, Wu YD. Chem Commun. 2006:215. doi: 10.1039/b511992h. [DOI] [PubMed] [Google Scholar]
- 171.Gu LQ, Yu ML, Wu XY, Zhang YZ, Zhao G. Adv Synth Catal. 2006;348:2223. [Google Scholar]
- 172.Gu Q, Wang XF, Wang L, Wu XY, Zhou QL. Tetrahedron: Asymmetry. 2006;17:1537. [Google Scholar]
- 173.Raj M, Vishnumaya SKG, Singh VK. Org Lett. 2006;8:4097. doi: 10.1021/ol0616081. [DOI] [PubMed] [Google Scholar]
- 174.Chen JR, Li XY, Xing XN, Xiao WJ. J Org Chem. 2006;71:8198. doi: 10.1021/jo0615089. [DOI] [PubMed] [Google Scholar]
- 175.Guizzetti S, Benaglia M, Pignataro L, Puglisi A. Tetrahedron: Asymmetry. 2006;17:2754. [Google Scholar]
- 176.Guillena G, del Carmen Hita M, Najera C. Tetrahedron: Asymmetry. 2006;17:1493. [Google Scholar]
- 177.Jiang M, Zhu SF, Yang Y, Gong LZ, Zhou XG, Zhou QL. Tetrahedron: Asymmetry. 2006;17:384. [Google Scholar]
- 178.Hayashi Y, Sumiya T, Takahashi J, Gotoh H, Urushima T, Shoji M. Angew Chem, Int Ed. 2006;45:958. doi: 10.1002/anie.200502488. [DOI] [PubMed] [Google Scholar]
- 179.Russo A, Botta G, Lattanzi A. Synlett. 2007:795. [Google Scholar]
- 180.Russo A, Botta G, Lattanzi A. Tetrahedron. 2007;63:11886. [Google Scholar]
- 181.Huang WP, Chen JR, Li XY, Cao YJ, Xiao WJ. Can J Chem. 2007;85:208. [Google Scholar]
- 182.He L, Jiang J, Tang Z, Cui X, Mi AQ, Jiang YZ, Gong LZ. Tetrahedron: Asymmetry. 2007;18:265. [Google Scholar]
- 183.Puleo GL, Masi M, Iuliano A. Tetrahedron: Asymmetry. 2007;18:1364. [Google Scholar]
- 184.Yu G, Ge ZM, Cheng TM, Li RT. Chin J Chem. 2008;26:911. [Google Scholar]
- 185.Sato K, Kuriyama M, Shimazawa R, Morimoto T, Kakiuchi K, Shirai R. Tetrahedron Lett. 2008;49:2402. [Google Scholar]
- 186.Zhao JF, He L, Jiang J, Tang Z, Cun LF, Gong LZ. Tetrahedron Lett. 2008;49:3372. [Google Scholar]
- 187.Chen JR, An XL, Zhu XY, Wang XF, Xiao WJ. J Org Chem. 2008;73:6006. doi: 10.1021/jo800910s. [DOI] [PubMed] [Google Scholar]
- 188.Zhao J, Chen A, Liu Q. Chin J Chem. 2009;27:930. [Google Scholar]
- 189.Almasi D, Alonso DA, Najera C. Adv Synth Catal. 2008;350:2467. [Google Scholar]
- 190.Jia YN, Wu FC, Ma X, Zhu GJ, Da CS. Tetrahedron Lett. 2009;50:3059. [Google Scholar]
- 191.Zhang SP, Fu XK, Fu SD. Tetrahedron Lett. 2009;50:1173. [Google Scholar]
- 192.Kano T, Takai J, Tokuda O, Maruoka K. Angew Chem Int Ed. 2005;44:3055. doi: 10.1002/anie.200500408. [DOI] [PubMed] [Google Scholar]
- 193.Kano T, Tokuda O, Maruoka K. Tetrahedron Lett. 2006;47:7423. [Google Scholar]
- 194.Teo YC. Tetrahedron: Asymmetry. 2007;18:1155. [Google Scholar]
- 195.Luo SZ, Xu H, Li JY, Zhang L, Cheng JP. J Am Chem Soc. 2007;129:3074. doi: 10.1021/ja069372j. [DOI] [PubMed] [Google Scholar]
- 196.Zheng BL, Liu QZ, Guo CS, Wang XL, He L. Org Biomol Chem. 2007;5:2913. doi: 10.1039/b711164a. [DOI] [PubMed] [Google Scholar]
- 197.Liu QZ, Wang XL, Luo SW, Zheng BL, Qin DB. Tetrahedron Lett. 2008;49:7434. [Google Scholar]
- 198.Liu J, Yang ZG, Wang Z, Wang F, Chen XH, Liu XH, Feng XM, Su ZS, Hu CW. J Am Chem Soc. 2008;130:5654. doi: 10.1021/ja800839w. [DOI] [PubMed] [Google Scholar]
- 199.Peng FZ, Shao ZH, Pu XW, Zhang HB. Adv Synth Catal. 2008;350:2199. [Google Scholar]
- 200.Da CS, Che LP, Guo QP, Wu FC, Ma X, Jia YN. J Org Chem. 2009;74:2541. doi: 10.1021/jo802758b. [DOI] [PubMed] [Google Scholar]
- 201.Miura T, Yasaku Y, Koyata N, Murakami Y, Imai N. Tetrahedron Lett. 2009;50:2632. [Google Scholar]
- 202.Guillena G, Hita MD, Najera C. Tetrahedron: Asymmetry. 2006;17:729. [Google Scholar]
- 203.Tanimori S, Naka T, Kirihata M. Synth Commun. 2004;34:4043. [Google Scholar]
- 204.Maya V, Raj M, Singh VK. Org Lett. 2007;9:2593. doi: 10.1021/ol071013l. [DOI] [PubMed] [Google Scholar]
- 205.Gandhi S, Singh VK. J Org Chem. 2008;73:9411. doi: 10.1021/jo8019863. [DOI] [PubMed] [Google Scholar]
- 206.Okuyama Y, Nakano H, Watanabe Y, Makabe M, Takeshita M, Uwai K, Kabuto C, Kwon E. Tetrahedron Lett. 2009;50:193. [Google Scholar]
- 207.Wang Y, Wei SY, Sun J. Synlett. 2006:3319. [Google Scholar]
- 208.Cheng CL, Wei SY, Sun J. Synlett. 2006:2419. [Google Scholar]
- 209.Sathapornvajana S, Vilaivan T. Tetrahedron. 2007;63:10253. [Google Scholar]
- 210.Li XJ, Zhang GW, Wang L, Hua MQ, Ma JA. Synlett. 2008:1255. [Google Scholar]
- 211.Chimni SS, Singh S, Mahajan D. Tetrahedron: Asymmetry. 2008;19:2276. [Google Scholar]
- 212.Chimni SS, Singh S, Kumar A. Tetrahedron: Asymmetry. 2009;20:1722. [Google Scholar]
- 213.Han J, Wu H, Teng M, Li Z, Wang Y, Wang L, Pan Y. Synlett. 2009:933. [Google Scholar]
- 214.Tzeng ZH, Chen HY, Reddy RJ, Huang CT, Chen K. Tetrahedron. 2009;65:2879. [Google Scholar]
- 215.Liu XW, Le TN, Lu YP, Xiao YJ, Ma JM, Li XW. Org Biomol Chem. 2008;6:3997. doi: 10.1039/b811581h. [DOI] [PubMed] [Google Scholar]
- 216.Doherty S, Knight JG, McRae A, Harrington RW, Clegg W. Eur J Org Chem. 2008:1759. [Google Scholar]
- 217.Danishefsky S, Cain P. J Am Chem Soc. 1976;98:4975. doi: 10.1021/ja00432a044. [DOI] [PubMed] [Google Scholar]
- 218.Agami C, Meynier F, Puchot C, Guilhem J, Pascard C. Tetrahedron. 1984;40:1031. [Google Scholar]
- 219.Wu XY, Jiang ZQ, Shen HM, Lu YX. Adv Synth Catal. 2007;349:812. [Google Scholar]
- 220.Nakayama K, Maruoka K. J Am Chem Soc. 2008;130:17666. doi: 10.1021/ja807807p. [DOI] [PubMed] [Google Scholar]
- 221.Li L, Xu LW, Ju YD, Lai GQ. Synth Commun. 2009;39:764. [Google Scholar]
- 222.Jiang Z, Liang Z, Wu X, Lu Y. Chem Commun. 2006:2801. doi: 10.1039/b606154k. [DOI] [PubMed] [Google Scholar]
- 223.Amedjkouh M. Tetrahedron: Asymmetry. 2007;18:390. [Google Scholar]
- 224.Dziedzic P, Zou WB, Hafren J, Córdova A. Org Biomol Chem. 2006;4:38. doi: 10.1039/b515880j. [DOI] [PubMed] [Google Scholar]
- 225.Guizzetti S, Benaglia M, Raimondi L, Celentano G. Org Lett. 2007;9:1247. doi: 10.1021/ol070002p. [DOI] [PubMed] [Google Scholar]
- 226.Guillena G, Hita MD, Najera C. Tetrahedron: Asymmetry. 2007;18:1031. [Google Scholar]
- 227.Guillena G, Hita MD, Najera C. Tetrahedron: Asymmetry. 2006;17:1027. [Google Scholar]
- 228.Chen XH, Luo SW, Tang Z, Cun LF, Mi AQ, Jiang YZ, Gong LZ. Chem Eur J. 2007;13:689. doi: 10.1002/chem.200600801. [DOI] [PubMed] [Google Scholar]
- 229.Tang Z, Yang ZH, Cun LF, Gong LZ, Mi AQ, Jiang YZ. Org Lett. 2004;6:2285. doi: 10.1021/ol049141m. [DOI] [PubMed] [Google Scholar]
- 230.Wu X, Ma Z, Ye Z, Qian S, Zhao G. Adv Synth Catal. 2009;351:158. [Google Scholar]
- 231.Li J, Luo S, Cheng JP. J Org Chem. 2009;74:1747. doi: 10.1021/jo802557p. [DOI] [PubMed] [Google Scholar]
- 232.Utsumi N, Imai M, Tanaka F, Ramasastry SSV, Barbas CF., III Org Lett. 2007;9:3445. doi: 10.1021/ol701467s. [DOI] [PubMed] [Google Scholar]
- 233.Guillena G, Hita MD, Najera C, Viozquez SF. Tetrahedron: Asymmetry. 2007;18:2300. [Google Scholar]
- 234.Luo S, Xu H, Zhang L, Li J, Cheng JP. Org Lett. 2008;10:653. doi: 10.1021/ol703023t. [DOI] [PubMed] [Google Scholar]
- 235.Ramasastry SSV, Albertshofer K, Utsumi N, Barbas CF., III Org Lett. 2008;10:1621. doi: 10.1021/ol8002833. [DOI] [PubMed] [Google Scholar]
- 236.Aratake S, Itoh T, Okano T, Nagae N, Sumiya T, Shoji M, Hayashi Y. Chem Eur J. 2007;13:10246. doi: 10.1002/chem.200700363. [DOI] [PubMed] [Google Scholar]
- 237.He L, Tang Z, Cun LF, Mi AQ, Jiang YZ, Gong LZ. Tetrahedron. 2006;62:346. [Google Scholar]
- 238.Guillena G, Hita MD, Najera C. Tetrahedron: Asymmetry. 2007;18:1272. [Google Scholar]
- 239.Xu XY, Wang YZ, Cun LF, Gong LZ. Tetrahedron: Asymmetry. 2007;18:237. [Google Scholar]
- 240.Mei K, Zhang S, He S, Li P, Jin M, Xue F, Luo G, Zhang H, Song L, Duan W, Wang W. Tetrahedron Lett. 2008;49:2681. [Google Scholar]
- 241.Zhang FL, Peng YY, Liao SH, Gong YF. Tetrahedron. 2007;63:4636. [Google Scholar]
- 242.Luo SZ, Xu H, Chen LJ, Cheng JP. Org Lett. 2008;10:1775. doi: 10.1021/ol800471b. [DOI] [PubMed] [Google Scholar]
- 243.Silva F, Sawicki M, Gouverneur V. Org Lett. 2006;8:5417. doi: 10.1021/ol0624225. [DOI] [PubMed] [Google Scholar]
- 244.Luppi G, Cozzi PG, Monari M, Kaptein B, Broxterman QB, Tomasini C. J Org Chem. 2005;70:7418. doi: 10.1021/jo050257l. [DOI] [PubMed] [Google Scholar]
- 245.Xu XY, Tang Z, Wang YZ, Luo SW, Cun LF, Gong LZ. J Org Chem. 2007;72:9905. doi: 10.1021/jo701868t. [DOI] [PubMed] [Google Scholar]
- 246.Tang Z, Cun LF, Cui X, Mi AQ, Jiang YZ, Gong LZ. Org Lett. 2006;8:1263. doi: 10.1021/ol0529391. [DOI] [PubMed] [Google Scholar]
- 247.Wang F, Xiong Y, Liu X, Feng X. Adv Synth Catal. 2007;349:2665. [Google Scholar]
- 248.Nakamura S, Hara N, Nakashima H, Kubo K, Shibata N, Toru T. Chem Eur J. 2008;14:8079. doi: 10.1002/chem.200800981. [DOI] [PubMed] [Google Scholar]
- 249.Mangion IK, Northrup AB, MacMillan DWC. Angew Chem, Int Ed. 2004;43:6722. doi: 10.1002/anie.200461851. [DOI] [PubMed] [Google Scholar]
- 250.Mase N, Tanaka F, Barbas CF., III Angew Chem, Int Ed. 2004;43:2420. doi: 10.1002/anie.200353546. [DOI] [PubMed] [Google Scholar]
- 251.Aratake S, Itoh T, Okano T, Usui T, Shoji M, Hayashi Y. Chem Commun. 2007:2524. doi: 10.1039/b702559a. [DOI] [PubMed] [Google Scholar]
- 252.Kano T, Yamaguchi Y, Tanaka Y, Maruoka K. Angew Chem Int Ed. 2007;46:1738. doi: 10.1002/anie.200604640. [DOI] [PubMed] [Google Scholar]
- 253.Hayashi Y, Itoh T, Aratake S, Ishikawa H. Angew Chem Int Ed. 2008;47:2082. doi: 10.1002/anie.200704870. [DOI] [PubMed] [Google Scholar]
- 254.Hayashi Y, Samanta S, Itoh T, Ishikawa H. Org Lett. 2008;10:5581. doi: 10.1021/ol802438u. [DOI] [PubMed] [Google Scholar]
- 255.Cortez G, Tennyson RL, Romo D. J Am Chem Soc. 2001;123:7945. doi: 10.1021/ja016134+. [DOI] [PubMed] [Google Scholar]
- 256.Cortez G, Oh SH, Romo D. Synthesis. 2001;11:1731. [Google Scholar]
- 257.Oh SH, Cortez G, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]
- 258.Mukaiyama T, Usui M, Saigo K. Chem Lett. 1976;5:49. [Google Scholar]
- 259.Wynberg H, Staring EGJ. J Am Chem Soc. 1982;104:166. [Google Scholar]
- 260.Wynberg H, Staring EGJ. J Org Chem. 1985;50:1977. [Google Scholar]
- 261.Tennyson RL, Romo D. J Org Chem. 2000;65:7248. doi: 10.1021/jo001010l. [DOI] [PubMed] [Google Scholar]
- 262.Purohit VC, Richardson RD, Smith JW, Romo D. J Org Chem. 2006;71:4549. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- 263.Gasparski CM, Miller MJ. Tetrahedron. 1991;47:5367. [Google Scholar]
- 264.Ma B, Parkinson JL, Castle SL. Tetrahedron Lett. 2007;48:2083. doi: 10.1016/j.tetlet.2007.01.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Mettath S, Srikanth GSC, Dangerfield BS, Castle SL. J Org Chem. 2004;69:6489. doi: 10.1021/jo049283u. [DOI] [PubMed] [Google Scholar]
- 266.Ooi T, Taniguchi M, Kameda M, Maruoka K. Angew Chem, Int Ed. 2002;41:4542. doi: 10.1002/1521-3773(20021202)41:23<4542::AID-ANIE4542>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 267.Ooi T, Kameda M, Taniguchi M, Maruoka K. J Am Chem Soc. 2004;126:9685. doi: 10.1021/ja048865q. [DOI] [PubMed] [Google Scholar]
- 268.Kano T, Lan Q, Wang XS, Maruoka K. Adv Synth Catal. 2007;349:556. [Google Scholar]
- 269.Kitamura M, Shirakawa S, Arimura Y, Wang X, Maruoka K. Chem Asian J. 2008;3:1702. doi: 10.1002/asia.200800107. [DOI] [PubMed] [Google Scholar]
- 270.Paradowska J, Rogozinska M, Mlynarski J. Tetrahedron Lett. 2009;50:1639. [Google Scholar]
- 271.Arai S, Hasegawa K, Nashida A. Tetrahedron Lett. 2004;45:1023. [Google Scholar]
- 272.Hasegawa K, Arai S, Nishida A. Tetrahedron. 2006;62:1390. [Google Scholar]
- 273.Calter MA, Phillips RM, Flaschenrien C. J Am Chem Soc. 2005;127:14566. doi: 10.1021/ja055752d. [DOI] [PubMed] [Google Scholar]
- 274.Ogawa S, Shibata N, Inagaki J, Nakamura S, Toru T, Shiro M. Angew Chem, Int Ed. 2007;46:8666. doi: 10.1002/anie.200703317. [DOI] [PubMed] [Google Scholar]
- 275.Shibasaki M, Matsunaga S. Chem Soc Rev. 2006;35:269. doi: 10.1039/b506346a. [DOI] [PubMed] [Google Scholar]
- 276.Matsunaga S, Shibasaki M. Bull Chem Soc Jpn. 2008;81:60. [Google Scholar]
- 277.Saito S, Yamamoto H. Acc Chem Res. 2004;37:570. doi: 10.1021/ar030064p. [DOI] [PubMed] [Google Scholar]
- 278.Matsunaga S, Ohshima T, Shibasaki M. Adv Synth Catal. 2002;344:3. [Google Scholar]
- 279.Ito Y, Sawamura M, Hayashi T. J Am Chem Soc. 1986;108:6405. [Google Scholar]
- 280.Ito Y, Sawamura M, Hayashi T. Tetrahedron Lett. 1987;28:6215. [Google Scholar]
- 281.Hayashi T, Sawamura M, Ito Y. Tetrahedron. 1992;48:1999. [Google Scholar]
- 282.Bachi MD, Melman A. J Org Chem. 1997;62:1896. [Google Scholar]
- 283.Ito Y, Sawamura M, Hamashima H, Emura T, Hayashi T. Tetrahedron Lett. 1989;30:4681. [Google Scholar]
- 284.Ito Y, Sawamura M, Hayashi T. Tetrahedron Lett. 1988;29:239. [Google Scholar]
- 285.Ito Y, Sawamura M, Kobayashi M, Hayashi T. Tetrahedron Lett. 1988;29:6321. [Google Scholar]
- 286.Ito Y, Sawamura M, Shirakawa E, Hayashizaki K, Hayashi T. Tetrahedron. 1988;44:5253. [Google Scholar]
- 287.Ito Y, Sawamura M, Shirakawa E, Hayashizaki K, Hayashi T. Tetrahedron Lett. 1988;29:235. [Google Scholar]
- 288.Pastor SD. Tetrahedron. 1988;44:2883. [Google Scholar]
- 289.Pastor SD, Togni A. J Am Chem Soc. 1989;111:2333. [Google Scholar]
- 290.Pastor SD, Togni A. Tetrahedron Lett. 1990;31:839. [Google Scholar]
- 291.Pastor SD, Togni A. Helv Chim Acta. 1991;74:905. [Google Scholar]
- 292.Sawamura M, Ito Y. Chem Rev. 1992;92:857. [Google Scholar]
- 293.Sawamura M, Ito Y, Hayashi T. Tetrahedron Lett. 1989;30:2247. [Google Scholar]
- 294.Sawamura M, Ito Y, Hayashi T. Tetrahedron Lett. 1990;31:2723. [Google Scholar]
- 295.Soloshonok VA, Hayashi T. Tetrahedron Lett. 1994;35:2713. [Google Scholar]
- 296.Soloshonok VA, Hayashi T. Tetrahedron: Asymmetry. 1994;5:1091. [Google Scholar]
- 297.Soloshonok VA, Kacharov AD, Hayashi T. Tetrahedron. 1996;52:245. [Google Scholar]
- 298.Togni A, Hausel R. Synlett. 1990:633. [Google Scholar]
- 299.Togni A, Pastor SD. Helv Chim Acta. 1989;72:1038. [Google Scholar]
- 300.Togni A, Pastor SD. Tetrahedron Lett. 1989;30:1071. [Google Scholar]
- 301.Togni A, Pastor SD. J Org Chem. 1990;55:1649. [Google Scholar]
- 302.Togni A, Pastor SD, Rihs G. Helv Chim Acta. 1989;72:1471. [Google Scholar]
- 303.Togni A, Pastor SD, Rihs G. J Organomet Chem. 1990;381:C21. [Google Scholar]
- 304.Hayashi T, Uozumi Y, Yamazaki A, Sawamura M, Hamashima H, Ito Y. Tetrahedron Lett. 1991;32:2799. [Google Scholar]
- 305.Sawamura M, Hamashima H, Ito Y. J Org Chem. 1990;55:5935. [Google Scholar]
- 306.Guillena G, Rodriguez G, van Koten G. Tetrahedron Lett. 2002;43:3895. [Google Scholar]
- 307.Kleij AW, Gebbink R, van den Nieuwenhuijzen PAJ, Kooijman H, Lutz M, Spek AL, van Koten G. Organometallics. 2001;20:634. [Google Scholar]
- 308.Longmire JM, Zhang XM, Shang MY. Organometallics. 1998;17:4374. [Google Scholar]
- 309.Meijer MD, Ronde N, Vogt D, van Klink GPM, van Koten G. Organometallics. 2001;20:3993. [Google Scholar]
- 310.Motoyama Y, Kawakami H, Shimozono K, Aoki K, Nishiyama H. Organometallics. 2002;21:3408. [Google Scholar]
- 311.Nesper R, Pregosin PS, Puntener K, Worle M. Helv Chim Acta. 1993;76:2239. [Google Scholar]
- 312.Rodriguez G, Lutz M, Spek AL, van Koten G. Chem Eur J. 2002;8:46. [Google Scholar]
- 313.Schlenk C, Kleij AW, Frey H, van Koten G. Angew Chem, Int Ed. 2000;39:3445. [PubMed] [Google Scholar]
- 314.Stark MA, Richards CJ. Tetrahedron Lett. 1997;38:5881. [Google Scholar]
- 315.Kuwano R, Miyazaki H, Ito Y. J Organomet Chem. 2000;603:18. [Google Scholar]
- 316.Inoue H, Kikuchi M, Ito J, Nishiyama H. Tetrahedron. 2008;64:493. [Google Scholar]
- 317.Mizuno M, Inoue H, Naito T, Zhou L, Nishiyama H. Chem Eur J. 2009;15:8985. doi: 10.1002/chem.200901329. [DOI] [PubMed] [Google Scholar]
- 318.Sasai H, Suzuki T, Arai S, Arai T, Shibasaki M. J Am Chem Soc. 1992;114:4418. [Google Scholar]
- 319.Shibasaki M, Sasai H, Arai T. Angew Chem Int Ed. 1997;36:1236. [Google Scholar]
- 320.Yamada YMA, Yoshikawa N, Sasai H, Shibasaki M. Angew Chem Int Ed. 1997;36:1871. [Google Scholar]
- 321.Yoshikawa N, Yamada YMA, Das J, Sasai H, Shibasaki M. J Am Chem Soc. 1999;121:4168. [Google Scholar]
- 322.Sawada D, Kanai M, Shibasaki M. J Am Chem Soc. 2000;122:10521. [Google Scholar]
- 323.Yamada YMA, Shibasaki M. Tetrahedron Lett. 1998;39:5561. [Google Scholar]
- 324.Yamaguchi A, Matsunaga S, Shibasaki M. J Am Chem Soc. 2009;131:10842. doi: 10.1021/ja904575e. [DOI] [PubMed] [Google Scholar]
- 325.Yoshikawa N, Kumagai N, Matsunaga S, Moll G, Ohshima T, Suzuki T, Shibasaki M. J Am Chem Soc. 2001;123:2466. doi: 10.1021/ja015580u. [DOI] [PubMed] [Google Scholar]
- 326.Yoshikawa N, Suzuki T, Shibasaki M. J Org Chem. 2002;67:2556. doi: 10.1021/jo0162538. [DOI] [PubMed] [Google Scholar]
- 327.Kumagai N, Matsunaga S, Kinoshita T, Harada S, Okada S, Sakamoto S, Yamaguchi K, Shibasaki M. J Am Chem Soc. 2003;125:2169. doi: 10.1021/ja028926p. [DOI] [PubMed] [Google Scholar]
- 328.Yoshikawa N, Shibasaki M. Tetrahedron. 2001;57:2569. [Google Scholar]
- 329.Mahrwald R, Ziemer B. Tetrahedron Lett. 2002;43:4459. [Google Scholar]
- 330.Kumagai N, Matsunaga S, Yoshikawa N, Ohshima T, Shibasaki M. Org Lett. 2001;3:1539. doi: 10.1021/ol015878p. [DOI] [PubMed] [Google Scholar]
- 331.Yoshikawa N, Shibasaki M. Tetrahedron. 2002;58:8289. [Google Scholar]
- 332.Yao WG, Wang JB. Org Lett. 2003;5:1527. doi: 10.1021/ol0343257. [DOI] [PubMed] [Google Scholar]
- 333.Maki K, Motoki R, Fujii K, Kanai M, Kobayashi T, Tamura S, Shibasaki M. J Am Chem Soc. 2005;127:17111. doi: 10.1021/ja0562043. [DOI] [PubMed] [Google Scholar]
- 334.Silva F, Reiter M, Mills-Webb R, Sawicki M, Klar D, Bensel N, Wagner A, Gouverneur V. J Org Chem. 2006;71:8390. doi: 10.1021/jo061292a. [DOI] [PubMed] [Google Scholar]
- 335.Trost BM, Ito H. J Am Chem Soc. 2000;122:12003. [Google Scholar]
- 336.Trost BM, Silcoff ER, Ito H. Org Lett. 2001;3:2497. doi: 10.1021/ol0161211. [DOI] [PubMed] [Google Scholar]
- 337.Trost BM, Ito H, Silcoff ER. J Am Chem Soc. 2001;123:3367. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]
- 338.Ito H, Kawabe C, Iguchi K. Heterocycles. 2006;67:695. [Google Scholar]
- 339.Trost BM, Fettes A, Shireman BT. J Am Chem Soc. 2004;126:2660. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]
- 340.Trost BM, Frederiksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J Am Chem Soc. 2005;127:3666. doi: 10.1021/ja042435i. [DOI] [PubMed] [Google Scholar]
- 341.Trost BM, Shin SH, Sclafani JA. J Am Chem Soc. 2005;127:8602. doi: 10.1021/ja051526s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Li H, Da CS, Xiao YH, Li X, Su YN. J Org Chem. 2008;73:7398. doi: 10.1021/jo801182n. [DOI] [PubMed] [Google Scholar]
- 343.Trost BM, Malhotra S, Fried BA. J Am Chem Soc. 2009;131:1674. doi: 10.1021/ja809181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Suzuki T, Yamagiwa N, Matsuo Y, Sakamoto S, Yamaguchi K, Shibasaki M, Noyori R. Tetrahedron Lett. 2001;42:4669. [Google Scholar]
- 345.Calter MA, Orr RK. Tetrahedron Lett. 2003;44:5699. [Google Scholar]
- 346.Darbre T, Machuqueiro M. Chem Commun. 2003:1090. [PubMed] [Google Scholar]
- 347.Fernandez-Lopez R, Kofoed J, Machuqueiro M, Darbre T. Eur J Org Chem. 2005:5268. [Google Scholar]
- 348.Gathergood N, Juhl K, Poulsen TB, Thordrup K, Jorgensen KA. Org Biomol Chem. 2004;2:1077. doi: 10.1039/b316092k. [DOI] [PubMed] [Google Scholar]
- 349.Zhou Y, Shan ZX. Tetrahedron. 2006;62:5692. [Google Scholar]
- 350.Paradowska J, Stodulski M, Mlynarski J. Adv Synth Catal. 2007;349:1041. [Google Scholar]
- 351.Mouri S, Chen Z, Matsunaga S, Shibasaki M. Chem Commun. 2009:5138. doi: 10.1039/b912380f. [DOI] [PubMed] [Google Scholar]
- 352.Casas J, Sundén H, Cordóva A. Tetrahedron Lett. 2004;45:6117. [Google Scholar]
- 353.Fujii M, Sato Y, Aida T, Yoshihara M. Chem Express. 1992;7:309. [Google Scholar]
- 354.Kuwano R, Miyazaki H, Ito Y. Chem Commun. 1998:71. [Google Scholar]
- 355.Fukuchi I, Hamashima H, Sodeoka M. Adv Synth Catal. 2007;349:509. [Google Scholar]
- 356.Wang W, Shen K, Hu X, Wang J, Liu X, Feng X. Synlett. 2009;10:1655. [Google Scholar]
- 357.Xu Z, Daka P, Budik I, Wang H, Bai FQ, Zhang HX. Eur J Org Chem. 2009:4581. [Google Scholar]