

Abstract
Protein diffusion in lipid membranes is a key aspect of many cellular signaling processes. To quantitatively describe protein diffusion in membranes, several competing theoretical models have been proposed. Among these, the Saffman-Delbrück model is the most famous. This model predicts a logarithmic dependence of a protein’s diffusion coefficient on its inverse hydrodynamic radius (D ∝ ln 1/R) for small radius values. For large radius values, it converges toward a D ∝ 1/R scaling. Recently, however, experimental data indicate a Stokes-Einstein-like behavior (D ∝ 1/R) of membrane protein diffusion at small protein radii. In this study, we investigate protein diffusion in black lipid membranes using dual-focus fluorescence correlation spectroscopy. This technique yields highly accurate diffusion coefficients for lipid and protein diffusion in membranes. We find that despite its simplicity, the Saffman-Delbrück model is able to describe protein diffusion extremely well and a Stokes-Einstein-like behavior can be ruled out.
Introduction
Diffusion in lipid membranes plays a key role in the interaction of membrane-associated proteins. Many signal transduction cascades are initialized by ligand binding and subsequent oligomerization of the activated receptors, which is a diffusion-limited process. In 1975, Philip Saffman and Max Delbrück developed a model to describe protein diffusion in lipid bilayers (1). They considered cylindrical membrane inclusions of radius R diffusing in an infinite two-dimensional bilayer of height h and viscosity μm that is surrounded by a solvent of viscosity μs. Saffman and Delbrück then predicted a logarithmic dependence of the protein’s diffusion coefficient, DSD, on its hydrodynamic radius according to
| (1) |
where kB denotes the Boltzmann constant and T the temperature, and γ ≈ 0.5772 is Euler’s constant. The variable ε is defined as ε = Rμs/(hμm). Recently, Gambin et al. challenged the Saffman-Delbrück model by observing a Stokes-Einstein-like behavior of the diffusion (2):
| (2) |
All parameters have the same meanings as above, and λ is a characteristic length introduced for dimensional reasons. This inverse linear dependence between the protein’s diffusion coefficient and its radius differs substantially from the prediction of Saffman and Delbrück. In contrast, Ramadurai et al. (3) found protein diffusion in membranes to comply with the Saffman-Delbrück model.
These contradictory experimental results have evoked an abundance of theoretical studies that reflect the experimental contradiction by either justifying the Stokes-Einstein-like behavior or supporting the Saffman-Delbrück theory.
Naji et al. present two explanations for the observed D ∝ 1/R scaling. On the one hand, changes in bulk hydrodynamics due to a height mismatch between the membrane and the embedded protein could explain the findings. On the other hand, additional dissipative stresses due to lipid chain stretching, lipid tilt, or local demixing in the case of lipid mixtures could also yield the D ∝ 1/R scaling (4). Overall, they argue that the Saffman-Delbrück model fails because the protein carries a deformed membrane patch that changes its diffusive behavior. Guigas and Weiss investigated the influence of hydrophobic mismatch on protein diffusion in more detail, arguing that the previous study by Naji et al. relied on ad hoc assumptions and might not be applicable to describe the diffusion of membrane inclusions with hydrophobic mismatch (5). Guigas and Weiss’s calculations showed that although hydrophobic mismatch changes the effective membrane viscosity, this does not alter the crude scaling behavior of protein diffusion. Therefore, the Saffman-Delbrück model should still be applicable even when accounting for mobility changes due to hydrophobic mismatch. The D ∝ 1/R scaling emerges, according to their calculations, only for large membrane inclusions (Rc ≈ 10 nm).
Addressing the problem that the Saffman-Delbrück model cannot be applied for larger membrane inclusions, Petrov and Schwille derived an analytical expression for the model suggested by Hughes, Pailthorpe, and White (6–8):
| (3) |
The variables have the same meanings as above, and a1, a2, b1, and b2 are constants given in Guigas and Weiss (5). In the size range relevant for proteins, the HPW approximation mostly reproduces the Saffman-Delbrück result.
The ambiguity in previous experimental and theoretical work reflects the urgent need for accurate, quantitative data on protein diffusion in membranes. We therefore combined highly stable black lipid membranes (BLMs) with calibration-free dual-focus fluorescence correlation spectroscopy (2fFCS) to obtain precise and absolute values for diffusion coefficients.
BLMs constitute free-standing lipid bilayers spanned over a pore, thereby circumventing the use of solid supports, which can severely influence diffusion processes inside the membrane (7). Another advantage of BLMs is their stability, as they are less prone to undulations than are giant unilamellar vesicles (GUVs) and are easy to prepare at high ionic strength, making it possible to work under physiological conditions.
2fFCS (9) is an extension of conventional FCS, employing two overlapping foci with known and fixed lateral distance. Aside from the autocorrelation functions for each focus, the cross-correlation functions between the two foci are also calculated. A global fit of all correlation curves with the exact distance between the centers of the two foci known yields an absolute value for the diffusion coefficient. Another significant advantage of 2fFCS over FCS is its higher accuracy and much reduced sensitivity to refractive index mismatch, optical aberrations, and coverslide thickness deviations.
To accurately measure membrane diffusion with 2fFCS, precise alignment of the foci with the lipid bilayer is essential. This can be achieved by z-scan FCS (10). Z-scan FCS measures correlation curves at different positions of the focal plane along the optical axis, which subsequently allows for determination of the relative position of the membrane with respect to the focal plane. Although this is a very precise method of measuring diffusion coefficients in membranes, it is rather time-consuming. We therefore used the maximum molecular brightness for positioning the foci, which is just as precise as z-scan FCS but much faster, as previously reported (11).
The proteins chosen for diffusion measurements differ in size by more than one order of magnitude. Much care has been taken to use proteins that do not aggregate inside the membrane. We hope that the covered size range of proteins combined with the superior accuracy of our measurement results will set a new benchmark for protein diffusion and lipid bilayer viscosity.
Materials and Methods
Chemicals
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) as sodium salt (DOPG), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) sodium salt were purchased from Avanti Polar Lipids (Alabaster, AL). Atto655-labeled 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DPPEAtto655) was provided by Christian Eggeling (Oxford University, Oxford, United Kingdom).
Dodecane, streptavidin, and chloroform were obtained from Sigma Aldrich (Traufkirchen, Germany). Octyl glycoside was purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Decylmaltoside (DM) was obtained from Genaxxon Bioscience (Ulm, Germany). Alexa647 succinimidyl ester and Alexa647 maleimide were obtained from Life Technologies (Darmstadt, Germany).
Bilayer preparation
Lipids were dissolved in chloroform and mixed at a ratio of 60 wt % POPE and 40 wt % POPC. After solvent evaporation for 30 min in vacuum, dodecane was added to the dry lipid mixture to yield a final lipid concentration of 10 mg/mL. BLMs were then generated using a commercially available setup (Ionovation Bilayer Explorer, Ionovation, Osnabrück, Germany) which has been described in detail in Weiß and Enderlein (11). Briefly, 0.2 μL of the lipid solution was injected into a chip filled with phosphate-buffered saline (PBS; 136.9 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, and 8.1 mM Na2HPO4 × 12 H2O, pH 7.4). The lipids were then painted over a pore of 120 μm diameter using an automated pumping cycle. Bilayer formation was monitored via capacitance measurements with a patch-clamp amplifier (EPC10, HEKA, Lambrecht, Germany).
GUV preparation
POPC and POPE were dissolved in chloroform and mixed in a ratio of 60 wt % POPE and 40 wt % POPC to a final lipid concentration of 1 mg/mL. We added 1 μL 1 mg/mL 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) sodium salt and 0.07 μL 5 × 10−5 mg/mL DPPEAtto655 in chloroform to give the stock solution. For electroformation, 60 μL of the stock solution was added to the lower electrode of a home-built electroformation chamber and evaporated for 30 min under vacuum. Then, 500 μL 100 mM sucrose solution was added to the dry lipid film and electroformation was performed for 3 h at 15 Hz. Afterward, the GUVs were collected in a 1.5 mL reaction tube and diluted 1:2 with 100 mM glucose solution. Due to the density difference between the sucrose and glucose solution, the GUVs sink to the bottom of the tube.
A two-sided adhesive spacer was attached to a 24 × 50 mm glass coverslide and 300 μL 0.02 mg/mL streptavidin solution in PBS (pH 7.4) was incubated inside the spacer for 1 h. The coverslide was then washed carefully with 100 mM glucose solution, and 200 μL of the GUV solution from the bottom of the reaction tube was added to the coverslide. The chamber was closed by attaching an 18 × 18 mm coverslide on the top part of the spacer. The solution was incubated for 2 h at room temperature to allow for sufficient biotin-streptavidin binding.
Protein expression, purification, and labeling
TRC40/cyctochorme-B5 complex in HEPES buffer (50 mM HEPES KOH (pH 7.4), 150 mM potassium acetate, 10 mM magnesium acetate, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, and 20 mM maltose) was provided by Fabio Villardi and Blanche Schwappach (Georg-August-University, Göttingen, Germany). Fluorescent labeling with Alexa647 succinimidyl ester was performed at pH 8.3 in NaHCO3 buffer for 3 h in the dark at room temperature with a twofold excess of fluorescent dye with regard to the amount of lysines in the protein. The labeled protein was then purified by size-exclusion chromatography using a self-packed Sephadex G-25 column (GE Healthcare, Munich, Germany).
The cytochrome B5 crystal structure was taken from the RCSB Protein Data Bank, file 2i96.
KcsA from Streptomyces lividans with a C-terminal hexahistidine tag in pQE60 vector was provided by Hildgund Schrempf (University of Osnarbrück, Osnarbrück, Germany) and transformed into E. coli BL21 gold bacteria (Agilent Technologies, Waldbronn, Germany). The bacteria were grown in lysogeny broth with 0.1 g/L ampicillin at 37°C. At OD600 = 0.4, expression was induced by addition of isopropyl-P-D-thiogalactoside to a final concentration of 1 mM and incubated for an additional 2 h at 37°C before cells were collected by centrifugation. After resuspension of the cells in lysis buffer (50 mM tricine (pH 7.5), 150 mM NaCl, 5 mM KCl, 0.02 mg/mL DNase, 0.2 mg/mL Lysozym, 1:100 protease inhibitor mix (50 mg/mL Tame, 1.33 mg/mL trypsin, 1 mg/mL pepstatin A, 5 mg/mL leupeptin, and 1 mg/mL aprotinin)), the cells were disrupted by sonification and pelleted by ultracentrifugation (100,000 × g, 20 min at 4°C). The pellet was resuspended in 4 mL buffer (50 mM tricine (pH 7.5), 150 mM NaCl, 5 mM KCl, and protease inhibitor mix (1:100)). Then, 2 mL solubilization buffer (50 mM tricine (pH 7.5), 150 mM NaCl, 5 mM KCl, and 20 mM DM) was added and the mixture was incubated on a tilting table for 2 h at 10°C. After centrifugation at 5000 × g for 30 min at 4°C, the supernatant was loaded on a self-packed Ni-NTA column of 3 mL bed volume equilibrated with 30 mL washing buffer 1 (50 mM tricine (pH 7.5), 150 mM NaCl, 5 mM KCl, 5 mM DM, and 50 mM imidazole). The protein was allowed to bind to Ni-NTA for 40 min. Afterward, the column was washed with 30 mL of washing buffer 2 (50 mM tricine (pH 7.5), 150 mM NaCl, 5 mM KCl, 5 mM DM, and 100 mM imidazole) and eluted with 6 mL elution buffer (50 mM tricine (pH 7.5), 150 mM NaCl, 5 mM KCl, 5 mM DM, and 500 mM imidazole).
For labeling, the buffer was exchanged with PBS (136.9 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4 × 12 H2O, and 5 mM DM, pH 7.4) using Vivaspin columns (Satorius, Göttingen, Germany) with a membrane of 10 kDa molecular weight cutoff (MWCO). For efficient succinimidyl ester formation, the pH was adjusted to pH 8.3 by adding aqueous NaHCO3 solution to the PBS buffer before labeling. Alexa647 succinimidyl ester was added in a 10-fold molar excess with respect to the amount of lysines present and incubated for 3 h in the dark at room temperature. The remaining free dye was removed via size-exclusion chromatography (Sephadex G-25, GE Healthcare, Munich, Germany).
The crystal structure of KcsA was taken from the RCSB Protein Data Bank, file 1BL8.
EcClC from E. coli with C-terminal hexahistidine tag in pET28 vector was provided by Raimund Dutzler (University of Zurich, Zurich, Switzerland) and transformed into E. coli BL21 gold. The protein was expressed as reported (12).
For purification, the pellet was resuspended in lysis buffer (50 mM Tris HCl (pH 7.5), 150 mM NaCl, 0.02 mg/mL DNase, 0.2 mg/mL Lysozym, 1:100 protease inhibitor mix, 1 mM phenylmethylsulfonyl fluoride, and 0.1% Triton X-100) and the cells were disrupted by sonification. DM was added to the solution to a final concentration of 50 mM. The mixture was incubated for 2 h at room temperature on a tilting table and then centrifuged at 40,000 × g for 30 min at 4°C. The pellet was discarded and the supernatant loaded on a self-packed Ni-NTA column of 3 mL bed volume that was previously washed with 30 mL Tris buffer (50 mM Tris HCl (pH 7.5) and 150 mM NaCl). The protein was allowed to bind to the column for 45 min. The column was washed with 20 mL washing buffer (PBS (pH 7.4), 10 mM DM, and 100 mM imidazole) and eluted with 6 mL elution buffer (PBS (pH 7.4), 10 mM DM, and 400 mM imidazole).
The protein was then labeled in elution buffer with Alexa647NHS with a fivefold molar excess of dye with respect to the amount of lysines present. The pH of the elution buffer was adjusted to 8.3 with 1 M aqueous NaHCO3 solution. The remaining free dye was removed as described via size-exclusion chromatography.
The crystal structure of EcClC was taken from the RCSB Protein Data Bank, file 1OTS.
AcrB from E. coli with a C-terminal histidine tag in pET24 vector was provided by Klaas Martinus Pos (J.-W.-Goethe-University, Frankfurt, Germany) and transformed into E. coli BL21 gold. The bacteria were grown in lysogeny broth with 0.1 g/L kanamycine at 37°C to an OD600 of 0.9. The culture was cooled to 4°C for 20 min before expression was induced by addition of isopropyl-P-D-thiogalactoside to a final concentration of 0.5 mM and incubated for an additional 2 h at 37°C. The cells were collected by centrifugation (5000 × g, 30 min at 4°C). All purification steps were carried out on ice. The pellet was resuspended in lysis buffer (20 mM Tris (pH 8), 500 mM NaCl, 2 mM MgCl2, 0.02 mg/mL DNase, and 0.2 mg/mL Lysozym). The cells were disrupted by sonification and collected by centrifugation at 9000 × g for 10 min at 4°C. The pellet was discarded and the supernatant subjected to ultracentrifugation at 45,000 × g for 1 h at 4°C. The resulting pellet was resuspended in 1 mL 20 mM Tris HCl (pH 8) and 500 mM NaCl. Then, 1 mL 2 mol/L DM solution and 2.2 mL buffer A (10 mM NaPipes (pH 8), 190 mM NaCl, 10 mM KCl, 10 mM imidazole, 10% glycerol, 1 mM DM) was added and the mixture was rotated slowly for 2 h on ice in the cold room (10°C). Afterward, the sample was spun at 45,000 × g at 4°C for 1 h. The pellet was discarded and the supernatant was loaded on a self-packed Ni-NTA column of 3 mL bed volume equilibrated with 10 mL of buffer A. Because AcrB labeling was performed during Ni-NTA affinity chromatography, 75 μg Alexa647 maleimide (50-fold molar excess with respect to the number of cysteines present) was added to the solution on the Ni-NTA resin. The pH was adjusted to 7.2 by addition of HCl to allow for efficient labeling of the cysteine groups. The mixture was incubated overnight on a tilting table on ice in the cold room (10°C).
Excess free dye was removed by washing the column with 60 mL washing buffer (10 mM NaPipes, pH 8, 190 mM NaCl, 10 mM KCl, 10% glycerol, and 1 mM DM, pH 8). Afterward, nonspecifically bound proteins were removed with 40 mL washing buffer A (same composition as washing buffer + 10 mM imidazole, pH 8) and 30 mL washing buffer B (200 mM NaCl, 10% glycerol, 1 mM DM, and 50 mM imidazole (pH 7)). The protein was eluted with 6 mL elution buffer (200 mM NaCl, 10% glycerol, 1 mM DM, and 200 mM imidazole (pH 5)).
The crystal structure of AcrB was taken from the RCSB Protein Data Bank, file 2GIF.
LUV preparation and vesicle fusion
25 μL of 25 mg/mL POPE and 16 μL of 25 mg/mL POPC solution in chloroform were mixed and the solvent was evaporated. The lipids were then resuspended in 800 μL PBS (pH 7.4) under vigorous shaking for 60 min. Afterward, the mixture was extruded for 350 cycles using a lipid extruder from Avanti Polar Lipids (Alabaster, AL) with a 100 nm polycarbonate membrane (GE Healthcare/Whatman, Piscataway, NJ).
To incorporate fluorescently labeled KcsA into large unilamellar vesicles (LUVs), the protein was subjected to a buffer exchange from tricine (50 mM tricine, 150 mM NaCl, 5 mM KCl, 500 mM Imidazole, and 5 mM DM (pH 7.5)) to PBS (150 mM NaCl and 50 mM octyl glycoside (pH 7.4)) before labeling using Vivaspin columns with 10 kDa MWCO membrane. Labeling was done as described. Then, 1 mL of a 10 μM fluorescently labeled protein solution was mixed with the LUVs, and 20 μL of a 156 μM unlabeled Synaptobrevin-2 (Syb) solution provided by Geert van den Bogaart and Reinhard Jahn (Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany) was added. The mixture was dialyzed overnight at 10°C in a dialysis tube with 3 kDa pore size against PBS containing Biobeads SM-2 (Biorad, Munich, Germany) to remove the detergent. Afterward, the solution was purified via size-exclusion chromatography to remove the remaining detergent and free dye.
Reconstitution of fluorescently labeled EcClC into POPC/POPE LUVs was done as described for KcsA. AcrB was reconstituted according to the protocol of Zgurskava and Nikaido (13). In addition, Syb was added to the membrane protein/LUV mixture and the vesicles were purified via size-exclusion chromatography after dialysis as described previously for KcsA reconstitution.
Fluorescently labeled cytochrome B5 (CytB5) was reconstituted into LUVs by combining 200 μL of 0.3 μM CytB5 solution with 10 μL of 156 μM Syb to 400 μL LUV solution. The mixture was incubated overnight in the dark at 4°C.
ΔN complex (14) provided by Geert van den Bogaart and Reinhard Jahn was added to the chip to a final concentration of 0.44 μM. It was allowed to incorporate into the bilayer for 15 min before 20 μL of the vesicle solution was added. The mixture was then equilibrated for another 10 min to allow for SNARE fusion. This time was found to be sufficient to yield a single-molecule concentration of labeled proteins in the BLM.
Dual-focus fluorescence correlation spectroscopy
A principal scheme of the setup and the measurement details has been described in detail elsewhere (9,11). Briefly, two linearly polarized pulsed diode lasers of 640 nm wavelength were used for excitation. The pulse duration was 50 ps (full width at half-maximum). Both lasers were pulsed alternately with a repetition rate of 40 MHz (pulsed interleaved excitation (15)). The (continuous-wave) laser power was adjusted to 3 μW. The beams are combined by a polarizing beam splitter and coupled into a polarization-maintaining single-mode fiber. At the fiber output, the light is collimated and reflected by a dichroic mirror (FITC/TRITC, Chroma Technology, Rockingham, VT). Before entering the objective (UPLSAPO 60×W, 1.2 NA, Olympus Deutschland, Hamburg, Germany), the light passes through a Nomarski prism, which deflects the beams into different directions according to their polarization. After focusing by the objective, two laterally shifted overlapping excitation foci are generated. The distance between the foci is determined by the Nomarski prism and the wavelength of the lasers and was 450 nm in our case.
Fluorescence from the sample is collected by the same objective. The light then passes through a dichroic mirror and is focused onto a pinhole (150 μm diameter) after which it is collimated, split by a 50/50 beam splitter and focused onto two single-photon avalanche diodes (APDs) (SPCM-AQR-13, PerkinElmer Optoelectronics, Wiesbaden, Germany). The detected photons of both APDs are recorded independently by single-photon counting electronics (HydraHarp 400, PicoQuant, Berlin, Germany) with an absolute temporal resolution of 2 ps on a common time frame.
Each fluorescence photon can be associated with the laser pulse that excited it, i.e., in whose focus it was excited. Thus, autocorrelation functions for each focus and a cross-correlation function between the foci can be calculated with a dedicated software algorithm (16). To avoid corruption of the calculations by afterpulsing effects, only photon pairs that have been detected in both APDs are correlated. To ensure correct positioning on the BLM, each measurement was performed for 10 min before readjusting the foci. Diffusion coefficients were extracted as described in Weiß and Enderlein (11). All measurements were performed at 22°C. Bilayer rupture and movement of the BLM out of focus could be seen by a drastic decrease of the amplitude in the correlation curves. The respective curves were not included in the evaluation.
Lipid diffusion measurements in GUVs were performed in a 3.4% sucrose/glucose solution. The viscosity of this solution is 0.99 mPa·s and was determined by measuring Alexa647 succinimidyl ester in the respective aqueous sucrose/glucose mixture. The obtained viscosity essentially matches the viscosity of water (0.96 mPa·s at 22°C), which means that the results for lipid diffusion in BLMs and GUVs can be directly compared.
For protein diffusion measurements in solution, the respective samples were diluted 1:100 with PBS buffer (pH 7.4) to decrease the surfactant concentration below its critical micelle concentration (cmc). Alternatively, size-exclusion chromatography (Sephadex G-25) using PBS buffer was performed. The latter method was especially useful for EcClC and AcrB, since they were very unstable upon surfactant removal and size-exclusion chromatography resulted in samples of higher homogeneity. 2fFCS measurements were performed for 10 min at 3 μW. Because the samples were highly unstable, they were prepared freshly before each measurement and only used for one single 10-min measurement.
Electrophysiology
KcsA was purified, labeled, and reconstituted into vesicles with Syb as described above. A BLM with 4 mg/mL POPC, 6 mg/mL POPE, and 3 mg/mL 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) as sodium salt was formed as described using the Ionovation Bilayer Explorer in a PBS buffer with 400 mM KCl (pH 4). The choice of lipid mixture and buffer was based on the methods of others (17–19). ΔN complex was added and incubated with the bilayer for 15 min. Vesicles were added and fused with the bilayer as described. For electrophysiology measurements, a HEKA EPC10 patch-clamp amplifier was used.
Membrane voltages were clamped to +200 mV or −200 mV. Currents were filtered at 2 kHz and sampled at 10 kHz. For display, traces were low-pass filtered with a digital Bessel filter (250–200 Hz cutoff) with a notch at 50 Hz to remove line noise. Afterward, 1 mM tetrabutyl ammonium (TBA) was added to block the KcsA channels. In control experiments, a POPC/POPE BLM in the same buffer without protein was measured. BLM and blocked KcsA were sampled as described before using +100 mV and −100 mV pulses.
Results
For bilayer preparation, POPE and POPC were used because they are abundant in biological membranes and form a homogeneous lipid system with a phase-transition temperature below 22°C. Since the bilayers were formed using a painting technique, the influence of potentially remaining solvent inside the BLM was checked by measuring lipid diffusion in BLMs and in solvent-free GUVs. In BLMs, a diffusion coefficient of DBLM = 11.6 (± 0.6) μm2 s−1 was obtained. In GUVs, we measured DGUV = 11.5 (± 0.6) μm2 s−1. The viscosity-corrected results for BLMs and GUVs match, indicating that the effect of the remaining solvent inside the BLM is negligibly small.
For the diffusion measurements, we have chosen proteins that are well characterized in terms of crystal structure and functionality, do not aggregate inside the membrane, and cover a large size range. In particular, we studied the heme protein CytB5, which is involved in electron transport, the potassium channel KcsA, the chloride channel EcClC, and the multidrug efflux pump AcrB. All proteins and their structural information are listed in Table 1. The cylindrical radii of the transmembrane domains were estimated from their respective crystal structures. For CytB5, the published crystal structures omit the protein’s transmembrane domain. It is known, however, that this domain consists of one α-helix (20). The cylindrical radius of CytB5 was therefore estimated to be 0.7 nm, i.e., 0.5 nm for the fixed backbone and two C-C distances for the side chains.
Table 1.
Investigated proteins and lipids
| Protein | Structure | RLit (nm) | D (± SD) μm2 s−1 (direct addition) | D (± SD) (μm2 s−1) (SNARE-mediated fusion) |
|---|---|---|---|---|
| DPPE (lipid) | monomer | 0.4 | 11.58 (± 0.57) | — |
| Cytochrome B5 | monomer | 0.7 | 10.24 (± 0.63) | 10.43 (± 0.76) |
| KcsA | monomer | 1.2 | 9.27 (± 0.53) | — |
| tetramer | 2.4 | — | 7.96 (± 0.53) | |
| EcClC | monomer | 1.8 | 8.49 (± 0.46) | — |
| dimer | 2.8 | — | 7.51 (± 0.84) | |
| AcrB | monomer | 2.1 | 8.47 (± 0.75) | — |
| trimer | 3.6 | — | 7.23 (± 0.39) |
The estimated radii, RLit, were taken from the respective crystal structures. Diffusion coefficients, D, were determined by 2fFCS. All diffusion measurements were performed at 22°C. SD, standard deviation.
Two different approaches have been used to incorporate the proteins into the BLMs: direct addition to the buffer surrounding the membrane and SNARE-mediated vesicle fusion. The latter was performed by reconstituting the fluorescently labeled protein of interest together with Synaptobrevin-2 into LUVs. ΔN complexes, consisting of Syntaxin and SNAP-25, were incorporated into the BLM. This combination leads to a significantly increased fusion speed compared to regular SNARE complexes where Syntaxin and SNAP-25 are not previously bound. Employing the ΔN complex yields efficient vesicle fusion within 1–2 min (14).
Using 2fFCS measurements in solution, we tested the behavior of surfactant-stabilized protein solutions after dilution below the surfactant’s cmc. This recreates the experimental conditions of direct addition to the BLM just before protein insertion into the membrane. Diffusion measurements of fluorescently labeled KcsA in surfactant solution before dilution yielded correlation curves that could not be fitted in a meaningful way (see Fig. S1 in the Supporting Material), indicating an extremely polydisperse sample. When the surfactant was diluted below its cmc, however, the curves became well defined and could be fitted perfectly with a single diffusion coefficient (see Fig. S2) representing a homogeneous solution of molecules. The obtained value corresponds to the diffusion of KcsA monomers. The same measurements were performed with EcClC and AcrB, also yielding diffusion coefficients that indicate the presence of the respective monomers (Table 2). All measurements were done immediately after dilution, and all samples were only used for one measurement, since they are highly unstable and the protein precipitates shortly after surfactant removal.
Table 2.
Protein diffusion in solution after dilution below surfactant cmc
| Protein | RLit (nm) | D (μm2 s−1) | R (nm) |
|---|---|---|---|
| KcsA | 1.2 | 280 (± 7) | 0.8 |
| EcClC | 1.8 | 120 (± 3) | 1.9 |
| AcrB | 2.1 | 107 (± 6) | 2.1 |
The radii, R, were calculated using the Stokes-Einstein equation for three-dimensional diffusion with T = 295 K and μ22°C = 0.96 mPa·s for the aqueous PBS buffer. The literature values, RLit, were taken from the respective crystal structures.
These results show that we could incorporate proteins in the monomeric form (CytB5, KcsA, EcClC, and AcrB) by direct addition. SNARE-mediated vesicle fusion incorporates the oligomeric proteins (KcsA tetramers, EcClC dimers, and AcrB trimers).
Because the state of the proteins in the membrane is a crucial factor of this study, further tests have been done to confirm these findings.
KcsA monomers were generated by heating the proteins after purification, i.e., in the presence of surfactant, based on the work of Valiyaveetil et al. (17). Monomer formation was shown by SDS PAGE (Fig. S4). Addition to the BLM leads to incorporation of the protein. The measured diffusion coefficient was 9.1 (± 0.2) μm2 s−1. Without heating, a diffusion coefficient of 9.3 (± 0.5) μm2 s−1 for KcsA was obtained after direct addition.
Using KcsA as a representative protein, incorporation of functional oligomeric proteins upon SNARE-mediated vesicle fusion was checked with electrophysiological measurements and yielded multiple channel openings. The measured conductance matched the value expected for KcsA (see Supporting Material). Direct addition of KcsA did not yield conductance steps, indicating that only monomers were present in the bilayer.
The obtained membrane diffusion data for all proteins were fitted with the classical Saffman-Delbrück model (see Eq. 1) and the Stokes-Einstein-like model suggested by Gambin et al. (see Eq. 2). The results are shown in Fig. 1. The product of membrane viscosity and membrane thickness, μmh, was used as the fit parameter.
Figure 1.
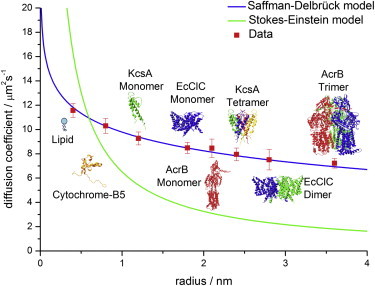
Saffman-Delbrück versus Stokes-Einstein model. The investigated species are DPPE, cytochrome B5 (depicted without transmembrane domain), KcsA, EcClC, and AcrB. The monomeric forms of membrane proteins were directly added to the BLM. The oligomeric forms were reconstituted via SNARE-mediated vesicle fusion. DPPE was labeled with Atto655, and all proteins were labeled with Alexa647. In addition, we fitted the HPW-based model suggested by Petrov and Schwille (6), which can reproduce the classical Saffman-Delbrück model in the size range investigated. The fit parameter for all fits was the product of membrane viscosity and thickness, μmh. The temperature was set to 295 K and the viscosity of the surrounding buffer was μs = 0.96 mPa·s.
Our diffusion data are almost perfectly described by the ln 1/R scaling suggested by the Saffman-Delbrück model. The 1/R scaling fails completely in describing protein diffusion. As expected and previously pointed out theoretically (6), the HPW-based model coincides with the Saffman-Delbrück model within the size range investigated.
The Saffman-Delbrück model becomes invalid for larger protein radii. In particular, the reduced radius ε′ is defined as
with μs the solvent viscosity, μm the membrane viscosity, h the membrane height, and R the protein radius, determines the crossover from ln 1/R to 1/R scaling. The Saffman Delbrück model is valid for ε′ < 0.1, whereas for ε′ > 1 the Saffman-Delbrück scaling fails completely (6). For our system, this means that the Saffman-Delbrück model is valid up to a protein radius of R ≈ 8 nm when substituting the specific parameters of our system, μs = 0.96 mPa·s, μm = 39.5 mPa·s, h = 3.8 nm, and ε′ = 0.1, into Eq. 4. This radius is well above the protein size range investigated in our study. Thus, the Saffman-Delbrück model indeed yields an accurate description of protein diffusion in our study.
Discussion
Using 2fFCS for measuring protein diffusion in membranes yielded highly accurate quantitative results and verified the Saffman-Delbrück model. Although the values for the diffusion coefficients are very precise, deviations might occur in the radii since the radius values were taken from the protein crystal structures and might not match the hydrodynamic radii exactly. This should be a minor problem though, because even rather large deviations in R up to 0.5 nm in either direction do not influence the scaling behavior significantly.
The Saffman-Delbrück model can be used to determine the membrane viscosity. Assuming a bilayer thickness of 3.8 nm (21), we obtain a membrane viscosity of 39.5 mPa·s from the Saffman-Delbrück equation (Eq. 1) with T = 295 K. This is smaller than the membrane viscosities reported, which are in the range 75–150 mPa·s (22). This discrepancy could be attributed to differences in lipid mixtures and measurement techniques. We can rule out that the smaller membrane viscosity is due to effects of organic solvent inside the BLM, because we found exactly the same diffusion coefficient in BLMs and solvent-free GUVs prepared with the same lipid mixture.
A more recent study (23) finds membrane viscosities in the range 3–150 mPa·s in the liquid-disordered phase of domain-forming lipid mixtures. Our result fits well into this range.
Comparing the obtained data with the study by Ramadurai et al. (3), we observe a striking discrepancy in protein diffusion coefficients. The values found by Ramadurai et al. are ∼45% smaller than ours, whereas their obtained lipid diffusion coefficient of 11.4 ± 0.7 μm2 s−1 corresponds perfectly to our lipid diffusion value. This makes their lipid diffusion coefficient about twice as high as the diffusion coefficient of their smallest investigated protein, Syb, whereas the difference in radii is only 0.1 nm. These deviations may be caused by aggregation of the proteins within the membrane during GUV formation by drying and rehydration, which is likely, since exact adjustment of the sucrose level in the rehydration buffer is challenging. Moreover, the diffusion values for larger proteins scatter significantly and the Saffman-Delbrück scaling is indicated only by comparing these values to Syb and WALP23 diffusion. Another problem could be the use of FCS, which is very sensitive to optical imperfections such as refractive index mismatch, laser beam astigmatism, or coverslide thickness deviations. Absolute values for diffusion coefficients can only be obtained by calibrating the FCS system with a dye of known diffusion coefficient, which can be problematic, and precise alignment of the focal plane with the GUV membrane is difficult.
To avoid any aggregation problems, proteins in this study were incorporated either directly or upon SNARE-mediated vesicle fusion without drying or rehydration. The differences in reconstitution behavior between the proteins can be explained by the differences in experimental conditions, in particular the surfactant concentration. When directly added to the chip, the protein solution is diluted below the surfactant’s cmc. Therefore, the proteins are no longer stabilized by surfactant micelles. We hypothesize that only a few surfactant molecules remain attached to the proteins due to the favorable hydrophobic interactions. This seems to be enough to stabilize the monomers in solution for the time span needed for insertion. The protein-lipid interactions inside membranes should be much more favorable than the interaction with single surfactant molecules, resulting in monomer incorporation into the bilayer. This theory is supported by protein diffusion measurements in solution before and after dilution. Before dilution, the sample is very polydisperse because the proteins are stabilized by surfactant micelles of different sizes. After dilution, the micelles vanish and the sample becomes monodisperse. The resulting diffusion coefficients correspond to the monomer radii, which indicates that the surfactants remaining on the protein are closely attached and do not change their size significantly.
After incorporation of the monomers inside the bilayer, the proteins could theoretically reassemble. This was not observed in our system, which is probably due to the extremely small concentration (≈1 protein/μm2).
In contrast, SNARE-mediated vesicle fusion leads to incorporation of the proteins in their oligomeric form because they are highly stabilized the entire time either by surfactant micelles or by lipid bilayers (LUVs and BLMs) upon surfactant removal. The incorporation of proteins into the LUVs before membrane fusion proceeds at much higher concentrations (μM) compared to the actual measurement and the direct-addition pathway (nM to pM). Therefore, disassembly of the oligomeric proteins is unlikely.
Conclusion
We investigated lipid and protein diffusion in homogeneous POPC/POPE BLMs with 2fFCS and obtained highly accurate results. Comparing the observed diffusion coefficients, we find a D ∝ ln 1/R scaling. The Saffman-Delbrück model is therefore able to describe diffusion for proteins of various sizes and shapes despite its simplicity. Furthermore, the accurately obtained diffusion values allow for a precise calculation of the membrane viscosity.
Acknowledgments
The authors thank Christian Eggeling for providing fluorescently labeled lipid and Fabio Villardi and Blanche Schwappach for providing the TRC40/cytochrome-B5 complex. We also thank Geert van den Bogaart and Reinhard Jahn for the SNARE proteins. The authors are grateful to Raimund Dutzler, Christopher Miller, Geert van den Bogaart, Fabio Villardi, and Blanche Schwappach for helpful discussions. The authors are thankful to Robin Padilla for proofreading.
Funding by the German Science Foundation (DFG, SFB 803, project A10) is gratefully acknowledged.
Supporting Material
References
- 1.Saffman P.G., Delbrück M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gambin Y., Lopez-Esparza R., Urbach W. Lateral mobility of proteins in liquid membranes revisited. Proc. Natl. Acad. Sci. USA. 2006;103:2098–2102. doi: 10.1073/pnas.0511026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramadurai S., Holt A., Poolman B. Lateral diffusion of membrane proteins. J. Am. Chem. Soc. 2009;131:12650–12656. doi: 10.1021/ja902853g. [DOI] [PubMed] [Google Scholar]
- 4.Naji A., Levine A.J., Pincus P.A. Corrections to the Saffman-Delbruck mobility for membrane bound proteins. Biophys. J. 2007;93:L49–L51. doi: 10.1529/biophysj.107.119222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guigas G., Weiss M. Influence of hydrophobic mismatching on membrane protein diffusion. Biophys. J. 2008;95:L25–L27. doi: 10.1529/biophysj.108.136069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrov E.P., Schwille P. Translational diffusion in lipid membranes beyond the Saffman-Delbruck approximation. Biophys. J. 2008;94:L41–L43. doi: 10.1529/biophysj.107.126565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dertinger T., von der Hocht I., Enderlein J. Surface sticking and lateral diffusion of lipids in supported bilayers. Langmuir. 2006;22:9339–9344. doi: 10.1021/la061389s. [DOI] [PubMed] [Google Scholar]
- 8.Hughes B.D., Pailthorpe B.A., White L.R. The translational and rotational drag on a cylinder moving in a membrane. J. Fluid Mech. 1981;110:349–372. [Google Scholar]
- 9.Dertinger T., Pacheco V., Enderlein J. Two-focus fluorescence correlation spectroscopy: a new tool for accurate and absolute diffusion measurements. ChemPhysChem. 2007;8:433–443. doi: 10.1002/cphc.200600638. [DOI] [PubMed] [Google Scholar]
- 10.Benda A., Beneš M., Hof M. How to determine diffusion coefficients in planar phospholipid systems by confocal fluorescence correlation spectroscopy. Langmuir. 2003;19:4120–4126. [Google Scholar]
- 11.Weiß K., Enderlein J. Lipid diffusion within black lipid membranes measured with dual-focus fluorescence correlation spectroscopy. ChemPhysChem. 2012;13:990–1000. doi: 10.1002/cphc.201100680. [DOI] [PubMed] [Google Scholar]
- 12.Dutzler R., Campbell E.B., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 13.Zgurskaya H.I., Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. USA. 1999;96:7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pobbati A.V., Stein A., Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313:673–676. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- 15.Müller B.K., Zaychikov E., Lamb D.C. Pulsed interleaved excitation. Biophys. J. 2005;89:3508–3522. doi: 10.1529/biophysj.105.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wahl M., Gregor I., Enderlein J. Fast calculation of fluorescence correlation data with asynchronous time-correlated single-photon counting. Opt. Express. 2003;11:3583–3591. doi: 10.1364/oe.11.003583. [DOI] [PubMed] [Google Scholar]
- 17.Valiyaveetil F.I., Zhou Y., MacKinnon R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry. 2002;41:10771–10777. doi: 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- 18.Heginbotham L., LeMasurier M., Miller C. Single streptomyces lividans K+ channels: functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 1999;114:551–560. doi: 10.1085/jgp.114.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faraldo-Gómez J.D., Kutluay E., Roux B. Mechanism of intracellular block of the KcsA K+ channel by tetrabutylammonium: insights from x-ray crystallography, electrophysiology and replica-exchange molecular dynamics simulations. J. Mol. Biol. 2007;365:649–662. doi: 10.1016/j.jmb.2006.09.069. [DOI] [PubMed] [Google Scholar]
- 20.Clarke T.A., Im S.C., Waskell L. The role of the length and sequence of the linker domain of cytochrome b5 in stimulating cytochrome P450 2B4 catalysis. J. Biol. Chem. 2004;279:36809–36818. doi: 10.1074/jbc.M406055200. [DOI] [PubMed] [Google Scholar]
- 21.Chen R., Poger D., Mark A.E. Effect of high pressure on fully hydrated DPPC and POPC bilayers. J. Phys. Chem. B. 2011;115:1038–1044. doi: 10.1021/jp110002q. [DOI] [PubMed] [Google Scholar]
- 22.Vaz W.L.C., Goodsaid-Zalduondo F., Jacobson K. Lateral diffusion of lipids and proteins in bilayer membranes. FEBS Lett. 1984;174:199–207. [Google Scholar]
- 23.Cicuta P., Keller S.L., Veatch S.L. Diffusion of liquid domains in lipid bilayer membranes. J. Phys. Chem. B. 2007;111:3328–3331. doi: 10.1021/jp0702088. [DOI] [PubMed] [Google Scholar]
- 24.Robertson J.L., Kolmakova-Partensky L., Miller C. Design, function and structure of a monomeric ClC transporter. Nature. 2010;468:844–847. doi: 10.1038/nature09556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutzler R. A structural perspective on ClC channel and transporter function. FEBS Lett. 2007;581:2839–2844. doi: 10.1016/j.febslet.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 26.Seeger M.A., Schiefner A., Pos K.M. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 27.Blunck R., McGuire H., Bezanilla F. Fluorescence detection of the movement of single KcsA subunits reveals cooperativity. Proc. Natl. Acad. Sci. USA. 2008;105:20263–20268. doi: 10.1073/pnas.0807056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.