

Abstract
WASp-interacting protein (WIP) is a 503-residue proline-rich polypeptide expressed in human T cells. The WIP C-terminal domain binds to Wiskott-Aldrich syndrome protein (WASp) and regulates its activation and degradation, and the WIP-WASp interaction has been shown to be critical for actin polymerization and implicated in the onset of WAS and X-linked thrombocytopenia. WIP is predicted to be an intrinsically disordered protein, a class of polypeptides that are of great interest because they violate the traditional structure-function paradigm. In this first (to our knowledge) study of WIP in its unbound state, we used NMR to investigate the biophysical behavior of WIPC, a C-terminal domain fragment of WIP that includes residues 407–503 and contains the WASp-binding site. In light of the poor spectral dispersion exhibited by WIPC and the high occurrence (25%) of proline residues, we employed 5D-NMR13C-detected NMR experiments with nonuniform sampling to accomplish full resonance assignment. Secondary chemical-shift analysis, 15N relaxation rates, and protection from solvent exchange all concurred in detecting transient structure located in motifs that span the WASp-binding site. Residues 446–456 exhibited a propensity for helical conformation, and an extended conformation followed by a short, capped helix was observed for residues 468–478. The 13C-detected approach allows chemical-shift assignment in the WIPC polyproline stretches and thus sheds light on their conformation and dynamics. The effects of temperature on chemical shifts referenced to a denatured sample of the polypeptide demonstrate that heating reduces the structural character of WIPC. Thus, we conclude that the disordered WIPC fragment is comprised of regions with latent structure connected by flexible loops, an architecture with implications for binding affinity and function.
Introduction
Wiskott-Aldrich syndrome protein (WASp) is expressed predominantly in hematopoietic cells and is a key participant in actin polymerization, which accompanies activation of T cells. Its cellular levels are closely controlled by a combination of activation and degradation mechanisms. Mutations in the WASp-encoding gene that disturb this delicate balance have been implicated in Wiskott-Aldrich syndrome (WAS) and X-linked thrombocytopenia (XLT) (1), both of which are hereditary immunodeficiency conditions characterized by impaired cytoskeleton formation and an increased incidence of autoimmune diseases and malignancies (2). WASp-interacting protein (WIP) acts as the cellular chaperone of WASp, conveys WASp to areas of active actin assembly after antigen-receptor and chemokine receptor signaling (3,4), and stabilizes WASp by shielding it from cellular degradation systems (5). The pathological effects of WAS-inducing mutations located in the N-terminal EVH1 domain of WASp result from a reduced affinity to WIP, suggesting that WIP’s chaperone function is essential for WASp stability. Supporting this notion is the fact that in one study (6), WIP-deficient mice had normal levels of WASp messenger RNA (mRNA) levels but suffered from decreased WASP activity, which could be rescued by exogenous addition of WIP. In resting lymphocytes, >95% of WASp is complexed with WIP, which inhibits Cdc42-induced activation of WASp (7).
As a member of the mammalian verprolin family, the 503-residue WIP is comprised of an N-terminal actin-binding WAS-homology (WH2) domain, proline-rich domains (PRDs) that bind profilin and SH3 domains, and a C-terminal segment capable of binding the N-terminal EVH1 domain of WASp (8). Disruption of the latter interface is the cause of WAS. The C-terminal region contains a consensus phosphorylation motif (9) that is recognized by the θ isoform of protein kinase C (PKCθ), and this phosphorylation leads to activation of WASp (7,10). A 35-mer peptide consisting of WIP residues 451–485 fused to a neuronal WASp homolog (N-WASp) was demonstrated to be an N-WASp-binding determinant comprised of three well-conserved epitopes, spanning residues 454–478. The WIP peptide, including a central polyproline motif, was shown to wrap around and bind to an elongated interaction surface on N-WASp that is best described as a concave spool (11,12). To date, the unbound state of WIP has not been structurally investigated.
WIP (particularly its C-terminal domain) is predicted to be an intrinsically disordered protein (IDP). IDPs fail to adopt a well-defined structure under biologically native conditions, and are best described as an ensemble of unfolded and partially folded states that contribute to their overall behavior in solution (13,14). IDPs are involved in protein aggregation, translocation, and degradation, are quite common in eukaryotic systems, and are closely related to human disease (15,16), accounting for the desire to understand their conformation and dynamics. It is now clear that transient structural elements observed in these polypeptides are associated with their biological function, often reflecting a latent structural propensity that may be evoked and stabilized by formation of a complex with a binding partner (17,18). The multiconformational nature of IDPs makes them flexible biological entities that are capable of interacting with—and thus being regulated by—a variety of biological effectors in response to changing cellular conditions. Located at the crossroads of T cell activation and regulated by its interaction with several cellular factors, WIP appears to fit this paradigm well.
Crystallographic studies of IDPs have been frustrated by the lack of permanent structure, since crystals are difficult to obtain, and any information extracted cannot correctly represent the flexible nature of this class of proteins. In contrast, it is precisely this attribute of IDPs that emphasizes the inherent advantages of high-resolution NMR over other structural methods, explaining why NMR has emerged as the preferred approach for investigating nascent structure in IDPs. Chemical shifts measured by NMR are highly sensitive probes of polypeptide secondary structure, representing an average over multiple populations and allowing the detection of minor contributions (19,20). NMR methods are also capable of detecting molecular motions over a wide range of timescales (21,22), as well as identifying long-range intramolecular contacts and intermolecular interaction surfaces that exhibit intermediate and even low affinity (23,24). The backbone of IDPs exhibits considerable motion on the picosecond-to-nanosecond timescale, resulting in favorable nuclear relaxation properties for NMR measurements, and recent methodological advances have alleviated the difficulties associated with the low spectral dispersion characteristic of IDPs (25–29). These two factors enable resonance assignment and measurement of structural parameters for this class of proteins possible.
Despite its central role in activation-induced actin polymerization in T cells, only limited structural information is available for WIP, and its biological activity is poorly understood on the molecular level. In particular, WIP has not been studied in the unbound state, which may be biologically significant once activation has occurred. In this work, we used NMR methods to determine the structural propensity and dynamics of the C-terminal domain of WIP. Secondary chemical-shift analysis, 15N relaxation measurements, solvent exchange of amide protons, and temperature-induced chemical-shift effects all identified within the WIP C-terminal domain short segments with transient structure connected by unstructured loops. The 13C′-based assignment provided new (to our knowledge) insight into the behavior of polyproline segments, which are usually inaccessible to HN-based assignment experiments. The alternating pattern of structured and unstructured regions may give WIP the flexibility to bind multiple cellular factors in its native biological setting.
Materials and Methods
Acquisition of NMR data
The construct GSSHHHHHH-WIP(407–503)-LEHHHHHH (WIPC) was expressed in M9 minimal medium containing 15NH4Cl (1 g/L), 13C6-D-glucose (2.5 g/L). Detailed expression and purification protocols are described in full in the Supporting Material. All NMR samples (unless stated otherwise) were prepared in 20 mM phosphate buffer, pH 5.0–5.2, 20 mM NaCl, 10 mM βME and 7% 2H2O, and placed in a Shigemi tube (Shigemi, Allison, PA). All 2D- and 3D-NMR measurements were conducted on a DRX700 Bruker spectrometer using a cryogenic triple-resonance TCI probe head equipped with z-axis pulsed field gradients. 4D and 5D experiments were performed on a DRX600 Bruker spectrometer using a cryogenic triple-resonance TCI probe head equipped with z-axis pulsed field gradients.
For backbone assignment we utilized a 13C′-detected strategy (CON spectrum as readout) based on the 3D experiments CANCO, CBCACON, CBCANCO, and C-(CC-TOCSY)-CON (25,30), and the 5D experiments CACONCACO and NCOCANCO (27,28). Details of the NMR experiments performed for purposes of resonance assignment appear in the Supporting Material. All chemical shifts were referenced directly to an external 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) reference measured under sample conditions identical to those used for WIPC. Assigned chemical shifts were deposited in the BioMagResBank (BMRB entry 18265).
15N relaxation data were acquired for a 0.9 mM sample of WIPC on the 700 MHz spectrometer and at 283 K. Previously reported heteronuclear single quantum coherence (HSQC)-based experiments were used to acquire R1, R2, and 15N-{1H}-nuclear Overhauser effect (NOE) parameters for the backbone 15N nuclei (31). Cross-correlated (CC) relaxation ηxy was estimated by acquiring two tr-HSQC spectra in interleaved fashion, differing by a time period of 2Δ during which the N+Hβ and N+Hα operators were interchanged, resulting in a intensity ratio of exp(−4Δηxy) between peaks in the two spectra (32). Details of the experiments conducted to measure WIPC relaxation rates appear in the Supporting Material. The exchange rates of amide protons with solvent at 298 K were estimated using a CLEANEX-PM-based experiment (33) modified to allow for a CON-type readout. Sample pH was raised to 6.0 for purposes of these measurements, with negligible effects on the spectrum, and amide recovery rates after 10 and 20 ms of mixing time were measured. A 13C-detected HNCO experiment served as the reference spectrum.
Analysis of multidimensional spectra and resonance assignment
We processed and visualized 2D- and 3D-NMR spectra using TopSpin2.1 (Bruker, Karlsruhe, Germany). In the 5D-NMR experiments, we processed preliminary 3D (CACO)NCACO and (NCO)CANCO hyperplanes using spectral processing and the analysis system NMRPipe/NMRDraw 3.0 (34), and processed the full 5D experiment using the sparse multidimensional Fourier transform (SMFT) algorithm (35). The procedure for extracting spectral information from these spectra was previously described (28). All of the spectra were analyzed with the use of the NMR assignment and integration software Sparky 3.115 (T.D. Goddard and D.G. Kneller, University of California, San Francisco, CA). Relaxation rates were extracted by least-square fitting of the decay curve using in-house algorithms, and errors in relaxation rates were estimated from the signal/noise ratio in the relevant experiment.
Sedimentation equilibrium experiments
Sedimentation equilibrium (SE) experiments on 15N,13C-WIPC (molecular mass 13,770 g/mol) in 20 mM phosphate buffer (pH 5.2), 20 mM NaCl, 10 mM βME were performed using an XL-I analytical ultracentrifuge equipped with An-60Ti rotor and absorbance optics (Beckman-Coulter, Brea, CA). Data (absorption at 280 nm) were collected at 298 K in double-sector cells of 12 mm thickness. For three different WIPC concentrations (22, 44, and 66 μM), sedimentation curves were recorded after spinning at 30,000 rpm for 10, 12, and 14 h, providing triplicate data with typical standard deviations (SDs) of 0.004 A280 units, or 0.5–2%. We analyzed the average curve using in-house-written scripts based on MATLAB (The MathWorks, Natick, MA) with a nonlinear least-squares approach to extract molecular weight information (36), using a value of 0.68 cm3/g for protein specific volume (37). Selection among the models tested (monomer, monomer-dimer equilibrium, and monomer with high MW aggregate) was based on an F statistic obtained from comparison of residuals.
Circular dichroism experiments
Circular dichroism (CD) experiments were performed on a 6.7 μM sample of 13C,15N-labeled WIPC in the NMR buffer (vide infra) on a Chirascan polarimeter (Applied Photophysics, Surrey, United Kingdom). Experiments were conducted at 278–308 K at 5 K intervals, with each measurement repeated three times and subtracted from a measurement of an identical buffer sample. Results were analyzed using the CDSSTR module of the DichroWeb platform for the 190–240 nm range (38) (http://dichroweb.cryst.bbk.ac.uk).
Results
The C-terminal domain of WIP is a disordered protein
Heterologous expression of large IDPs is challenging to achieve due to their sensitivity to proteolytic degradation in the host cell. We therefore focused on the WASp-binding C-terminal domain of WIP as our structural target, and explored the possibility of expressing C-terminal fragments at sufficient levels. The best results were obtained for a polypeptide corresponding to residues 407–503 of WIP (WIPC; Fig. 1 A), which yielded a sample amenable to NMR study. This segment includes the central verprolin conserved region (VCR; residues 444–478), which is a WASp-binding domain that is common to the veprolin family, exhibiting 60–65% homology among WIP, WIP-related protein, and CR16). The VCR is flanked by two PRDs, the N-terminal segment of residues 413–433 (PRD1), and the C-terminal segment of residues 495–503 (PRD2). The PKCθ consensus sequence R485SGSNR490 is located immediately before the PRD2 sequence (39).
Figure 1.
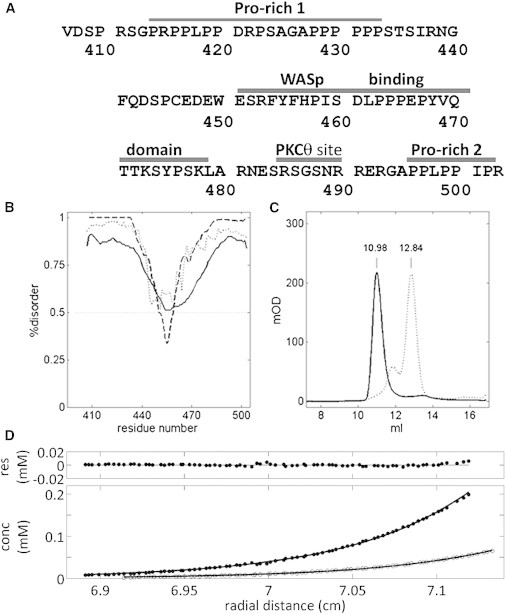
WIPC. (A) Sequence of WIPC showing the WASp-binding epitopes (as defined in Volkman et al. (11)), polyproline stretches, and PKCθ phosphorylation site. (B) Predictions of disorder along WIPC by IUPRED (solid line), RONN (dotted line), and ISUnstruct2.0 (dashed line). (C) Size-exclusion chromatograms for WIPC (solid line) and ribonuclease A (ribA, dotted line), a 13.7 kDa protein. The smaller ribA peak at 11.9 ml behaves as a dimer of ribA (27 kDa) in SDS-PAGE. (D) SE curves for uniformly 15N, 13C-labeled WIPC under conditions identical to those used for the NMR samples at 298 K and 30,000 rpm. Results for WIPC concentrations of 22 and 66 μM are shown in open and solid circles, respectively, with fitted curves corresponding to a monomeric species of 15.2 kDa shown as solid lines. Residuals are shown in the upper panel for the 66 μM concentration.
Sequence analysis of WIPC using three prediction algorithms predicts this 12.9 kDa fragment (13,770 g/mol in its 13C,15N-labeled form) to be generally disordered, with an indication of a more structured region in the VCR segment (Fig. 1 B). This predicted structural propensity was strongest for the aromatic-rich sequence FYFH (residues 454–457), which coincides with one of the conserved WASp-binding epitopes. Indeed, WIPC migrated on a native polyacrylamide gel as a larger protein than expected (data not shown), and in size-exclusion chromatography (SEC) the protein preceded its expected elution volume on an analytical Superdex 75 column of 0.55 column volumes, appearing instead at 0.45 column volumes, corresponding to a protein of ∼45–50 kDa (Fig. 1 C). To ensure that these findings were not biased by aggregation of WIPC, we employed SE methods, which offer a quantitative estimation of protein size. SE curves of 13C,15N-WIPC suggested a mass of ∼15,200 g/mol, close to that expected for a monomer. This deviation from the expected mass may be attributed to the difficulty of accurately determining the specific volume of an IDP, which would cause an overestimation of the molecular mass (37). In any event, the curves were incompatible with the formation of a dimer above the detection limit of 1%, and, notably, the fitted values were independent of sample concentration (Fig. 1 D). Together, these findings suggest a larger-than-expected radius of gyration for WIPC and confirm its disordered character.
Resonance assignment of WIPC
A comparison of the 2D 1H,15N-HSQC, and 2D-IPAP 13C′-15N (CON) spectra of WIPC (Fig. 2) reveals a dual justification for employing 13C′-detected experiments in the case of WIPC: 1), they detect the otherwise invisible proline residues, which comprise 26% of the WIPC sequence; and 2), they display significantly advantageous spectral dispersion. Despite this spread of resonances, a set of 3D-NMR experiments, including CANCO, CBCANCO, and CBCACON (25), could achieve no more than 50% of the assignment. We attributed this to the highly repetitive WIPC sequence, which contains (in its 97 WIP-derived residues) 26% Pro, 15% Ser, and 17% Arg/Glu (with a highly similar 13Cα/13Cβ pair of shifts). To overcome this difficulty, we employed a recently proposed method for assigning unstructured, repetitive sequences based on a pair of 5D-NMR experiments, (H)NCOCANCO and (H)CACONCACO, which utilize nonuniform sampling (NUS) and T1-optimized excitation to correlate five different frequencies within a reasonable experimental time (27,28). This approach is demonstrated in Fig. 3 A. All WIPC residues, with the exception of the 450–456 segment, were successfully assigned using this approach. The absence of this missing region is attributable to its more structured nature (vide infra), with faster relaxation rates limiting the obtainable signal/noise ratio in the relaxation-sensitive 5D-NMR experiments. Supporting this explanation is the fact that these residues afforded significantly weaker peaks in the CON experiment, and signal intensity showed a gradual decrease for residues flanking this segment, pointing to a systematic structural effect rather than an effect of random spectral overlap. By the process of elimination, reconsideration of the above 3D spectra and proton-detected HNCACB spectrum identified a set of weaker peaks corresponding to the missing residues. Using this combined assignment strategy, we were able to fully assign the 97 WIPC segment and flanking residues (Fig. 3 B). In addition, Hα and 13Cβ resonances were obtained from a 1J(NCα)-selective HCBCANCO experiment (40). The complete set of backbone and 13Cβ frequencies lays the necessary groundwork for further characterization of the structure and dynamics of WIPC.
Figure 2.
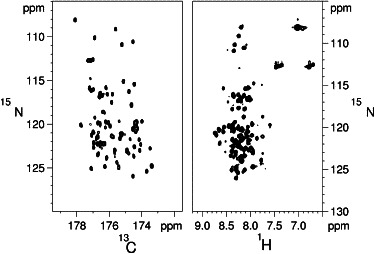
Comparison of fingerprint spectra of WIPC. 13C-15N CON-IPAP (left) and 1H-15N HSQC (right) spectra of WIPC, both acquired at 16.4 T and 298 K. The spectral width shown in the F2 dimension was chosen so that typical crosspeak line widths in this dimension (6 Hz and 23 Hz for the CON and HSQC, respectively) appear equal, demonstrating the advantageous spectral dispersion of the CON spectrum. Peaks emanating from proline residues (in the 134–141 15N ppm range) are not shown, but are clearly another advantageous feature of the CON experiment.
Figure 3.
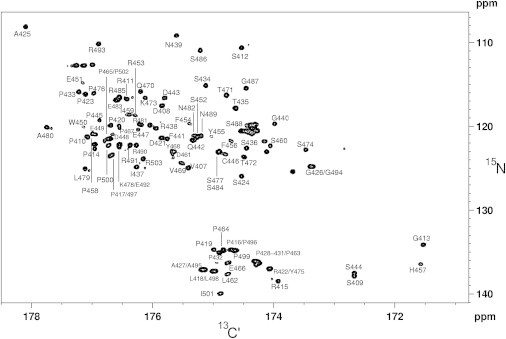
5D-NMR-based assignment of WIPC resonance frequencies. Full assignment of WIPC. Peaks are identified by their 13C′ residue. Unmarked peaks arise from side-chain, His6-tag, or linker-residue 13C′-15N pairs.
Structural features of WIPC
Backbone assignment immediately provides a view of secondary structure, even if it is transient, because chemical shifts are very sensitive to dihedral angles along the polypeptide backbone. Secondary chemical shifts, or the change in resonance frequency compared with those measured for a random coil conformation, have traditionally been relied upon as an indicator of secondary structure (41–44). This general relationship was recently refined for unstructured proteins based on a representative database of 14 IDPs (neighbor-corrected IDP (ncIDP) (45)) and an ensemble approach based on chemical shifts in folded proteins (δ2D (46)). Fig. 4 shows the results of these analyses for WIPC; secondary chemical shifts for WIPC 13C′, 13Cα, and 13Cβ nuclei (Δδ values) obtained from the ncIDP prediction; and secondary structure predictions from the δ2D database, revealing several intriguing features.
Figure 4.
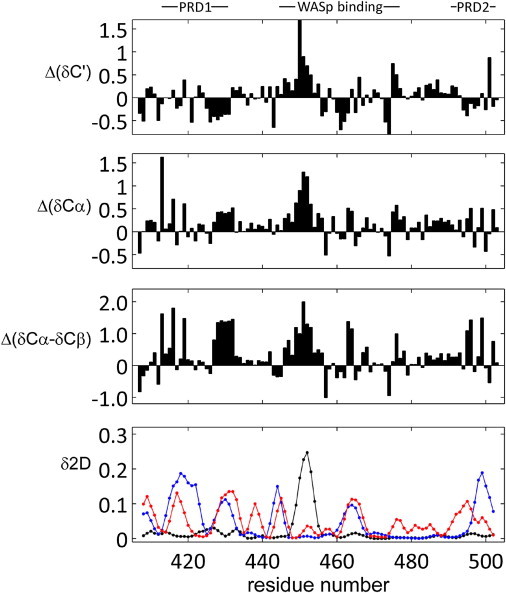
Secondary chemical-shift analysis for WIPC. Chemical-shift analysis using the ncIDP and δ2D databases, comparing WIPC chemical shifts with reference values in IDPs corrected for neighbor effects. Data are shown for 13C′ (first panel), 13Cα (second panel), and (13Cα −13Cβ) (third panel). Fourth panel: δ2D analysis of WIPC chemical shifts. Shown are the probabilities for α-helix (black), β-sheet/extended (dark grey, blue online), and polyproline II helix (light grey, red online). The remaining probabilities (not plotted) are for coil structure.
Most striking is the propensity for α-helical structure in the central core of WIPC at residues C446EDEWESRFYF456, for which all chemical-shift parameters are in agreement. The magnitude of the shifts observed is consistent with a 15–20% population of such a helical conformation, consistent with the value obtained in the secondary structure prediction. Supporting this conclusion is the fact that CON peaks emanating from these residues were relatively weak, typically exhibiting a 10-fold reduction in the signal/noise ratio. This behavior cannot be attributed to a dimerization of WIPC via the side chains of C446 residues, since spectra were recorded in the presence of 10 mM βME and were unaffected by addition of 5 mM dithiothreitol, and the C446 13Cβ chemical shift (28.2 ppm) is typical for a reduced thiol group. Another region with structural tendency is apparent at residues K473SYPS477, which exhibits the characteristics of a short N-capped helix, with a typical negative-positive motif for Δδ of 13C′ and 13Cα and an opposite effect for 13Cβ. Less pronounced is a marginal propensity for helical conformation in the S484RSGSNRRE492 segment.
Secondary shifts and structural propensity in polyproline segments
Of particular interest is the residual structure assumed by polyproline stretches. Proline-rich WIPC has a six-residue polyproline motif (P428PPPPP433) and three broken stretches (the N-terminal P414RPPLPP420, the central (WASp-binding) P463PPEP467, and the C-terminal P496PLPPIP502). The chemical shifts of these sequences are inaccessible to assignment approaches using amide 1H-15N moieties as the spectroscopic link between two adjacent amino acids. Consequently, these chemical shifts are poorly represented in resonance frequency databases, rendering secondary structure prediction for these regions only marginally accurate. In contrast, the 13C-detected approach used in this study is well suited for measuring chemical shifts in such regions. Internal proline residues within the hexa-proline segment exhibited shifts of 174.3, 61.2, and 30.5 ppm for the 13C′, 13Cα, and 13Cβ nuclei, respectively. These values represent Δδ(13C′,13Cα,13Cβ) secondary shifts (−0.5, +0.5, −0.8) when using the ncIDP database, and (−1.2, +0.2, −0.4) when compared with neighbor-corrected random-coil values calculated by SPARTA+ (47). The unique imino acid proline is known to exert significant effects on polypeptide backbone conformation (48–50). A closer look at ncIDP-derived secondary chemical shifts revealed that this characteristic down-up-down secondary shift motif is observed in Pro(i)-Pro(i + 1) dyads throughout the WIPC sequence for the 13Cα/13Cβ nuclei in residue Pro(i) and the 13C′ nucleus of the preceding (i − 1) residue.
In view of the shortage of polyproline segments with well-documented chemical shifts, the structural interpretation of these results is not straightforward. Of the 15 BioMagResBank entries containing polyproline segments, only seven provided chemical-shift data for the polyproline region, and only two provided 13Cα and 13Cβ chemical shifts; 13C′ data were unavailable. The pentaproline segment of the SPCp41 protein (BMRB #15013; PDB accession code 2JMC) (51) and the PPLPP motif of a tyrosine kinase-interacting peptide (BMRB #6456; PDB accession code 1WA7) (52) exhibit shifts that are highly similar to those obtained for residues 428–433 and 416–420/496–500, respectively, of WIPC. They also both adopt helical polyproline-II (PII) structures, characterized by a trans configuration of the peptide bonds and a left-handed helical arrangement, creating dihedral angles of (ϕ, ψ) = (−75, 140) and a near alignment of each third residue in an axial view of the helix. In light of this similarity, we conclude that polyproline stretches in WIPC do contain a significant measure of PII structure.
Backbone dynamics in WIPC
We investigated the dynamics of WIPC by measuring 15N relaxation rates, which are well-established reporters of backbone motions on the picosecond-to-nanosecond timescale and a useful probe of transient structure in IDPs. A fully unstructured polypeptide conforming to the segmental motion model (53) should exhibit a flat, bell-shaped profile with a central plateau; however, this is not the case for relaxation rates in WIPC measured at 283 K (Fig. 5, top four panels). Relaxation rates are relatively uniform throughout WIPC, with R1 values of 1.5–1.6 s−1, R2 values of 6–8 s−1, and 1H-15N NOE (hetNOE) values of 0.15–0.30. Notably, nonproline residues within the PRDs and the short helical motif at residues 473–477 exhibit such typical relaxation rates as well. The exception is the VCR region, and particularly residues 445–460, where relaxation rates change to R1 values of 1.3–1.4 s−1, R2 values of 15–20 s−1, and hetNOE values of 0.40–0.60. Such changes are consistent with an increase in structural character in this region. Generally, in the WIPC VCR domain, R1 and R2 values are anticorrelated, a behavior that is typical of polypeptides in the slow-tumbling regime and is notably absent in the less-structured terminal regions. Cross-correlated relaxation rates, which are insensitive to exchange processes, exhibit a similar pattern, with peak values of ηxy = 9–14 s−1 in the core region, as opposed to ηxy = 2–5 s−1 in the unstructured regions. This clearly indicates that it is the more-structural nature of the VCR segment that causes elevated R2 values, rather than exchange on the microsecond-to-millisecond timescale. Although IDPs are incompatible with the standard Lipari-Szabo model-free approach (54), the observed patterns are consistent with the structural view offered by secondary chemical shifts, which show the central region containing the WASp binding epitopes to be more structured than the generally flexible terminal segments of WIPC.
Figure 5.
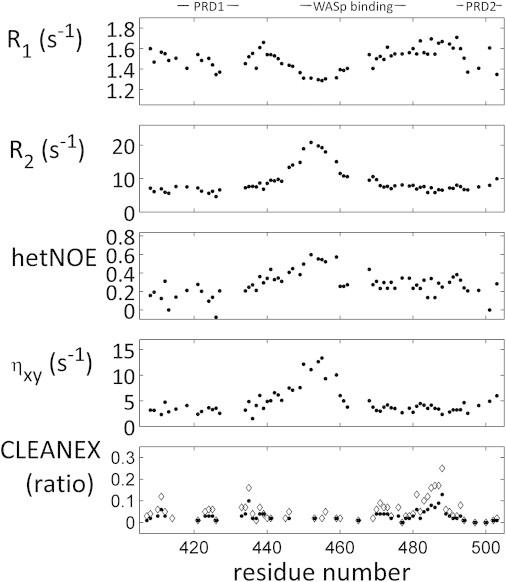
Backbone dynamics along the WIPC polypeptide. Relaxation rates were measured for WIPC backbone 15N nuclei at 16.4 T and 283 K. Top to bottom: 15N R1, 15N R2, 1H-15N NOE, 15N cross-correlated relaxation (ηxy), and rate of amide proton exchange with bulk solvent as measured in a CLEANEX-PM experiment with 10 (solid circles) and 20 (open diamonds) ms of exchange time. Residue numbers refer to the 13C′ nucleus of the residue preceding the amide proton for which the rate was measured.
Solvent accessibility along the WIPC backbone
To further characterize the degree of structure along the WIPC, we estimated the exchange rates of amide protons with the solvent using a CON-based CLEANEX-PM experiment (33) in which exchange-mediated recovery of magnetization was measured for WIPC at 298 K and pH 6 after 10 and 20 ms. Reduced WIPC solubility at pH values in the 6.5–7.5 range required these CLEANEX experiments to be conducted under conditions in which solvent exchange is inherently lower. Nevertheless, data acquired after t = 20 ms, which still met the necessary t ≪ 1/R1 condition, allowed us to discriminate among different protection factors along the WIPC backbone (Fig. 5, bottom panel). Regions that were most protected from solvent exchange, exhibiting negligible recovery after 20 ms, included residues 440–468, spanning the rigid VCR domain, and residues 475–479, both consistent with structural elements delineated by chemical-shift data. Notably, nonproline residues nested in proline-rich segments, such as R415, L418, E466, L498, and I501, exhibited low recovery rates as well. This may be due to the relatively hydrophobic character of the polyproline backbone environment. Low protection factors identified increased flexibility in two regions of WIPC, residues 470–474 and 481–490, which exhibited recovery rates of 5–10% and 10–25%, respectively. The former is the segment that links two structured regions, the WASp-binding poly-Pro sequence and the N-capped short helix, and the latter corresponds to the PKCθ phosphorylation sequence. Both relaxation and solvent-exchange results concur with the secondary structure tendencies identified by the chemical-shift data, together demarcating regions of relative rigidity, reflecting a structural propensity, and more flexible regions that are fully unfolded.
Effects of temperature on WIPC structure
As a disordered protein, WIPC exists in rapidly interchanging multiple conformations separated by low-activation energy barriers, and the effect of temperature on this conformational distribution is a reliable indicator of their relative stability. Increasing temperature caused a reduction and bathochromic shift of the polyproline-II helix (PII) signal and an apparent increase in the α-helical signal in the far-UV CD curve of WIPC, creating a characteristic isodichroic intersection of CD curves at 209 nm (Fig. 6 A). However, it was previously noted that a shift of residues from the PII to the extended region of the Ramachandran plot, rather than an increase in helical content, may account for this change (55). Whereas CD provides a macroscopic view of protein structure, chemical shifts offer more quantitative information with per-residue resolution, motivating us to use them to investigate temperature-dependent structure changes.
Figure 6.
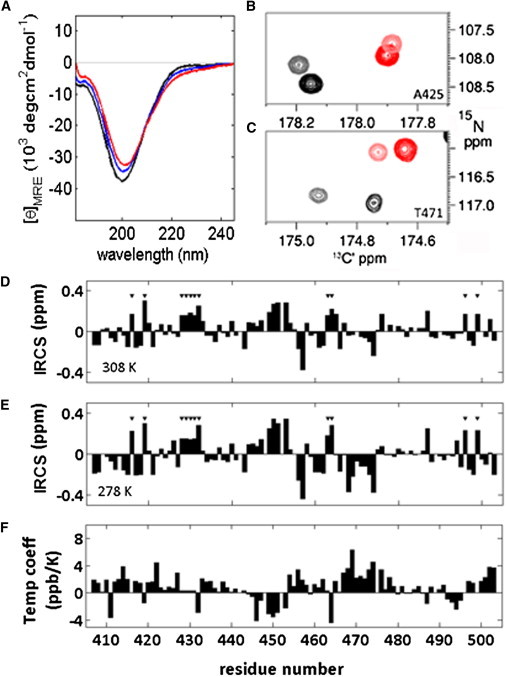
Effects of temperature on structural motifs in WIPC. (A) CD curves for a 6.7 μM WIPC sample at 278 (black), 293 (blue), and 308 (red) K. An increase (decrease) in signal is observed for the 215–230 (195–205) range. (B and C) Temperature effects on the intrinsic 13C′ shifts of WIPC as observed in CON-IPAP spectra. Black (red) peaks were obtained at 278 (308) K; dark (light) peaks were obtained for samples under normal (denaturing) conditions. Plots focus on residues A425 (B) and T471 (C). (D–F) 13C′ IRCSs at 308 K (D) and 278 K (E), and temperature factors (F) plotted against the WIPC sequence. The intrinsic temperature coefficient was calculated from the slope of IRCS versus temperature in the 278–308 range. The first residues of Pro-Pro dyads are designated by black triangles. Typical errors for temperature factors were 0.2 and 0.5 ppb/K for the unstructured and structured regions of WIPC, respectively.
Reference statistical coil chemical shifts are also affected by temperature and must be correctly estimated for accurate measurement of temperature-dependent secondary chemical shifts, particularly since IDPs with transient structure will typically exhibit only minor temperature effects. We accomplished this using intrinsic random coil referencing (56), comparing 13C′ and 13Cα resonances under normal and denaturing conditions at various temperatures. We acquired the fingerprint CON spectrum of WIPC at temperatures ranging from 278 to 308 K at 10 K intervals, and the 3D CACON spectrum, providing {13Cα(i),13C′(i),15N(i + 1)} correlations, at 288 and 308 K, repeating all measurements for a identical sample denatured by addition of 7.5 M urea. All spectra could be assigned by comparison with the data obtained for WIPC under normal conditions at 298 K; to facilitate this process, spectra were recorded for WIPC in 2.5 and 5 M urea as well. Intrinsically referenced chemical shifts (IRCSs) at each temperature were calculated as the difference between shifts in samples under normal and denaturing conditions. Generally, 13Cα IRCS values were less informative than their 13C′ counterparts, particularly since several shifts in more structured regions were missing due to increased relaxation losses from the low-temperature measurement. Therefore, the following analysis focuses on temperature effects on 13C′ shifts.
The derivation of 13C′ IRCS values at 308 K and 278 K for residues in random coil and transiently structured domains (A425 and T471, respectively) is shown in Fig. 6 B, and the data for the entire WIPC domain are presented in Fig. 6 C. IRCS values are residue specific and clearly distinguish between regions with undetectable transient structure, such as residues 422–427, 434–444, and 479–494, with values close to zero, and more structured segments as determined by previous experiments. Throughout the sequence and at all temperatures, Pro(i)-Pro(i + 1) dyads exhibit a pattern of negative 13C′(i − 1) and positive 13Cα(i − 1)/13C′(i) shifts. This observation highlights the effect of the constrained Xxx-Pro peptide bond on chemical shifts. The more-structured region of WIPC corresponding to the WASp-binding site (residues 446–478) exhibits the most significant IRCS values. Positive values for residues 446–453 and negative values for residues 460–475 (with the exception of the poly-Pro segment) are indicative of helical and extended conformations, respectively, in these regions.
Intrinsic temperature coefficients, defined as a slope of IRCS versus temperature, were calculated to assess the effects of temperature on WIPC transient structure. Cooling WIPC to 278 K caused an enhancement of the structural pattern, as shown by an analysis of 13C′ intrinsic temperature coefficients (Fig. 6 C, bottom panel). Negative values for the helical region (residues 446–453) and positive values in the extended region (residues 468–475) are consistent with loss of structure at higher temperatures. Similarly, positive values in the PRDs indicate that a decrease in temperature stabilized the proline-imposed rigidity in these regions. The small (<1.5 ppb/K) positive baseline in unstructured regions is consistent with the previously reported behavior of 13C′ IRCS values (55). We conclude that an increase in temperature induces an increase in flexibility and a loss of structural content, and that the CD results must be interpreted as a reduction in helical and PII conformations on the background of a redistribution of the statistical random coil ensemble.
Discussion
The flexibility and multiconformational nature of IDPs are inherent contributors to their functionality in biological systems. The ability of NMR to study structure and motions on a per-residue basis along the polypeptide backbone of IDPs is unique and accounts for its increasing utility in investigations of this class of proteins (20). The introduction of two recent breakthrough methodologies, 13C-detected backbone assignment experiments (25) and higher-dimensionality NMR experiments (57–59), supported by nonuniform data sampling techniques (28,35,60,61), has enhanced this capability by effectively addressing the difficulties presented by the low-spectral-dispersion characteristic of IDPs. In this study, these technologies were instrumental in completing backbone assignment of WIPC, allowing for structural interpretation of other experiments. The assignment process utilized a combination of two 13C′-detected approaches: 3D-NMR experiments correlating the backbone 13C′-15N group with the four adjacent 13Cα/13Cβ nuclei, and 5D-NMR with nonuniform sampling correlating among five backbone nuclei spanning two to three residues. As expected, the 5D experiments that required more magnetization-transferred steps were most challenged by the more structured regions of WIPC, where relaxation losses are most pronounced. Still, whereas the 3D-NMR experiments were unable to provide >50% of the assignment, >90% could be assigned by 5D-NMR, with only the most structured region defying assignment. Once the majority of residues were assigned, the more sensitive 3D-NMR experiments, both proton- and 13C′-detected, were successful in identifying the missing frequencies. Thus, in tandem the two methods performed in complementary fashion. In another methodological study (40), it was established that two 13C′-detected 4D experiments, HabCabCON and intra-HabCabNCO, could also provide sequential connectivities and allow assignment, although this approach was considerably more tedious than the 5D approach described above.
In this work we utilized chemical-shift information, 15N relaxation rates, and exposure to solvent exchange to characterize the unbound state of a WIPC fragment. The results of these measurements are consistent in identifying regions in which a higher contribution of structured conformations is observed. Structure is most tangible in the helical WASp-binding core domain including residues 446–456, which represents the main structural feature of WIPC. This conclusion is supported by secondary chemical shifts as well as relaxation rates in this region. It is unlikely that the helical conformation is affected or induced by intermolecular interactions, since 1), SE results did not reflect WIP oligomerization; and 2), chemical shifts exhibited only negligible concentration dependence. In addition, the similarity of ηxy/R2 ratios in this region to those predicted by theory rules out significant exchange contributions to relaxation. Additional regions with an appreciable population of structured conformation are the polyproline stretches, residues 414–423, 428–433, 463–467 (in the WASp-binding domain) and 496–502, and the 473–478 segment, which adopts a short N-capped helical conformation. A comparison of WIPC with other recently studied IDPs demonstrates a similarity between levels of transient structure observed for these proteins. The secondary shifts measured for WIPC were comparable to those observed for securin (62), tau protein (63), and others (64,65). The transiently structured regions are connected by unstructured linker segments, as could be deduced from minimal secondary structure, low transverse relaxation and hetNOE rates, minimal IRCS values, and exposure to solvent exchange. One such region of interest is the phosphorylation site of residues 484–492, whose flexibility is consistent with its need to be sufficiently exposed to allow access to the phosphorylating enzyme PKCθ. These findings are in agreement with the paradigm that IDPs, rather than being fully disordered, are actually comprised of flexible segments connecting regions of transient secondary structure. The latter sample a more restricted conformational space and may represent nuclei of emerging structure that can be evoked by interaction with binding partners.
A unique aspect of WIPC is the prevalence of proline residues. Proline-rich segments have proven roles in mediating protein-protein interactions (66–68), suggesting a biological role for such segments along WIPC. The use of 13C′-detected experiments is an attractive alternative to other approaches (69,70) that render such segments NMR visible, here allowing the first (to our knowledge) assignment of extensive poly-proline segments in the context of a larger protein, whereas previously this could be achieved only for small peptides. Chemical shifts observed for the P428PPPPP433 and P416PLPP420/ P496PLPP500 segments indicated that they adopted PII structures, which appeared to be relatively rigid as suggested by the fact that these segments were protected from solvent exchange. Pro-Pro dyads induced characteristic deviations from random coil chemical shifts, particularly the down-up-down motif for chemical shifts {13C′(i − 1),13Cα(i),13Cβ(i)} and the down-up-up motif for IRCS values of {13C′(i − 1),13Cα(i − 1),13C′(i)}. Regarding the former, however, we note that the ncIDP database (which lacks poly-Pro segments) does not consider two-residue neighbor effects, which are nonnegligible for proline residues (50). Adjusting for this additional effect would mitigate the observed chemical-shift deviations.
Characteristic temperature-dependent changes have been observed in several IDPs (55,71,72). Here, temperature-dependent chemical shifts, compared between normal and denaturing conditions to obtain the intrinsic structure-induced chemical shift, were acquired to follow the effects of temperature on the transient structure in WIPC. Generally, IRCS values in more structured regions were low compared with those reported in a previous study (55), indicating lower levels of transient structure, and temperature effects were commensurately reduced. However, a pattern of increasing structural content at lower temperatures was evident, in agreement with a previous study showing that IDPs did not undergo cold denaturation (73). Analysis of CD curves (in the 278–308 K range) and δ2D analysis of chemical shifts (in the 278–298 K range; data not shown) was consistent with these findings, with both failing to identify temperature-induced structural changes in excess of 5–10%. Nevertheless, the correlation between significant IRCS values and regions with transient structure as determined by other NMR parameters suggests that intrinsic shifts are useful for identifying structural traces in IDPs. However, such an analysis assumes that all structural elements are fully denatured in 7.5 M urea, which may be an overgeneralization. Thus, positive temperature coefficients for the PRDs (residues 407–423, 496–502) may result from a stabilization of the PII conformation (characterized by negative 13C′ shifts) in 7.5 M urea, as suggested in previous studies (74,75). Residues 428–431 from the hexa-proline segment exhibited negligible temperature effects, reflecting the fact that poly-Pro structures are stabilized by steric clashes rather than enthalpically favored interactions. These observations underline the fact that it is extremely difficult to establish a truly random peptide chain conformation. Whereas in folded proteins effects of this magnitude are usually negligible on the backdrop of secondary shifts of 2–3 ppm, they must be considered in the context of IDPs, where secondary chemical shifts are far less pronounced.
A schematic picture of structural propensity along the WIPC backbone summarizing our findings is shown in Fig. 7. It is instructive to compare the structural tendencies of the core region of WIPC in its unbound state with the conformation of a WIP-derived peptide (residues 451–485) when bound to a neuronal homolog of WASp, N-WASp (12). With the WIP peptide fused before the N-terminus of N-WASp, the three salient structural features observed were 1), a cis-peptide H457-P458 bond connecting the aromatic stretch F454YF456 to a tight turn at residues I459SD461; 2), a PII conformation for the polyproline segment of residues L462PPP465; and 3), a helical turn at residues S474YPSK478. The last two elements were connected by a flexible linker in which T471 was the least structured residue (12). Structural motifs 2 and 3 are paralleled by the findings presented here, and the 468–473 linker segment exhibits high-temperature coefficients although it lacks structure, indicating it may adopt a specific biologically relevant conformation in the bound form. In contrast, free WIPC does not foreshadow the cis H457-P458 bond or the turn at residues 459–461. Thus, contributing to the formation of the WIP/WASp complex are preformed motifs at residues 462–465 and 474–478, and a third motif (residues 457–461) whose structure is formed only in the presence of WASp. It is possible that the flexible segment connecting motifs 2 and 3 makes different interaction modes possible, as would be required in various cellular situations.
Figure 7.
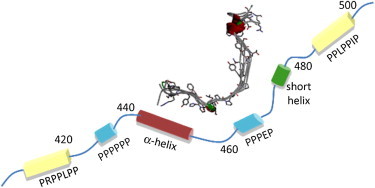
Comparison of WIPC in the WASp-bound and unbound states. In ribbons/stick style, the conformation of a WIP peptide (residues 451–485) bound to neuro-WASp (12) is shown as a superposition of five low-energy structures. For clarity, side chains are shown for one conformer only. Below is a schematic representation of unbound WIPC obtained in this study, showing unstructured linkers (thin lines) and regions of structural tendency (cylinders).
It is tempting to speculate about the contribution of helical WIPC residues 446–452 to the affinity of the WIP/WASp complex, since sequences of previously determined WIP/WASp structures (11,12) failed to include this structural element. Binding to WASp can be expected to stabilize this helical conformation because the structural propensities of long IDP segments (six to seven residues) are good predictors of the bound conformation (76). Using the known position of the shorter WIPC fragment in relation to N-WASp, it is possible to suggest potential intermolecular interactions contributed by the predicted helix. Specifically, N-WASp presents a positively charged surface consisting of residues K56, K57, K81, and K84, which could form contacts with the aforementioned putative helix, including four negatively charged residues. Notably, only residues K56 and K84 are conserved (replaced by Arg residues) in the homologous T-cell WASp, allowing the ubiquitous WIP to interact differently with its binding partners in different biological environments. Further studies will be necessary to determine the role of the helical segment at residues 446–452 in the context of WIP/WASp complex formation.
Conclusions
In this work, we used advanced NMR methods to fully assign the backbone resonances of the 97-residue intrinsically disordered fragment of WIP, which allowed us to study its structure and dynamics in its unbound state. The emerging picture is one of a pattern of alternating partially structured and fully unstructured regions, with the established WASp-binding epitope at the core of the more-structured domain. The WIP-WASp binding interface is a combination of preformed and WASp-induced structural motifs, and it is likely that this pattern is closely related to its function. This study also lays the foundation for investigating the interaction between the extended WIPC fragment and WASp, as well as other cellular binding partners, which has important biological and pharmaceutical implications.
Acknowledgments
We thank Drs. Hugo Gottlieb and Keren Keinan-Adamsky for spectrometer assistance, Prof. Isabella Felli (CERM, University of Florence) for the CON-based CLEANEX-PM experiment, members of the Barda-Saad laboratory (Bar Ilan University) for assistance in cloning of WIPC, and Dvir Doron (Bar Ilan University) for support in modeling the bound WIP conformation.
This work was supported by the Access to Research Infrastructures activity in the 7th Framework Programme of the European Commission (contract 228461, EAST-NMR), the Czech Science Foundation (grant P206/11/0758), and the Heritage Legacy fund (award 491/10). The 700 MHz spectrometer was purchased with the assistance of a Converging Technologies award and a generous donation by Fundacion Adar.
Supporting Material
References
- 1.Derry J.M., Kerns J.A., Francke U. WASP gene mutations in Wiskott-Aldrich syndrome and X-linked thrombocytopenia. Hum. Mol. Genet. 1995;4:1127–1135. doi: 10.1093/hmg/4.7.1127. [DOI] [PubMed] [Google Scholar]
- 2.Thrasher A.J., Burns S.O. WASP: a key immunological multitasker. Nat. Rev. Immunol. 2010;10:182–192. doi: 10.1038/nri2724. [DOI] [PubMed] [Google Scholar]
- 3.Antón I.M., Jones G.E. WIP: a multifunctional protein involved in actin cytoskeleton regulation. Eur. J. Cell Biol. 2006;85:295–304. doi: 10.1016/j.ejcb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Quiles N., Rohatgi R., Ramesh N. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat. Cell Biol. 2001;3:484–491. doi: 10.1038/35074551. [DOI] [PubMed] [Google Scholar]
- 5.de la Fuente M.A., Sasahara Y., Ramesh N. WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP) Proc. Natl. Acad. Sci. USA. 2007;104:926–931. doi: 10.1073/pnas.0610275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konno A., Kirby M., Candotti F. The expression of Wiskott-Aldrich syndrome protein (WASP) is dependent on WASP-interacting protein (WIP) Int. Immunol. 2007;19:185–192. doi: 10.1093/intimm/dxl135. [DOI] [PubMed] [Google Scholar]
- 7.Sasahara Y., Rachid R., Geha R.S. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol. Cell. 2002;10:1269–1281. doi: 10.1016/s1097-2765(02)00728-1. [DOI] [PubMed] [Google Scholar]
- 8.Ramesh N., Antón I.M., Geha R.S. WIP, a protein associated with wiskott-aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc. Natl. Acad. Sci. USA. 1997;94:14671–14676. doi: 10.1073/pnas.94.26.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bi K., Altman A. Membrane lipid microdomains and the role of PKCθ in T cell activation. Semin. Immunol. 2001;13:139–146. doi: 10.1006/smim.2000.0305. [DOI] [PubMed] [Google Scholar]
- 10.Dong X., Patino-Lopez G., Shaw S. Structure-function analysis of the WIP role in T cell receptor-stimulated NFAT activation: evidence that WIP-WASP dissociation is not required and that the WIP NH2 terminus is inhibitory. J. Biol. Chem. 2007;282:30303–30310. doi: 10.1074/jbc.M704972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volkman B.F., Prehoda K.E., Lim W.A. Structure of the N-WASP EVH1 domain-WIP complex: insight into the molecular basis of Wiskott-Aldrich syndrome. Cell. 2002;111:565–576. doi: 10.1016/s0092-8674(02)01076-0. [DOI] [PubMed] [Google Scholar]
- 12.Peterson F.C., Deng Q., Volkman B.F. Multiple WASP-interacting protein recognition motifs are required for a functional interaction with N-WASP. J. Biol. Chem. 2007;282:8446–8453. doi: 10.1074/jbc.M609902200. [DOI] [PubMed] [Google Scholar]
- 13.Dunker A.K., Silman I., Sussman J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Rezaei-Ghaleh N., Blackledge M., Zweckstetter M. Intrinsically disordered proteins: from sequence and conformational properties toward drug discovery. ChemBioChem. 2012;13:930–950. doi: 10.1002/cbic.201200093. [DOI] [PubMed] [Google Scholar]
- 15.Uversky V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 2009;14:5188–5238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 16.Babu M.M., van der Lee R., Gsponer J. Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 2011;21:432–440. doi: 10.1016/j.sbi.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Ward J.J., Sodhi J.S., Jones D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 19.Mittag T., Forman-Kay J.D. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009;19:23–30. doi: 10.1016/j.sbi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torchia D.A. Dynamics of biomolecules from picoseconds to seconds at atomic resolution. J. Magn. Reson. 2011;212:1–10. doi: 10.1016/j.jmr.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Mittermaier A.K., Kay L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009;34:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Zuiderweg E.R.P. Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry. 2002;41:1–7. doi: 10.1021/bi011870b. [DOI] [PubMed] [Google Scholar]
- 24.Clore G.M., Tang C., Iwahara J. Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr. Opin. Struct. Biol. 2007;17:603–616. doi: 10.1016/j.sbi.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bermel W., Bertini I., Pierattelli R. Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J. Am. Chem. Soc. 2006;128:3918–3919. doi: 10.1021/ja0582206. [DOI] [PubMed] [Google Scholar]
- 26.Mäntylahti S., Hellman M., Permi P. Extension of the HA-detection based approach: (HCA)CON(CA)H and (HCA)NCO(CA)H experiments for the main-chain assignment of intrinsically disordered proteins. J. Biomol. NMR. 2011;49:99–109. doi: 10.1007/s10858-011-9470-z. [DOI] [PubMed] [Google Scholar]
- 27.Motáčková V., Nováček J., Sklenář V. Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J. Biomol. NMR. 2010;48:169–177. doi: 10.1007/s10858-010-9447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nováček J., Zawadzka-Kazimierczuk A., Sklenář V. 5D 13C-detected experiments for backbone assignment of unstructured proteins with a very low signal dispersion. J. Biomol. NMR. 2011;50:1–11. doi: 10.1007/s10858-011-9496-2. [DOI] [PubMed] [Google Scholar]
- 29.Bermel W., Bertini I., Stanek J. Speeding up sequence specific assignment of IDPs. J. Biomol. NMR. 2012;53:293–301. doi: 10.1007/s10858-012-9639-0. [DOI] [PubMed] [Google Scholar]
- 30.Bermel W., Bertini I., Pierattelli R. Exclusively heteronuclear NMR experiments to obtain structural and dynamic information on proteins. ChemPhysChem. 2010;11:689–695. doi: 10.1002/cphc.200900772. [DOI] [PubMed] [Google Scholar]
- 31.Farrow N.A., Muhandiram R., Kay L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 32.Chill J.H., Louis J.M., Bax A. Measurement of 15N relaxation in the detergent-solubilized tetrameric KcsA potassium channel. J. Biomol. NMR. 2006;36:123–136. doi: 10.1007/s10858-006-9071-4. [DOI] [PubMed] [Google Scholar]
- 33.Mori S., Berg J.M., van Zijl P.C. Separation of intramolecular NOE and exchange peaks in water exchange spectroscopy using spin-echo filters. J. Biomol. NMR. 1996;7:77–82. doi: 10.1007/BF00190459. [DOI] [PubMed] [Google Scholar]
- 34.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Kazimierczuk K., Zawadzka-Kazimierczuk A., Koźmiński W. Non-uniform frequency domain for optimal exploitation of non-uniform sampling. J. Magn. Reson. 2010;205:286–292. doi: 10.1016/j.jmr.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 36.Cole J.L., Hansen J.C. Analytical ultracentrifugation as a contemporary biomolecular research tool. J. Biomol. Tech. 1999;10:163–176. [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer H., Polikarpov I., Craievich A.F. Average protein density is a molecular-weight-dependent function. Protein Sci. 2004;13:2825–2828. doi: 10.1110/ps.04688204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 39.Ramesh N., Geha R.S. Recent advances in the biology of WASP and WIP. Immunol. Res. 2009;44:99–111. doi: 10.1007/s12026-008-8086-1. [DOI] [PubMed] [Google Scholar]
- 40.Novacek J., Haba N.Y., Zidek L. 4D non-uniformly sampled HABCABCON and 1J(NCα)-selective HCBCANCO experiments for the sequential assignment and chemical shift analysis of intrinsically disordered proteins. J. Biomol. NMR. 2012;53:139–148. doi: 10.1007/s10858-012-9631-8. [DOI] [PubMed] [Google Scholar]
- 41.Wishart D.S., Case D.A. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
- 42.Cavalli A., Salvatella X., Vendruscolo M. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. USA. 2007;104:9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen Y., Lange O., Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raman S., Lange O.F., Baker D. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamiola K., Acar B., Mulder F.A.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 2010;132:18000–18003. doi: 10.1021/ja105656t. [DOI] [PubMed] [Google Scholar]
- 46.Camilloni C., De Simone A., Vendruscolo M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry. 2012;51:2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- 47.Shen Y., Bax A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho B.K., Brasseur R. The Ramachandran plots of glycine and pre-proline. BMC Struct. Biol. 2005;5:14. doi: 10.1186/1472-6807-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacArthur M.W., Thornton J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 50.Schwarzinger S., Kroon G.J.A., Dyson H.J. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 51.Candel A.M., Conejero-Lara F., Bruix M. The high-resolution NMR structure of a single-chain chimeric protein mimicking a SH3-peptide complex. FEBS Lett. 2007;581:687–692. doi: 10.1016/j.febslet.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 52.Bauer F., Schweimer K., Sticht H. Structural characterization of Lyn-SH3 domain in complex with a herpesviral protein reveals an extended recognition motif that enhances binding affinity. Protein Sci. 2005;14:2487–2498. doi: 10.1110/ps.051563605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwalbe H., Fiebig K.M., Dobson C.M. Structural and dynamical properties of a denatured protein. Heteronuclear 3D NMR experiments and theoretical simulations of lysozyme in 8 M urea. Biochemistry. 1997;36:8977–8991. doi: 10.1021/bi970049q. [DOI] [PubMed] [Google Scholar]
- 54.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982;104:4546–4570. [Google Scholar]
- 55.Kjaergaard M., Nørholm A.-B., Kragelund B.B. Temperature-dependent structural changes in intrinsically disordered proteins: formation of α-helices or loss of polyproline II? Protein Sci. 2010;19:1555–1564. doi: 10.1002/pro.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Modig K., Jürgensen V.W., Poulsen F.M. Detection of initiation sites in protein folding of the four helix bundle ACBP by chemical shift analysis. FEBS Lett. 2007;581:4965–4971. doi: 10.1016/j.febslet.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 57.Atreya H.S., Eletsky A., Szyperski T. Resonance assignment of proteins with high shift degeneracy based on 5D spectral information encoded in G2FT NMR experiments. J. Am. Chem. Soc. 2005;127:4554–4555. doi: 10.1021/ja042562e. [DOI] [PubMed] [Google Scholar]
- 58.Hiller S., Wasmer C., Wüthrich K. Sequence-specific resonance assignment of soluble nonglobular proteins by 7D APSY-NMR spectroscopy. J. Am. Chem. Soc. 2007;129:10823–10828. doi: 10.1021/ja072564+. [DOI] [PubMed] [Google Scholar]
- 59.Wen J., Wu J., Zhou P. Sparsely sampled high-resolution 4-D experiments for efficient backbone resonance assignment of disordered proteins. J. Magn. Reson. 2011;209:94–100. doi: 10.1016/j.jmr.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kupce E., Freeman R. Projection-reconstruction of three-dimensional NMR spectra. J. Am. Chem. Soc. 2003;125:13958–13959. doi: 10.1021/ja038297z. [DOI] [PubMed] [Google Scholar]
- 61.Coggins B.E., Zhou P. High resolution 4-D spectroscopy with sparse concentric shell sampling and FFT-CLEAN. J. Biomol. NMR. 2008;42:225–239. doi: 10.1007/s10858-008-9275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Csizmok V., Felli I.C., Bertini I. Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J. Am. Chem. Soc. 2008;130:16873–16879. doi: 10.1021/ja805510b. [DOI] [PubMed] [Google Scholar]
- 63.Mukrasch M.D., Bibow S., Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009;7:e34. doi: 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rao J.N., Kim Y.E., Ulmer T.S. Effect of pseudorepeat rearrangement on α-synuclein misfolding, vesicle binding, and micelle binding. J. Mol. Biol. 2009;390:516–529. doi: 10.1016/j.jmb.2009.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marsh J.A., Singh V.K., Forman-Kay J.D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Williamson M.P. The structure and function of proline-rich regions in proteins. Biochem. J. 1994;297:249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ball L.J., Kühne R., Oschkinat H. Recognition of proline-rich motifs by protein-protein-interaction domains. Angew. Chem. Int. Ed. Engl. 2005;44:2852–2869. doi: 10.1002/anie.200400618. [DOI] [PubMed] [Google Scholar]
- 68.Ingham R.J., Colwill K., Pawson T. WW domains provide a platform for the assembly of multiprotein networks. Mol. Cell. Biol. 2005;25:7092–7106. doi: 10.1128/MCB.25.16.7092-7106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bottomley M.J., Macias M.J., Sattler M. A novel NMR experiment for the sequential assignment of proline residues and proline stretches in 13C/15N-labeled proteins. J. Biomol. NMR. 1999;13:381–385. doi: 10.1023/a:1008393903034. [DOI] [PubMed] [Google Scholar]
- 70.Kanelis V., Donaldson L., Kay L.E. Sequential assignment of proline-rich regions in proteins: application to modular binding domain complexes. J. Biomol. NMR. 2000;16:253–259. doi: 10.1023/a:1008355012528. [DOI] [PubMed] [Google Scholar]
- 71.Uversky V.N., Li J., Fink A.L. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 72.Sánchez-Puig N., Veprintsev D.B., Fersht A.R. Human full-length Securin is a natively unfolded protein. Protein Sci. 2005;14:1410–1418. doi: 10.1110/ps.051368005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tantos A., Friedrich P., Tompa P. Cold stability of intrinsically disordered proteins. FEBS Lett. 2009;583:465–469. doi: 10.1016/j.febslet.2008.12.054. [DOI] [PubMed] [Google Scholar]
- 74.Meier S., Strohmeier M., Grzesiek S. Direct observation of dipolar couplings and hydrogen bonds across a β-hairpin in 8 M urea. J. Am. Chem. Soc. 2007;129:754–755. doi: 10.1021/ja067522k. [DOI] [PubMed] [Google Scholar]
- 75.Whittington S.J., Chellgren B.W., Creamer T.P. Urea promotes polyproline II helix formation: implications for protein denatured states. Biochemistry. 2005;44:6269–6275. doi: 10.1021/bi050124u. [DOI] [PubMed] [Google Scholar]
- 76.Eliezer D. Characterizing residual structure in disordered protein states using nuclear magnetic resonance. Methods Mol. Biol. 2007;350:49–67. doi: 10.1385/1-59745-189-4:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cai M., Huang Y., Craigie R. An efficient and cost-effective isotope labeling protocol for proteins expressed in Escherichia coli. J. Biomol. NMR. 1998;11:97–102. doi: 10.1023/a:1008222131470. [DOI] [PubMed] [Google Scholar]
- 78.Kazimierczuk K., Zawadzka A., Koźmiński W. Optimization of random time domain sampling in multidimensional NMR. J. Magn. Reson. 2008;192:123–130. doi: 10.1016/j.jmr.2008.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.