

Abstract
The proteorhodopsin family consists of retinal proteins of marine bacterial origin with optical properties adjusted to their local environments. For green proteorhodopsin, a highly specific mutation in the EF loop, A178R, has been found to cause a surprisingly large redshift of 20 nm despite its distance from the chromophore. Here, we analyze structural and functional consequences of this EF loop mutation by time-resolved optical spectroscopy and solid-state NMR. We found that the primary photoreaction and the formation of the K-like photo intermediate is almost pH-independent and slower compared to the wild-type, whereas the decay of the K-intermediate is accelerated, suggesting structural changes within the counterion complex upon mutation. The photocycle is significantly elongated mainly due to an enlarged lifetime of late photo intermediates. Multidimensional MAS-NMR reveals mutation-induced chemical shift changes propagating from the EF loop to the chromophore binding pocket, whereas dynamic nuclear polarization-enhanced 13C-double quantum MAS-NMR has been used to probe directly the retinylidene conformation. Our data show a modified interaction network between chromophore, Schiff base, and counterion complex explaining the altered optical and kinetic properties. In particular, the mutation-induced distorted structure in the EF loop weakens interactions, which help reorienting helix F during the reprotonation step explaining the slower photocycle. These data lead to the conclusion that the EF loop plays an important role in proton uptake from the cytoplasm but our data also reveal a clear interaction pathway between the EF loop and retinal binding pocket, which might be an evolutionary conserved communication pathway in retinal proteins.
Introduction
Retinal proteins are photoreactive proteins found in various organisms, enabling light responses such as vision in animals, phototaxis in flagellates, triggering ion channels in algae, and proton pumps in bacteria. All these proteins have a common seven trans-membrane helix motif and a retinal cofactor for light reception. The characteristic optical absorption of the retinylidene chromophore is strongly influenced by the protein environment. This bathochromic (opsin) shift compared to retinal protonated Schiff base in methanol (λmax. = 440 nm) has been observed in several microbial rhodopsins such as channelrhodopsin-2 (λmax.= 480 nm) (1), sensory rhodopsin II (λmax. = 487 nm) (2), proteorhodopsin (PR) (λmax. = 518 nm) (3), and bacteriorhodopsin (BR) (λmax. = 568 nm) (4).
The existence of different opsin shifts shows that mechanisms for color tuning of microbial retinal proteins must exist. This is especially evident in the case of PR, an almost ubiquitous retinal protein occurring in gamma-proteobacteria in the photic zone of the oceans (5). PR has been found to be color tuned with respect to the water depth and two main families, green absorbing PR (GPR) (λmax.= 525 nm) close to the surface and blue absorbing PR (λmax. = 490 nm) at greater depths, have been identified (6). It is generally accepted that the spectral characteristics of the retinylidene chromophore is strongly affected by its interaction with amino acids located in its direct vicinity. Early reports stated that mainly the distance of the protonated Schiff base from the counterion complex affects its spectral characteristics (7), but interactions with polar and polarizable amino acid residues along the polyene chain, the distortion of the chromophore structure, and the relative orientation of the β-ionine ring to the polyene chain due to steric repulsions within the protein contribute significantly as well (8–12). For example, one single point mutation L105Q in GPR, which is close to the chromophore, shifts the absorption maximum toward blue, whereas the corresponding Q105L mutation in blue absorbing PR causes a green shift (6,13). The recent discovery of a 20 nm redshift of the GPR absorption spectrum upon a single mutation A178R in the EF loop came therefore as a great surprise as the mutation site is far away from the retinylidene (14). This effect is highly position specific and has not been observed in BR (15). Only for halorhodopsin, a similar long-range effect based on a BC-loop mutation has been reported (16). Based on their data and sequence alignments, Kandori and co-workers (17) have suggested that having a small residue such as alanine at position 178 is essential to blueshift the absorption of GPR at neutral pH into a color range compatible with a marine environment.
GPR works as a light-driven proton pump (3) but additional functions have been discussed (18). In addition to the color tuning, GPRs have also other unusual molecular properties: The primary proton acceptor D97 has an elevated pKa due to a pH-dependent coupling to highly conserved H75 (19), which also seems to be involved in proton transfer (20). GPR forms pentamers and hexamers in detergent and lipid bilayers (21,22). The favorable properties of GPR in terms of stability, activity, and spectral resolution have led to an extensive chemical shift assignment and secondary structure analysis by solid-state NMR (23-25). It was also shown that the chromophore assumes an almost 100 percent all-trans conformation in ground-state GPR (26). Hydrogen bonding and protonation states of H75 and D97 have been characterized at different pH and reveal a complex interaction pattern, which partially explains the complicated pH dependence of PR’s photocycle (19). Solution state NMR has been used to determine backbone structure and dynamic properties of cell free produced GPR within detergent micelles (27,28)
Here, we have resolved the molecular basis of the surprising color shift in GPRA178R. Time-resolved optical spectroscopy has been applied to show that the A178R mutation also affects significantly the kinetics of the primary photoreaction and elongates the duration of the photocycle by a factor of 10. Using solid-state NMR, we have identified mutation-induced structural changes based on chemical shift perturbations. Dynamic nuclear polarization (DNP), a novel hybrid-technique combining electron paramagnetic resonance (EPR) and solid-state NMR to enhance sensitivity of solid-state NMR by orders of magnitude through polarization transfer from stable radicals to nuclei (29), was used to probe retinylidene chromophore and Schiff base conformation. These structural data allow the observed effects on the photocycle dynamics to be explained and reveal a distinct communication pathway between the chromophore and EF loop. Our data indicate that the EF loop plays an essential role for proton uptake in GPR and regulates helical movements during late stages of the photocycle.
Materials and Methods
Sample preparation
Samples were essentially prepared as described previously (25): GPR and GPRA178R were cloned in a pET27b(+)-plasmid and expressed in Escherichia coli C43-cells. Cells were grown in minimal medium with 13C-labeled glucose and 15NH4Cl at 37°C. To reduce spectral overlap, amino acids F, L, W, Y, and V were added to the medium to suppress isotope labeling of these residues. When an OD578 ∼0.7 was reached, 200 mg IPTG and 2 mg retinal dissolved in ethanol were added. Cells were harvested 16–18 h after induction and broken by a Constant System cell disrupter by 1.85 bar. The protein was solubilized in 1.5% n-dodecyl-β-D-maltoside (DDM) at 4°C overnight. The solubilized protein (supernatant) was obtained by ultracentrifugation and incubated for 1 h with a Ni-NTA matrix at 4°C. The bound protein was washed and finally eluted with 500 mM imidazole in 0.05% DDM. Purity was checked by absorption spectroscopy and SDS-PAGE (Fig. S1 and Fig. S2 in the Supporting Material).
For optical spectroscopy, GPR and GPRA178R were used solubilized in 0.15% DDM, 150 mM NaCl, 50 mM Tris, pH 6 or pH 9. The sample was diluted to an OD of 0.5 (d = 0.1 cm) for pump-probe spectroscopy and to an OD of 0.7 (d = 1 cm) for flash photolysis.
For solid-state NMR experiments GPR and GPRA178R were reconstituted in DMPC/DMPA (9:1) liposomes in a protein/lipid ratio of 2:1 (w/w). Lipids were resolved in methanol/chloroform (1:2). After evaporation of the solvent, lipids were dissolved in 100 mM NaCl, 50 mM MES, pH 7 and liposomes were formed by passage through a 2 μm pore sized membrane by an extruder with 5–7 bar. Protein and liposomes were mixed slowly and incubated for 0.5 h. Finally, detergent was removed by repeated incubation with biobeads. Proteoliposomes in 50 mM Tris, 5 mM MgCl2, pH 9 were sedimented by ultracentrifugation for 1 h and packed into a magic-angle spinning (MAS) rotor.
Preparing [14-15-13C-all-trans-retinal]-PRA178R for DNP-enhanced solid-state NMR
All-trans retinal 13C-labeled at positions 14 and 15 was synthesized by adaptation of reported procedures (for the synthetic scheme see the Supporting Material) (30). It was incorporated into GPR by adding it to the growth medium as described previously. Reconstituted samples were incubated overnight with 10% H2O, 30% D8-Glycerol and 60% D2O, and 20 mM TOTAPOL (31).
Time-resolved optical spectroscopy
Pump-probe-spectroscopy
A CPA 2001 was used as the laser source operating at a repetition rate of 1 kHz, delivering laser pulses with a central wavelength of 775 nm. The pump-pulse was converted to the absorption maximum using a home-built noncollinear optical parametric amplifier (20 nJ/pulse). Afterward, the pump-pulse was temporally compressed using a prism compressor. The probe-pulse consists of a spectrally broad pulse (white light continuum) and was generated by focusing a part of the laser fundamental into a sapphire crystal. The pump- and probe-pulses were spatially overlapped in the sample and the probe-pulse was detected on a photodiode array with a spectral resolution of Δλres. = 4 nm. The time resolution varied between 50 and 90 fs.
Flash photolysis
The samples were excited at the particular absorption maximum (pulse width: 20 ns; intensity: 3 mJ/cm2). The laser pulses were generated using an optical parametric oscillator (OPO, GWU Laser Technics) that is pumped by the frequency tripled of a Nd:YAG laser (Spitlight 600 Innolas Laser GmbH). For the probe beam the output of a xenon lamp (LC8; Hamamatsu) is guided through a monochromator (Photon Technologies International) for wavelength selection. Afterward, this beam is focused into a fused quartz cuvette (d = 1 cm) and guided through a second monochromator to get rid of scattering light, due to laser excitation. The probe beam is detected by a photomultiplier tube (Photosensor H6780-02, Hamamatsu) and analyzed by a digital storage oscilloscope (LeCroy, Waverunner 62 Xi).
Simulation of the transient data
The flash photolysis data were fitted to a reaction scheme that was slightly altered compared to an earlier one proposed by Varo et al. (32). The following reaction sequence was assumed: . The spectra of M1 and M2, as well as PR(O) and PR are assumed to be equal. A Levenberg-Marquardt algorithm was used to calculate photointermediate rate constants and absorption spectra using data from Varo et al. as starting values (32). The system of linear differential equations is solved numerically.
Solid-state NMR experiments
DNP-enhanced MAS-NMR
Experiments were performed on a Bruker Avance II 393 MHz spectrometer equipped with a modified Bruker 3.2 mm low-temperature probehead and connected to a 259 GHz gyrotron (Gycom, Russia). The effective microwave power applied to our NMR samples was ∼2 W. A sample of [14-15-13C-all-trans-retinal]-GPRA178R (∼2 mg) was packed into a 3.2 mm ZrO2 rotor and sealed with a homemade tapered rubber disk. The rotor was closed by a Vespel cap and flash frozen in liquid nitrogen before inserting it into the low-temperature probehead. For all measurements, a MAS frequency of 8000 ± 2 Hz was used. The temperature inside the MAS rotor was kept at 103.0 ± 0.2 K. All DNP experiments started from a 1H hard pulse (2.2 μs) followed by 1H-13C cross-polarization (CP) with a contact time of 400 μs and 1H and 13C spin lock fields of 56.8 and 65.4 kHz, respectively. The POST-C7 scheme was used for exciting and reconverting DQ (double-quantum) coherence (33). A continuous wave decoupling at 112 kHz and a SPINAL64 (34) decoupling at 105 kHz were applied during POST-C7 pulses and acquisition time, respectively. HCCH torsion angles were measured by separated local field (SLF) experiments, essentially as described by Levitt and co-workers (35): DQ coherences are excited, evolve under homonuclear proton decoupling, and are detected after a reconversion step. Here, two complete POST-C7 cycles were used for both DQ excitation and reconversion. A PMLG (phase modulated Lee-Goldburg) homonuclear decoupling step (36) with an RF field of 112 kHz was applied during the DQ-evolution time, which was incremented by multiple integers of 18 PMLG cycles (1/9th of one rotor period). Two equal cw proton-decoupling periods (112 kHz) were applied before and after the evolution time to keep the total evolution time constant (two rotor periods, 250 μs). The simulation of spin dynamics was performed with SIMPSON (37).
Multidimensional MAS-NMR experiments for identifying chemical shift changes
All MAS-NMR experiments were conducted using a 3.2 mm triple-resonance DVT HCN probehead on a Bruker wide bore Avance III solid-state NMR spectrometer with 1H frequency of 850.32 MHz. A sample spinning rate of 14 kHz and SPINAL64 proton decoupling of 83 kHz was used during evolution and acquisition. Typical 1H and 13C 90° pulse lengths were 3 and 4 μsec, respectively. For all experiments the acquisition time in the direct dimension was 20 ms and sample temperature was set to 270 K. 13C and 15N chemical shift referencing was carried out with respect to DSS through Alanin (179.85 ppm). 13C-13C correlation spectra were acquired using proton-driven spin diffusion (PDSD) (see Fig. 3) with a mixing time of 20 ms. A CP contact time of 1.5 ms and a 2.5 sec recycle delay time were used. The acquisition time was set to 10 ms in the indirect dimension and 1024 increments with 80 scans each were recorded. For NCA/NCO/NCACX/NCOCX experiments, initial magnetization was created by a 1H-15N CP step of 1.25 ms. A recycle delay of 3 s was used. Specific CP with a 90–100 linear ramp on the 13C channel was used to create 13C magnetization from 15N using a contact time of 3.5 ms and 5 ms for NCO and NCA transfer, respectively. For NCACX and NCOCX spectra, a 30 ms dipolar-assisted rotational resonance mixing step was used for 13C-13C transfers. Spectra of wild-type (WT) GPR were assigned using published chemical shift data (23). Comparing the PDSD, NCA, and NCO spectra of GPRA178R with GPR data enabled identifying significant chemical shift changes. Peak assignment was confirmed using the NCACX and NCOCX spectra. Data analysis was done using TOPSPIN 2.1 and CCPN 2.2.2.
Figure 3.
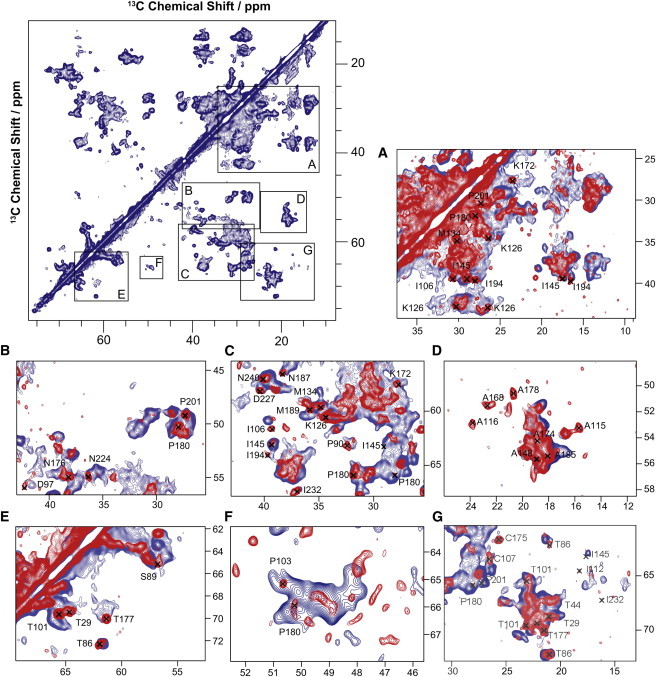
13C-13C-PDSD spectra of GPR (red) and GPRA178R (blue) at pH 9 reconstituted in lipid bilayers. A mixing time of 20 ms was used. A large number of chemical shift differences can be identified as highlighted in the regions A–G. Affected are for example D97 (B) and D227 (C) and alanines in the C-helix and EF loop (D). Chemical shift changes were also identified in NCA and NCO spectra (Fig. S4 and Fig. S5) and resonance assignment was verified by NCOCX and NCACX experiments. A complete list of chemical shift changes is given in Table S1 and highlighted in the topology plot shown in Fig. 7 and in the 3D structure model in Fig. 8.
Results
UV/Vis spectroscopy
A series of pH-dependent absorption spectra of GPR and GPRA178R solubilized in DDM have been recorded (Fig. 1, A–C). The absorption maximum λmax depends on the protonation state of the primary proton acceptor D97 (38). For PR, λmax shifts from 520 to 540 nm when lowering the pH from 9 to 6. GPRA178R follows this trend but shows a significant redshift with changes from 533 to 550 nm within the studied pH range (Fig. 1, A and B). The pKa of D97 can be obtained from the inflection point of the sigmoidal titration curves and was found at 7.0 for GPR and 7.7 for GPRA178R (Fig. 1 C). These data agree with those reported by Kandori and co-workers (17) for a cysteine-less mutant of GPR in which the A178R mutation was introduced.
Figure 1.

pH-dependent stationary absorption spectra of GPR (A) and GPRA178R (B). The single point mutation A178R causes a general redshift of up to 13 nm (14). The pKa of the primary proton acceptor D97 changes from 7.0 for GPR to 7.7 for GPRA178R as determined by analyzing the absorption maximum upon titration of the primary proton acceptor (C). Correlation between λmax and 15N chemical shifts of the pSB obtained from retinal derivatives with all-trans polyene chains with different halide counterions (39) in comparison to GPR and GPRA178R (D). The mutant deviates more from model behavior than GPR. 15N-MAS NMR spectra are shown in Fig. S3.
Solid-state NMR on GPRA178R
Solid-state NMR experiments on GPRA178R reconstituted into DMPC/DMPA lipid bilayers have been carried out to probe structural effects of the A178R EF loop mutation onto Schiff base environment, retinylidene conformation, and protein structure.
The 15N chemical shift of the protonated Schiff base at pH 9 is not affected by the A178R mutation (Fig. S3), but a comparison with the corresponding values from Schiff base—counter ion model complexes and their correlation with λmax shows that GPRA178R deviates from model behavior more than GPR (Fig. 1 D) (39).
To probe mutation-induced effects on the chromophore itself, we have incorporated 14-15-13C-all-trans retinal into GPRA178R ([14-15-13C-retinal]-GPRA178R). Retinal 13C chemical shifts are sensitive indicators for conformational changes. In particular, double quantum spectroscopy has been shown to offer unique insight: Chemical shift data can be extracted more easily by natural background suppression and CC bond lengths and HCCH torsion angles can be obtained from DQ built-up curves (40) and SLF experiments (35). Such experiments will also benefit from signal enhancement provided by DNP (29). DNP-enhanced DQ filtered 13C spectra are shown in Fig. 2 A. A DNP enhancement of 20 (microwave on/off) was obtained, which accelerates data acquisition 400-fold. Both peaks, C14 and C15, appear broader than in GPR and are slightly shifted. C14 is detected at 122.1 ppm (120.2 ppm in GPR) and C15 at 160.7 ppm (161.7 ppm in GPR). A small additional subpopulation is observed at 111 and 165 ppm, which is not detectable in WT GPR. The evolution of DQ coherences between C14 and C15 over two rotor periods (including a centering π-pulse) under homonuclear proton decoupling is shown in Fig. 2 B. Simulations illustrate that the evolution curve depends on the HCCH torsion angle around the C14-C15 bond, but our data show that it remains almost unchanged in GPRA178R compared to GPR (161°).
Figure 2.
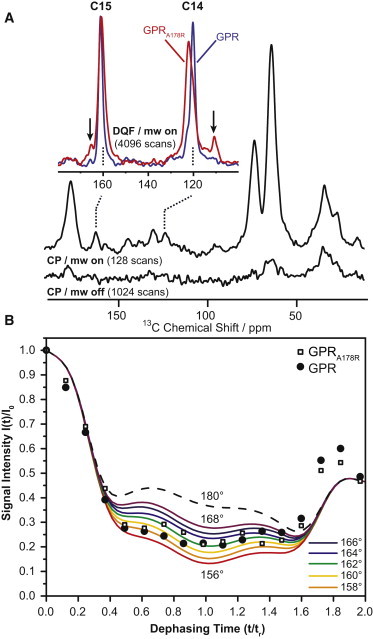
DNP-enhanced 13C-CP MAS NMR spectra of 14-15-13C-all-trans-retinal GPR and GPRA178R (A). Using DNP, a 20-fold signal enhancement is achieved at 100 K. 13C natural abundance is suppressed by double-quantum filtering. In GPR, the 13C chemical shifts of C14 and C15 are found at 120.2 and 161.1 ppm and change in GPRA178R to 122.1 and 160.7 ppm, respectively. The linewidth increases from 160 to 290 Hz. In GPRA178R, small additional peaks at 110.7 and 165.6 ppm are detected (arrows). The HCCH torsion angle at position C14-C15 has been determined by DQ-SLF experiments (B). Experimental and simulated DQ signal intensities under homonuclear decoupling are plotted as a function of time t in units of rotor periods tr and reveal a torsion angle of 161° in GPRA178R, which is similar to GPR. Further details are given in the Materials and Methods section.
The observed effects on color and retinylidene chemical shifts must be induced by structural changes triggered by the EF loop and transferred through the protein into the retinal binding pocket. We have therefore analyzed mutation-induced 13C and 15N chemical shift perturbations by comparing CC, NCA, and NCO spectra of GPRA178R with those obtained from GPR. Samples were uniformly 13C and 15N labeled except for amino acids F, L, W, Y, and V to reduce spectral overlap. In a first step, peaks shifted upon mutation were identified and tentatively assigned based on published chemical shift assignment (23). In a second step, assignments were supported by two-dimensional NCOCX/NCACX experiments on GPR and GPRA178R. We found 63 residues in which either backbone and/or side-chain resonances show chemical shift changes larger than 0.2 ppm (Table S1). A CC correlation spectrum (13C-13C-PDSD) of GPR and GPRA178R is shown in Fig. 3. This experiment displays through-space correlations but a short mixing time of 20 ms was used emphasizing spin-spin distances in the range of single bonds. A number of crosspeaks shift upon mutation such as D97 (Fig. 3 B) and D227 (Fig. 3 C) or A115, 116, 143, 168, 174, and 178 (Fig. 3 D). Altogether, 31 affected residues can be identified from this spectrum. Changes mainly in CA-CB crosspeaks are detected but in 11 cases, chemical shift perturbations in side chains were monitored as well. These data are complemented by NCA (Fig. S4) and NCO (Fig. S5) spectra, which confirmed our findings from Fig. 3 and also allowed identification of an additional 32 chemical shift perturbations. The location of all affected residues is plotted in a topology diagram (see Fig. 7). The observed chemical shift perturbations progress from the mutation site in the EF loop through helices E and F into all other helices as well as into the BC and DE loops.
Figure 7.

The A178R mutation in the EF loop induces backbone and/or side-chain chemical shift changes in 63 residues (colored dark, see also Fig. 3, Fig. S4, Fig. S5, and Table S1). They are located in all seven helices as well as in the BC, DE, and EF loops. Residues of direct functional relevance are labeled. The EF loop contains a short interfacial helix E′ followed by a turn structure (23,27).
Probing the primary reaction in GPRA178R by pump-probe spectroscopy
Our solid-state NMR data show remarkable structural effects on the photoactive site caused by the distant EF loop. We have therefore probed its consequences for the dynamics of the primary photoreaction by time-resolved optical pump-probe spectroscopy in the ps - ns time range. The photoisomerization around the C13-C14 double bond and the formation of the first ground state intermediate (K intermediate in the photocycle of BR) occur on this timescale. For BR and GPR it is well known that these processes are very sensitive toward the structural environment of the primary proton acceptor (D85 in BR, D97 in GPR) (41–44).
The photoexcitation-induced time-resolved absorbance changes in GPRA178R are compared to data obtained for GPR as reported earlier (44,45) (Fig. 4). Experiments were performed at pH 6 and pH 9, which is below and above the pKa of the primary proton acceptor. For GPR at alkaline pH, four spectral features can be observed. These regions can be attributed to the absorption of the excited state, the stimulated emission, the depletion of the ground state, and the absorption of the photoproduct. All spectral features are redshifted for GPRA178R according to the bathochromic shift observed in the stationary absorption spectrum (Fig. 1). The primary reaction is significantly prolonged for the GPRA178R mutant illustrated by the lifetime of the S1-state, represented by the decay of the excited state and stimulated emission (for a comparison of the temporal evolution of the different spectral features see also the single transients at distinct probe wavelengths in Fig. S6). However, the amplitude of the photoproduct at longer delay times is lower for GPRA178R, suggesting either a reduced quantum yield for photoisomerization or extinction coefficient of the photoproduct.
Figure 4.

Transient absorption changes of GPR compared to GPRA178R at pH 6 and pH 9 after photoexcitation at their particular absorption maximum. The time axis is scaled linear until 1 ps and logarithmic until 1 ns. The amplitudes are color-coded: red indicates a positive absorption, blue negative, and green marks areas where no absorption change could be detected. Abbreviations: ESA (excited state absorption), GSB (ground state bleach), PA (photoinduced absorption), and SE (stimulated emission). Individual time traces at 451, 579, and 707 nm are given in Fig. S6. Time constants obtained via a global fit analysis are given in Table 1.
The transient data set is simulated by a global fit analysis consisting of a sum of exponential decays convoluted with the instrument response function. The time constants for all wavelengths are the same, whereas the amplitudes are varied (Table 1). The corresponding decay-associated spectra are plotted in Fig. S7. The global fit analysis reveals four time constants for the GPR and an additional time constant τx for GPRA178R. The time constants can be assigned to similar processes described for GPR earlier (44,45). τ1 describes the motion of the initially generated wave packet out of the Franck-Condon region and contains contributions from stimulated emission and excited state population, respectively. τx can be assigned to the same processes as τ1 (see the Supporting Material). The time constants τ2 and τ3 can be assigned to the decay of the excited state population.
Table 1.
Global fit analyses of the data set shown in Fig. 4
| τ1/ps | τx/ps | τ2/ps | τ3/ps | τ4 | |
|---|---|---|---|---|---|
| GPR/pH 6.4 | 0.15 | – | 1 | 16.2 | ∞ |
| GPR/pH 9 | 0.14 | – | 0.28 | 9.5 | ∞ |
| GPRA178R/pH 6 | <0.1 | 0.6 | 3.7 | 24 | ∞ |
| GPRA178R/pH 9 | <0.1 | 0.7 | 3.6 | 20 | ∞ |
The data in Fig. 4 were analyzed by a global fit routine using a sum of exponentials with the same time constants for all wavelengths but different amplitudes. As reported earlier (19,44), the primary reaction in GPR can be described by four time constants (τ1–τ4), whereas one additional constant (τx) was needed for GPRA178R. Furthermore, a time constant that is set to infinite is necessary to describe the constant amplitude for longer delay times. This time constant represents the difference spectrum between the first photointermediate and the initial state.
Obviously, the mechanism of photoisomerization seems to be affected upon A178R mutation. For GPR, the corresponding time constants τ2 and τ3 exhibit similar amplitude spectra suggesting that the initial and final states of these transitions are equal (45). In contrast, the amplitude spectra of τ2 and τ3 obtained for GPRA178R differ significantly, illustrating that τ2 and τ3 describe different reaction paths on the S1 potential energy surface. Moreover, the ratio of the amplitudes is similar at both pH-values, whereas for GPR it was shown that at alkaline pH the faster reaction channel is preferred.
The obtained time constants in Table 1 confirm that all processes are prolonged for GPRA178R. The comparison of the obtained time constants and the corresponding decay-associated spectra at different pH for GPR and GPRA178R suggests that photoisomerization becomes nearly pH-independent in GPRA178R. For GPR, τ2 is increased by a factor of 3 and τ3 by a factor of 2 upon lowering the pH from 9 to 6. In contrast to this the obtained time constants are similar for GPRA178R at both pH-values. Only for τ3 a small but significant increase can be observed upon lowering the pH from 9 to 6. Again, the effect is not as pronounced as for the WT GPR.
Probing the photocycle in GPRA178R by flash photolysis
Our solid-state NMR data show mutation-inducing effects at the retinylidene but also on residues such as D97, which are directly involved in proton transfer. We have therefore analyzed mutation-induced alterations in the photocycle dynamics after photoexcitation on a timescale of 1 μs to 10 s. Transient absorption changes for GPR and GPRA178R after photoexcitation are shown in Fig. 5.
Figure 5.

Laser-flash-induced transient absorption changes at pH 9 for GPRA178R and GPR. The transients are representative for the dynamics of the ground state population (500 nm), the K-decay at early delay times and the formation of the N/O-intermediates at late delay times (590 nm) and the formation and decay of the M-intermediate (400 nm). Time constants obtained via a global fit analysis are given in Table S2.
At 590 nm, a decrease of the positive absorption signal in GPR is observed due to the decay of the K-intermediate, which is the photoproduct of the primary reaction observed in the pump-probe measurements discussed previously. This decay is accompanied by an increasing absorption monitored at 400 nm typically found for a deprotonated Schiff base species (M-intermediate). The M-intermediate is followed by an absorption increase at 590 nm, caused by the formation of late N/O-intermediates. These species decay simultaneously with the growth of the signal at 500 nm arising from the repopulation of the initial state. The GPR photocycle is completed within hundreds of ms. In contrast, the photodynamics of GPRA178R is strongly altered: The K-intermediate has almost completely decayed after 1 μs, whereas at the same time the M-state at 400 nm appears already completely populated. The subsequent formation of the N/O-intermediates at 590 nm and the repopulation of the initial state monitored at 500 nm are delayed compared to GPR.
We have analyzed the transient data set using a reaction scheme proposed earlier (32). However, an additional photointermediate was necessary to satisfactorily simulate the experimental data. The additional photointermediate is populated with a time constant of 48 ms. Considering the obviously multiexponential rise of the transient monitored at 590 nm in Fig. 5, the additional intermediate is attributed to an interconversion of several N-intermediates (termed N1 and N2). Fig. 6 shows an overview of the photocycle dynamics of GPR and GPRA178R. The decay of the K-intermediate and the formation of the M-intermediate are dramatically accelerated for GPRA178R. In contrast to this the later steps of the photocycle, mainly the lifetimes of the N1, N2, and O intermediate are prolonged, leading to an overall photocycle duration, that is 10-fold higher for GPRA178R compared to GPR. Fig. 6 also contains the time-dependent population changes of the different intermediates, as well as the calculated absorption spectra of the different intermediates. The N1 to N2 interconversion is accompanied by a small bathochromic shift and an increase of the extinction coefficient.
Figure 6.

The experimental data is fitted to a reaction scheme described in the Materials and Methods (A). For simulating our experimental data we have varied the extinction coefficients of the photointermediates (B) and the time-dependent population changes (C). Multiplying the time-dependent population changes and the extinction coefficients reestablishes the experimental data obtained by flash photolysis.
Discussion
Kandori and co-workers (14) have shown that a single mutation A178R in the EF loop of GPR, which is far away from the retinal binding site, causes a surprisingly large redshift and changes the pKa of the primary proton acceptor D97 (Fig. 1). They have also shown that this effect is strongly position dependent (15) and depends merely on the side-chain volume and less on the charge of the amino acid at position 178 (17). Here, we demonstrate that this mutation not only causes a redshift but also significantly modulates the photocycle dynamics of GPR. The photoisomerization is prolonged and nearly independent on pH. The latter is surprising because it was shown that this process is twofold accelerated at alkaline pH compared to acidic pH. Overall, the photocycle takes ∼10 times longer due to a longer lifetime of the N/O intermediate. In the following, we will explain the molecular basis for these surprising effects based on our solid-state NMR data.
We have identified mutation-induced chemical shift changes in 63 residues spread out across the protein sequence as illustrated in the topology plot in Fig. 7. Affected residues are found in all seven helices and in the BC, DE, and EF loops. The location of these residues is visualized in a structural model of GPR in Fig. 8 A. Replacing A178 with R in the EF loop triggers chemical shift changes, which progress from the EF loop through helices E and F across the whole protein. The chemical shifts of resonances arising from CA, CO, CB, and N sites in the protein backbone depend on dihedral angle Φ and Ψ and alterations indicate small structural changes.
Figure 8.

Residues for which mutation-induced chemical shift changes are observed and are highlighted in a simplified structure model, which is consistent with NMR data and based on homology to BR (19,24,25,27,65) (A). An interaction pathway from the EF loop to the retinal binding pocket can be envisaged. The observed chemical shift changes (B) indicate that the secondary structure in the EF loop is distorted (C). A number of seven residues out of 63 affected by the A178R mutation (Fig. 7) are directly located within or close to the retinal binding pocket (D). These include G203, C156, M134, and G138 in close proximity to the retinal ring as well as the primary proton acceptor D97. D227 is assumed to play a role in retinal isomerization. Chemical shift changes within these residues indicated slight structural rearrangements, which can explain the significant mutation-induced changes in GPR’s optical and kinetic properties.
It seems that incorporating a larger side-chain volume at position 178 (Arg is twice as large as Ala) disrupts the EF loop with consequences for the whole GPR structure including the retinal binding pocket and subsequently also photocycle dynamics. Kandori and co-workers had already speculated that the EF loop must contain specific structural elements, which are disturbed upon mutation. Indeed, a previous solid-state NMR study by Brown, Ladizhanski and co-workers has identified a kink at the cyotoplasmatic end of helix E at positions A168 and G169 preceding a short α-helix E170-N176 (E′), which is followed by a β-turn formed by T177, A178, and S179 (23). The existence of a kink at position G169 and the helical segment E′ was later also observed in the 3D backbone structure of cell-free produced GPR determined by liquid-state NMR in DHPC micelles (27) and is also consistent with EPR/DNP studies (46). Interestingly, a structured EF loop is not exclusive to GPR but is also found in other retinal proteins and GPCRs (47–51). Our data clearly show that the NMR signals of almost all residues between A168 and S179 in this kink-helix-β-turn region are affected by the A178R mutation: We have identified chemical shift changes larger than 0.2 ppm for A168, G169, E170, G171, K172, S173, A174, C175, N176, and P180 (Table S1). The CA chemical shift differences are shown in Fig. 8 B and reveal a trend toward smaller values upon mutation indicating a distortion of the E′ helical structure between residues 168 and 179 as illustrated in the cartoon in Fig. 8 C. This loosened loop structure must trigger structural and dynamic changes in GPR explaining the observed color shift and altered photocycle dynamics.
In or close to the retinal binding pocket, we have identified seven residues, which show clear chemical shift changes larger than 0.2 ppm (Fig. 8 A, color-coded in red). These are D97, M134, G138, C156, G203, D227, and I232. Fig. 8 C shows the approximate location of those seven residues with respect to the retinal. The primary proton acceptor D97 and residue D227 are located on both sides of the retinal polyene chain, close to C14, C15, and the protonated Schiff base. The pSB 15N resonance is not affected, whereas the 13C chemical shifts of retinal carbons C14 and C15 move by +1.9 and −0.4 ppm, respectively, upon mutation. D97 is considered as primary counterion of the protonated Schiff base and has a strong influence on the photokinetics either for the ultrafast timescales (from femtoseconds to 1 ns) (44,45) or on the later stages of the photocycle (1 μs to 1 s) (3,32,52). Residue D227 has been suggested to play a role in the photocycle and retinylidene isomerization (53).
Our observations allow the unusual redshift in GPRA178R to be explained. The chemical shift changes in the retinylidene polyene chain indicate alterations in pSB/counterion distances and relative orientation. A weakening of pSB/counterion association has been shown to shift retinal absorption maxima to longer wavelengths (54). Another contribution could be a change in the β-ionone ring/polyene chain coplanarity, which is mainly caused by protein-retinal contacts and contributes 20% to the opsin shift of BR (12). Direct structural changes within the polyene chain around C14 and C15 are not observed, as the HCCH torsion angle remains almost unaltered compared to the WT (Fig. 2). However, residues for which chemical shift changes have been observed surround the β-ionone ring. Therefore, the ring-protein interaction interface is altered, which could result in altered ring orientation and subsequent color change.
Restructuring effects of the retinal binding pocket also agree with the different pKa of D97 in the main titration curve (Fig. 1) and the loss of pH dependence of the primary reaction, which is also 2.5-fold slower. In GPR, pH affects the protonation state of the primary proton acceptor leading to a spectral shift in the absorption spectrum of the initial state (38) and a strong pH-dependent isomerization rate (44,45). In general, it is known that the first electronic state S1 exhibits a more pronounced charge-transfer character than the ground state S0. The transition into the first electronic state S1 is accompanied by an increase of the dipole moment (55) that was estimated to be 12 D (56). Theoretical studies revealed that electrons are shifted from the hydrocarbon tail toward the protonated Schiff base upon photoexcitation (57–59). Therefore, a negative charge placed in the vicinity of the Schiff base influences the electronic configuration of the retinal. The relative orientation of the primary proton acceptor to the Schiff base can therefore stabilize or destabilize the S1 and the S0 state. This directly affects the absorption maximum of the retinal, which reflects the energetic difference between the S1 and the S0 state. For GPRA178R a pH-dependent shift of the absorption maximum can be observed indicating that the electronic configuration of the retinal is affected by an either protonated or deprotonated primary proton acceptor D97. However, pump-probe spectroscopy reveals that the protonation state of D97 has nearly no influence on the photoisomerization process. For BR (60), GPR (44), and ChR2 (61), it was shown that a primary proton acceptor functions as an efficient catalyst for photoisomerization, which is not the case in GPRA178R. It is known from quantum mechanical calculations that the slopes on the S1 potential energy surfaces of the S1 state are significantly affected by the relative orientation of an acetate residue with respect to the protonated Schiff base resulting in altered isomerization rates (62). A steeper slope on the S1-potential energy surface leads to an accelerated relaxation from the Franck-Condon region to the S1/S0 conical intersection, therefore accelerating the decay of the S1-state (62). These findings support that the A178R mutation strongly affects the position of D97 with respect to the chromophore, which is also supported by our NMR data. D97 adopts a position in which the effect of deprotonation on the S1 potential surface is nearly absent.
Our 13C-DNP MAS NMR data show a small 13-cis subpopulation in GPRA178R in contrast to GPR (Fig. 2). Isomerization control in GPR has been linked to residue D227 (53,63), which is located in close proximity to the chromophore and is directly affected by the A178R mutation. A structural change in the retinal binding pocket could reduce the isomerization control of this residue. For example, it has been shown that PRD227N contains larger amounts of 13-cis retinal in its ground state and exhibits only minimal pH dependence for the primary reaction and a prolonged photocycle in contrast to GPR (64). Similar observations are made here. This means that restructuring the retinal binding pocket involves altered distances between Schiff base, retinal, and counterions including alteration in the hydrogen bonding network.
As discussed previously, the EF loop structure is distorted in GPRA178R resulting probably in weaker structured constraints of the EF loop onto helices E and F. Hence, structural changes propagate through both helices via DE and FG loops, via side-chain contacts, and through retinal-protein contacts to the rest of the protein (Fig. 8 A). The altered structure in the EF loop must also have consequences for the photocycle steps involving movements of helices E and F. The EF loop structures could restrain helix movements during the photocycle, which become more relaxed in the mutant. Indeed, the A178R mutation slows down the photocycle 10-fold despite the accelerated decay of the K-intermediate (<1 μs). For GPR, photocycles between 100 and 200 ms have been reported (3,32,52,65,66). The longer photocycle for GPRA178R is caused by the elongated lifetime of the N/O intermediate and the interconversion of two different N intermediates. The decay of these intermediates is correlated with the reprotonation of the primary proton donor. In BR at pH > 9, a reaction scheme M1→M2→N1 →N2→BR was found, where N1 and N2 are referred to as the N intermediates before and after reprotonation of D96 (67,68). In GPR, the primary proton donor E108 is found in helix C, in which a number of mutation-induced chemical shift changes were observed (Figs.7 and 8). It has been observed in BR that this step of proton uptake involves an outward motion of helix F facilitating the proton uptake from the cytoplasmic side, most probably due to the entrance of water molecules (69–72). After proton uptake from the cytoplasmic surface this outward movement of the F-helix is reversed (73). Assuming a similar mechanism in GPR leads to the conclusion that the restoring force imposed by the structured EF loop to helices E and F is significantly reduced in GPRA178R resulting in slower helix reorientation, hence an elongated lifetime of the N/O intermediate. Therefore, the native EF loop seems to play a major role in proton uptake from the cytoplasmic side of GPR. This conclusion is also supported by a combined EPR and DNP study in which altered hydration levels upon illumination indicated an EF loop movement (46). However, the capability of proton pumping is not affected by this mutation as shown by light-induced pH changes in spheroblast vesicles (15) and by black lipid membrane experiments (Fig. S9).
Conclusion
We have shown by time-resolved optical spectroscopy that the EF-loop mutation A178R in GPR does not only cause a redshift but slows down the primary reaction, weakens its pH dependence, and slows down the complete photocycle due to the elongated lifetime of the N/O intermediates. Using conventional and DNP-enhanced solid-state NMR, we have identified the molecular origin of these surprising effects. Introducing a bulky residue such as R at position 178 disrupts the secondary EF-loop structure. The resulting distorted loop structure imposes weaker structural constraints on E and F helices compared to GPR, which alters the structure of the retinal binding site. As a consequence, the chromophore color, the primary reaction as well as late stages of the photocycle, involving E and F helix movements during proton uptake, are affected. Our data indicate that the EF loop plays an important role for proton uptake similar to BR (69). On the other hand, our data also show that an interaction pathway exists between the EF loop and retinal binding pocket. One could speculate that in this way signals could be transmitted across the membrane needed for potentially additional functions of GPR such as sensing or signaling. Our study also illustrates that advanced methodology such as DNP/solid-state NMR and time-resolved optical spectroscopy provides highly compatible data sets if properly combined and enables obtaining unprecedented insight into the molecular mechanism of retinal proteins. A related study on mechanism of the major green/blue switch L105Q in GPR will be reported elsewhere.
Acknowledgments
Lenica Reggie, Vasyl Denysenkov, Jörn Plackmeyer, and Thomas Prisner, BMRZ Frankfurt are acknowledged for their support with our DNP setup. We are grateful to Daniel Gottstein and Peter Güntert, BMRZ Frankfurt, for helpful discussions and GPR structure modeling. Marie Concistre and Malcolm H. Levitt, Southampton are acknowledged for helpful advice on the HCCH data analysis. We thank Anja Becker, MPI Biophysics, Frankfurt, for help with BLM experiments.
This work was funded by SFB 807 Transport and communication across membranes. The DNP experiments were enabled through an equipment grant provided by the Deutsche Forschungsgemeinschaft (DFG) (Cluster of Excellence Macromolecular Complexes Frankfurt).
Contributor Information
Josef Wachtveitl, Email: wveitl@theochem.uni-frankfurt.de.
Clemens Glaubitz, Email: glaubitz@em.uni-frankfurt.de.
Supporting Material
References
- 1.Bamann C., Kirsch T., Bamberg E. Spectral characteristics of the photocycle of channelrhodopsin-2 and its implication for channel function. J. Mol. Biol. 2008;375:686–694. doi: 10.1016/j.jmb.2007.10.072. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi T., Yan B., Spudich J.L. Color regulation in the archaebacterial phototaxis receptor phoborhodopsin (sensory rhodopsin II) Biochemistry. 1990;29:8467–8474. doi: 10.1021/bi00488a038. [DOI] [PubMed] [Google Scholar]
- 3.Friedrich T., Geibel S., Bamberg E. Proteorhodopsin is a light-driven proton pump with variable vectoriality. J. Mol. Biol. 2002;321:821–838. doi: 10.1016/s0022-2836(02)00696-4. [DOI] [PubMed] [Google Scholar]
- 4.Oesterhelt D., Stoeckenius W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 1971;233:149–152. doi: 10.1038/newbio233149a0. [DOI] [PubMed] [Google Scholar]
- 5.Béjà O., Spudich E.N., DeLong E.F. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411:786–789. doi: 10.1038/35081051. [DOI] [PubMed] [Google Scholar]
- 6.Man D., Wang W., Béjà O. Diversification and spectral tuning in marine proteorhodopsins. EMBO J. 2003;22:1725–1731. doi: 10.1093/emboj/cdg183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grossjean M.F., Tavan P. Wavelength regulation in bacteriorhodopsin and halorhodopsin - a Pariser-Parr-Pople multireference double excitation configuration-interaction study of retinal dyes. J. Chem. Phys. 1988;88:4884–4896. [Google Scholar]
- 8.Sekharan S., Sugihara M., Buss V. Origin of spectral tuning in rhodopsin—it is not the binding pocket. Angew. Chem. Int. Ed. Engl. 2007;46:269–271. doi: 10.1002/anie.200603306. [DOI] [PubMed] [Google Scholar]
- 9.Kochendoerfer G.G., Lin S.W., Mathies R.A. How color visual pigments are tuned. Trends Biochem. Sci. 1999;24:300–305. doi: 10.1016/s0968-0004(99)01432-2. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann M., Wanko M., Elstner M. Color tuning in rhodopsins: the mechanism for the spectral shift between bacteriorhodopsin and sensory rhodopsin II. J. Am. Chem. Soc. 2006;128:10808–10818. doi: 10.1021/ja062082i. [DOI] [PubMed] [Google Scholar]
- 11.Wanko M., Hoffmann M., Elstner M. Calculating absorption shifts for retinal proteins: computational challenges. J. Phys. Chem. B. 2005;109:3606–3615. doi: 10.1021/jp0463060. [DOI] [PubMed] [Google Scholar]
- 12.Yan B., Spudich J.L., Nakanishi K. Spectral tuning in bacteriorhodopsin in the absence of counterion and coplanarization effects. J. Biol. Chem. 1995;270:29668–29670. doi: 10.1074/jbc.270.50.29668. [DOI] [PubMed] [Google Scholar]
- 13.Wang W.W., Sineshchekov O.A., Spudich J.L. Spectroscopic and photochemical characterization of a deep ocean proteorhodopsin. J. Biol. Chem. 2003;278:33985–33991. doi: 10.1074/jbc.M305716200. [DOI] [PubMed] [Google Scholar]
- 14.Yoshitsugu M., Shibata M., Kandori H. Color change of proteorhodopsin by a single amino acid replacement at a distant cytoplasmic loop. Angew. Chem. Int. Ed. Engl. 2008;47:3923–3926. doi: 10.1002/anie.200705989. [DOI] [PubMed] [Google Scholar]
- 15.Yoshitsugu M., Yamada J., Kandori H. Color-changing mutation in the E-F loop of proteorhodopsin. Biochemistry. 2009;48:4324–4330. doi: 10.1021/bi900228a. [DOI] [PubMed] [Google Scholar]
- 16.Otomo J. Influence exercised by histidine-95 on chloride transport and the photocycle in halorhodopsin. Biochemistry. 1996;35:6684–6689. doi: 10.1021/bi952853n. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K., Kawanabe A., Kandori H. Importance of alanine at position 178 in proteorhodopsin for absorption of prevalent ambient light in the marine environment. Biochemistry. 2010;49:2416–2423. doi: 10.1021/bi9020204. [DOI] [PubMed] [Google Scholar]
- 18.Fuhrman J.A., Schwalbach M.S., Stingl U. Proteorhodopsins: an array of physiological roles? Nat. Rev. Microbiol. 2008;6:488–494. doi: 10.1038/nrmicro1893. [DOI] [PubMed] [Google Scholar]
- 19.Hempelmann F., Hölper S., Glaubitz C. His75-Asp97 cluster in green proteorhodopsin. J. Am. Chem. Soc. 2011;133:4645–4654. doi: 10.1021/ja111116a. [DOI] [PubMed] [Google Scholar]
- 20.Bergo V.B., Sineshchekov O.A., Spudich J.L. His-75 in Proteorhodopsin, a Novel Component in Light-driven Proton Translocation by Primary Pumps. J. Biol. Chem. 2009;284:2836–2843. doi: 10.1074/jbc.M803792200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klyszejko A.L., Shastri S., Glaubitz C. Folding and assembly of proteorhodopsin. J. Mol. Biol. 2008;376:35–41. doi: 10.1016/j.jmb.2007.11.030. [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann J., Aslimovska L., Brutschy B. Studying the stoichiometries of membrane proteins by mass spectrometry: microbial rhodopsins and a potassium ion channel. Phys. Chem. Chem. Phys. 2010;12:3480–3485. doi: 10.1039/b924630d. [DOI] [PubMed] [Google Scholar]
- 23.Shi L., Ahmed M.A., Ladizhansky V. Three-dimensional solid-state NMR study of a seven-helical integral membrane proton pump—structural insights. J. Mol. Biol. 2009;386:1078–1093. doi: 10.1016/j.jmb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Shi L.C., Lake E.M.R., Ladizhansky V. Solid-state NMR study of proteorhodopsin in the lipid environment: secondary structure and dynamics. Biochim. Biophys. Acta. 2009;1788:2563–2574. doi: 10.1016/j.bbamem.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Yang J., Aslimovska L., Glaubitz C. Molecular dynamics of proteorhodopsin in lipid bilayers by solid-state NMR. J. Am. Chem. Soc. 2011;133:4874–4881. doi: 10.1021/ja109766n. [DOI] [PubMed] [Google Scholar]
- 26.Pfleger N., Lorch M., Glaubitz C. Characterisation of Schiff base and chromophore in green proteorhodopsin by solid-state NMR.J. Biomol. NMR. 2008;40:15–21. doi: 10.1007/s10858-007-9203-5. [DOI] [PubMed] [Google Scholar]
- 27.Reckel S., Gottstein D., Dötsch V. Solution NMR structure of proteorhodopsin. Angew. Chem. Int. Ed. Engl. 2011;50:11942–11946. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stehle J., Scholz F., Schwalbe H. Characterization of the ground state dynamics of proteorhodopsin by NMR and optical spectroscopies. J. Biomol. NMR. 2012;54:401–413. doi: 10.1007/s10858-012-9684-8. [DOI] [PubMed] [Google Scholar]
- 29.Maly T., Debelouchina G.T., Griffin R.G. Dynamic nuclear polarization at high magnetic fields. J. Chem. Phys. 2008;128:052211–052219. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pardoen J., Winkel C., Lugtenburg J. Synthesis of retinals labeled at positions 14 and 15 (with C-13 and or H-2) Recl. Trav. Chim. Pays-Bas. 1984;103:135–141. [Google Scholar]
- 31.Song C.S., Hu K.N., Griffin R.G. TOTAPOL: a biradical polarizing agent for dynamic nuclear polarization experiments in aqueous media. J. Am. Chem. Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 32.Váró G., Brown L.S., Lanyi J.K. Characterization of the photochemical reaction cycle of proteorhodopsin. Biophys. J. 2003;84:1202–1207. doi: 10.1016/S0006-3495(03)74934-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hohwy M., Jakobsen H.J., Nielsen N.C. Broadband dipolar recoupling in the nuclear magnetic resonance of rotating solids: a compensated C7 pulse sequence. J. Chem. Phys. 1998;108:2686–2694. [Google Scholar]
- 34.Fung B.M., Khitrin A.K., Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 35.Concistrè M., Johannessen O.G., Levitt M.H. A large geometric distortion in the first photointermediate of rhodopsin, determined by double-quantum solid-state NMR. J. Biomol. NMR. 2012;53:247–256. doi: 10.1007/s10858-012-9635-4. [DOI] [PubMed] [Google Scholar]
- 36.Vinogradov E., Madhu P.K., Vega S. High-resolution proton solid-state NMR spectroscopy by phase-modulated Lee-Goldburg experiment. Chem. Phys. Lett. 1999;314:443–450. [Google Scholar]
- 37.Bak M., Rasmussen J.T., Nielsen N.C. SIMPSON: a general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 38.Sharaabi Y., Brumfeld V., Sheves M. Binding of anions to proteorhodopsin affects the Asp97 pK(a) Biochemistry. 2010;49:4457–4465. doi: 10.1021/bi901746b. [DOI] [PubMed] [Google Scholar]
- 39.Hu J., Griffin R.G., Herzfeld J. Synergy in the spectral tuning of retinal pigments: complete accounting of the opsin shift in bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 1994;91:8880–8884. doi: 10.1073/pnas.91.19.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carravetta M., Zhao X., Levitt M.H. Protein-induced bonding perturbation of the rhodopsin chromophore detected by double-quantum solid-state NMR. J. Am. Chem. Soc. 2004;126:3948–3953. doi: 10.1021/ja039390q. [DOI] [PubMed] [Google Scholar]
- 41.Song L., El-Sayed M.A., Lanyi J.K. Effect of changing the position and orientation of Asp85 relative to the protonated Schiff base within the retinal cavity on the rate of photoisomerization in bacteriorhodopsin. J. Phys. Chem. 1996;100:10479–10481. [Google Scholar]
- 42.Song L., El-Sayed M.A., Lanyi J.K. Protein catalysis of the retinal subpicosecond photoisomerization in the primary process of bacteriorhodopsin photosynthesis. Science. 1993;261:891–894. doi: 10.1126/science.261.5123.891. [DOI] [PubMed] [Google Scholar]
- 43.Logunov S.L., El-Sayed M.A., Lanyi J.K. Photoisomerization quantum yield and apparent energy content of the K intermediate in the photocycles of bacteriorhodopsin, its mutants D85N, R82Q, and D212N, and deionized blue bacteriorhodopsin. J. Phys. Chem. 1996;100:2391–2398. [Google Scholar]
- 44.Huber R., Köhler T., Wachtveitl J. pH-dependent photoisomerization of retinal in proteorhodopsin. Biochemistry. 2005;44:1800–1806. doi: 10.1021/bi048318h. [DOI] [PubMed] [Google Scholar]
- 45.Lenz M.O., Huber R., Wachtveitl J. First steps of retinal photoisomerization in proteorhodopsin. Biophys. J. 2006;91:255–262. doi: 10.1529/biophysj.105.074690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hussain S., Franck J.M., Han S. Transmembrane protein activation refined by site-specific hydration dynamics. Angew. Chem. Int. Ed. Engl. 2013;52:1953–1958. doi: 10.1002/anie.201206147. [DOI] [PubMed] [Google Scholar]
- 47.Alexiev U., Rimke I., Pöhlmann T. Elucidation of the nature of the conformational changes of the EF-interhelical loop in bacteriorhodopsin and of the helix VIII on the cytoplasmic surface of bovine rhodopsin: a time-resolved fluorescence depolarization study. J. Mol. Biol. 2003;328:705–719. doi: 10.1016/s0022-2836(03)00326-7. [DOI] [PubMed] [Google Scholar]
- 48.Higman V.A., Varga K., Watts A. The conformation of bacteriorhodopsin loops in purple membranes resolved by solid-state MAS NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2011;50:8432–8435. doi: 10.1002/anie.201100730. [DOI] [PubMed] [Google Scholar]
- 49.Schubert M., Kolbe M., Schmieder P. Heteronuclear multidimensional NMR spectroscopy of solubilized membrane proteins: resonance assignment of native bacteriorhodopsin. ChemBioChem. 2002;3:1019–1023. doi: 10.1002/1439-7633(20021004)3:10<1019::AID-CBIC1019>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 50.Ulfers A.L., McMurry J.L., Mierke D.F. Structure of the third intracellular loop of the human cannabinoid 1 receptor. Biochemistry. 2002;41:11344–11350. doi: 10.1021/bi0259610. [DOI] [PubMed] [Google Scholar]
- 51.Yeagle P.L., Alderfer J.L., Albert A.D. Structure of the third cytoplasmic loop of bovine rhodopsin. Biochemistry. 1995;34:14621–14625. doi: 10.1021/bi00045a002. [DOI] [PubMed] [Google Scholar]
- 52.Lakatos M., Lanyi J.K., Váró G. The photochemical reaction cycle of proteorhodopsin at low pH. Biophys. J. 2003;84:3252–3256. doi: 10.1016/S0006-3495(03)70049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikeda D., Furutani Y., Kandori H. FTIR study of the retinal Schiff base and internal water molecules of proteorhodopsin. Biochemistry. 2007;46:5365–5373. doi: 10.1021/bi700143g. [DOI] [PubMed] [Google Scholar]
- 54.Blatz P.E., Mohler J.H., Navangul H.V. Anion-induced wavelength regulation of absorption maxima of Schiff bases of retinal. Biochemistry. 1972;11:848–855. doi: 10.1021/bi00755a026. [DOI] [PubMed] [Google Scholar]
- 55.Hufen J., Sugihara M., Buss V. How the counterion affects ground- and excited-state properties of the rhodopsin chromophore. J. Phys. Chem. B. 2004;108:20419–20426. [Google Scholar]
- 56.Schenkl S., van Mourik F., Chergui M. Probing the ultrafast charge translocation of photoexcited retinal in bacteriorhodopsin. Science. 2005;309:917–920. doi: 10.1126/science.1111482. [DOI] [PubMed] [Google Scholar]
- 57.Garavelli M., Celani P., Olivucci M. The C5H6NH2+ protonated Schiff base: an ab initio minimal model for retinal photoisomerization. J. Am. Chem. Soc. 1997;119:6891–6901. [Google Scholar]
- 58.Mathies R., Stryer L. Retinal has a highly dipolar vertically excited singlet state: implications for vision. Proc. Natl. Acad. Sci. USA. 1976;73:2169–2173. doi: 10.1073/pnas.73.7.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.González-Luque R., Garavelli M., Olivucci M. Computational evidence in favor of a two-state, two-mode model of the retinal chromophore photoisomerization. Proc. Natl. Acad. Sci. USA. 2000;97:9379–9384. doi: 10.1073/pnas.97.17.9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Logunov S.L., El-Sayed M.A., Lanyi J.K. Catalysis of the retinal subpicosecond photoisomerization process in acid purple bacteriorhodopsin and some bacteriorhodopsin mutants by chloride ions. Biophys. J. 1996;71:1545–1553. doi: 10.1016/S0006-3495(96)79357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scholz F., Bamberg E., Wachtveitl J. Tuning the primary reaction of channelrhodopsin-2 by imidazole, pH, and site-specific mutations. Biophys. J. 2012;102:2649–2657. doi: 10.1016/j.bpj.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cembran A., Bernardi F., Garavelli M. Counterion controlled photoisomerization of retinal chromophore models: a computational investigation. J. Am. Chem. Soc. 2004;126:16018–16037. doi: 10.1021/ja048782+. [DOI] [PubMed] [Google Scholar]
- 63.Imasheva E.S., Shimono K., Lanyi J.K. Formation of a long-lived photoproduct with a deprotonated Schiff base in proteorhodopsin, and its enhancement by mutation of Asp227. Biochemistry. 2005;44:10828–10838. doi: 10.1021/bi050438h. [DOI] [PubMed] [Google Scholar]
- 64.Herz J., Verhoefen M.K., Wachtveitl J. Critical role of Asp227 in the photocycle of proteorhodopsin. Biochemistry. 2012;51:5589–5600. doi: 10.1021/bi3003764. [DOI] [PubMed] [Google Scholar]
- 65.Ranaghan M.J., Schwall C.T., Birge R.R. Green proteorhodopsin reconstituted into nanoscale phospholipid bilayers (nanodiscs) as photoactive monomers. J. Am. Chem. Soc. 2011;133:18318–18327. doi: 10.1021/ja2070957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dioumaev A.K., Brown L.S., Lanyi J.K. Proton transfers in the photochemical reaction cycle of proteorhodopsin. Biochemistry. 2002;41:5348–5358. doi: 10.1021/bi025563x. [DOI] [PubMed] [Google Scholar]
- 67.Váró G., Needleman R., Lanyi J.K. Protein structural change at the cytoplasmic surface as the cause of cooperativity in the bacteriorhodopsin photocycle. Biophys. J. 1996;70:461–467. doi: 10.1016/S0006-3495(96)79589-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zimányi L., Cao Y., Lanyi J.K. Pathway of proton uptake in the bacteriorhodopsin photocycle. Biochemistry. 1993;32:7669–7678. doi: 10.1021/bi00081a010. [DOI] [PubMed] [Google Scholar]
- 69.Subramaniam S., Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 70.Lanyi J.K., Luecke H. Bacteriorhodopsin. Curr. Opin. Struct. Biol. 2001;11:415–419. doi: 10.1016/s0959-440x(00)00226-8. [DOI] [PubMed] [Google Scholar]
- 71.Hirai T., Subramaniam S. Protein conformational changes in the bacteriorhodopsin photocycle: comparison of findings from electron and X-ray crystallographic analyses. PLoS ONE. 2009;4:e5769. doi: 10.1371/journal.pone.0005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vonck J. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 2000;19:2152–2160. doi: 10.1093/emboj/19.10.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thorgeirsson T.E., Xiao W.Z., Shin Y.K. Transient channel-opening in bacteriorhodopsin: an EPR study. J. Mol. Biol. 1997;273:951–957. doi: 10.1006/jmbi.1997.1362. [DOI] [PubMed] [Google Scholar]
- 74.Bergen H., Furr H., Olson J. Synthesis of tri-deuterated tetra-deuterated and penta-deuterated forms of Vitamin-A. J. Labelled Comp. Radiopharm. 1988;25:11–21. [Google Scholar]
- 75.Magoulas G.E., Bariamis S.E., Maroulis G. Syntheses, antiproliferative activity and theoretical characterization of acitretin-type retinoids with changes in the lipophilic part. Eur. J. Med. Chem. 2011;46:721–737. doi: 10.1016/j.ejmech.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 76.McLean N., Gansmuller A., Brown R. Syntheses of C-13(2)-labelled 11Z-retinals. Tetrahedron. 2011;67:8404–8410. [Google Scholar]
- 77.Sadhukhan S., Han Y., Tochtrop G.P. Using isotopic tools to dissect and quantitate parallel metabolic pathways. J. Am. Chem. Soc. 2010;132:6309–6311. doi: 10.1021/ja100399m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanumihardjo S. Synthesis of 10,11,14,15-C-13(4)- and 14,15-C-13(2)-retinyl acetate. J. Labelled Comp. Radiopharm. 2001;44:365–372. [Google Scholar]
- 79.Wada A., Ieki Y., Ito M. Palladium-catalyzed coupling reaction of an enol nonaflate with (vinyl)tributylstannanes and acetylenes: a highly stereoselective synthesis of 8,18-C-13(2)-labeled retinal. Synthesis. 2005:1581–1588. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.