

Abstract
The iron-transport glycoprotein transferrin has recently been shown to serve as a potent inhibitor of Aβ self-association. Although this novel, to our knowledge, inhibitory function of transferrin is of potential therapeutic interest for the treatment of Alzheimer’s disease, the underlying mechanism is still not fully understood. Although it has been shown that the Fe(III) sequestration by transferrin reduces oxidative damage and Aβ aggregation, it is not clear whether transferrin is also able to inhibit Aβ self-association through direct binding of Aβ. Here, using saturation transfer and off-resonance relaxation NMR spectroscopy, we show that transferrin inhibits Aβ aggregation also by preferentially binding Aβ oligomers and outcompeting Aβ monomers that would otherwise cause the growth of the Aβ oligomers into larger assemblies. This inhibitory mechanism is different from the iron-sequestration model, but it is qualitatively similar to a mechanism previously proposed for the inhibition of Aβ self-association by another plasma and cerebrospinal fluid protein, i.e., human serum albumin. These results suggest that Aβ monomer competition through direct Aβ oligomer binding might be a general strategy adopted by proteins in plasma and cerebrospinal fluid to prevent Aβ aggregation.
Abbreviations used: AD, Alzheimer’s disease; CNS, central nervous system; CSF, cerebrospinal fluid; HSA, human serum albumin; ICP, inductively coupled plasma; ORR, off-resonance relaxation; SL, spin-lock; STD, saturation transfer difference; STR, saturation transfer reference; Tf, transferrin; WG, watergate water-suppression NMR technique
Introduction
Several plasma and CSF proteins have recently emerged as potent inhibitors of Aβ aggregation (1–11). The inhibition of Aβ self-association by plasma proteins is exploited in therapeutic approaches of AD that rely on the peripheral sink hypothesis (1,10,12). According to this hypothesis, agents that bind plasma Aβ without crossing the blood-brain barrier function as a sink for the brain Aβ and can therefore be effectively exploited to sequester the latter in peripheral tissues, minimizing the accumulation of Aβ in the CNS (1,10,12). The inhibition of Aβ self-association by CSF proteins is of potential therapeutic interest also because low levels of these proteins have been linked to increased odds of cognitive impairment (2,10,11,13).
Tf is one of the plasma and CSF proteins with significant Aβ self-association inhibitory potency (7,11). Tf is a 78 kDa glycoprotein with a concentration of 38 and 0.17 μM in plasma and CSF, respectively. Tf transports iron by binding Fe(III) with a 1:2 Tf:Fe(III) stoichiometry and high affinity (Kd ∼10−21 M) (11). Fe(III) in its free form is known to induce the generation of oxygen radicals, which in turn stimulate the production of toxic Aβ aggregates (14). In addition, the toxicity of free iron has been shown to arise also from the iron-induced stabilization of toxic, soluble intermediate oligomers of Aβ, which delays the formation of allegedly less toxic well-ordered aggregates (15). By binding Fe(III), Tf sequesters the ferric ions in a relatively nonreactive and inert state, which reduces both the oxidative damage and the stabilization of toxic Aβ oligomers caused by free iron. The Fe(III) sequestration by Tf thus provides a viable mechanism to explain the Aβ self-association inhibitory function observed for Tf (7,11). However, other plasma and CSF proteins have been shown to inhibit Aβ aggregation through a different and more direct mechanism, which does not necessarily rely on metal sequestration (1,4,5). For instance, HSA inhibits Aβ self-association by directly binding Aβ oligomers and outcompeting Aβ monomers that would otherwise cause the growth of the Aβ oligomers into larger assemblies (1,4,5). In the case of Tf, it is currently not known whether this HSA-like direct inhibition mechanism applies or iron sequestration is the only mechanism for inhibiting Aβ aggregation through Tf.
Although iron sequestration by Tf is an effective mechanism to inhibit Aβ self-association, here we hypothesize that additional inhibitory mechanisms should also be taken into consideration when explaining the effect of Tf on Aβ oligomerization. To test this hypothesis, we have investigated how Tf affects the self-association of the Aβ (12–28) peptide, which includes the central hydrophobic core of Aβ and is known to serve as a reproducible model system for soluble Aβ oligomers (1,4,5,16,17). Using a combination of NMR techniques, which include STD (18) and ORR (19–22), we show that, similarly to HSA, Tf preferentially binds Aβ (12–28) oligomers and competes with their interaction with monomeric Aβ (12–28). These results suggest that iron sequestration (23–26) is not the only mechanism through which Tf inhibits Aβ aggregation and provide a new framework to interpret the role of Tf in AD.
Materials and Methods
Peptide sample preparation
Aβ (12–28) (+H3N-V12HHQKLVFFAEDVGSNK28-COO−) was purchased from EZBiolab, Westfield, IN, as a lyophilized powder with a minimum purity of 96.5%. The purity and the molecular weights were checked through HPLC and mass spectrometry by EZBiolab. Each experimental trial that involved a comparison between Aβ (12–28) solutions was based on the same batch of peptides to avoid any bias due to potential sample variability. For the NMR samples, a 50 mM deuterated (d3) sodium acetate buffer was prepared from a mixture of deuterated acetic acid (d4) and sodium hydroxide used to adjust the pH to 4.7. 10% D2O (Cambridge Isotopes, Andover, MA) was added to the acetate buffer for NMR locking purposes. The buffer was then subjected to a syringe-driven filtration unit with a pore size of 0.22 μm from Millipore (Etobicoke, ON). One-dimensional (1D) 1H-WG NMR spectra of the buffer was acquired before its use for sample preparation to confirm its purity. Lyophilized Aβ (12–28) was dissolved in the original vials to minimize peptide losses and left to stand on ice for 10–20 min until the peptide was fully dissolved. The uniformly dissolved sample of Aβ (12–28) in 50 mM acetate-d3, 10% D2O at pH 4.7 was purified to its largely monomeric form through filtration. Ultrafree 30 kDa filter units were used to ensure minimal binding of the peptide. An Allegra 25R Centrifuge was used and run at 4000 rpm and 4°C for 5 min. Before sample filtration, potential residual glycerol from the filter units was removed from the filter through centrifugal washing with 50 mM deuterated (d3) sodium acetate buffer for 5–7 cycles. Aβ (12–28) samples were spun repeatedly with stabilization every 5 min in ice to minimize the heating of the sample. After filtration, aggregation was reintroduced in a controlled manner through the addition of NaCl. The salt was added using aliquots from concentrated stock solutions (1 and 5 M NaCl), which were added to the filtered Aβ (12–28) sample. All volumes added to the peptide samples were in the 1–15 μL range, resulting in negligible dilution factors. The Aβ (1–42) peptide samples were prepared as previously described (1,4).
Protein sample preparation
Tf was purchased from Sigma-Aldrich, Oakville, ON, as lyophilized powder of human serum Tf with >98% purity and 20–40% saturation with ferric irons, similar to human serum. Human serum Tf is here referred as Tf. 500 μM samples of Tf were prepared by dissolving a weighted amount of the powder in 50 mM deuterated (d3) sodium acetate buffer, pH 4.7 with 90:10 doubly distilled H2O:D2O. After dissolving the protein in the acetate buffer, the solution was kept on ice and/or refrigerated at +4°C. Typically, the total protein solution volume added to the peptide samples was within the range of 1–8 μL, again resulting in negligible dilution factors.
NMR spectroscopy
All NMR data were collected at 293 K using either a Bruker Avance 600 MHz NMR spectrometer with a 5 mm inverse triple resonance multinuclear TBI-Z probe or a Bruker Avance 700 MHz NMR with a TCI cryoProbe. All 1D 1H NMR spectra acquired were recorded with 128 scans, 32 dummy scans, and a spectral width of 8389.26 Hz sampled with 4096 complex points and a repetition delay of 1 s. The WG by gradient-tailored excitation with a binomial 3-9-19 pulse train was applied to all pulse sequences used throughout the analysis (21). The self-association of Aβ (1–42) was monitored though signal losses over time in a 1D 1H-WG NMR experiment incorporating a 30 ms long SL pulse with 2.6 kHz strength before acquisition to suppress the residual protein signal. The 1D time-profiles for Aβ (1–42) were acquired at 600 MHz using 128 scans and 64 dummy scans.
STD experiments were used to monitor the effect of Tf on the aggregated Aβ (12–28) samples (1,5,18). To achieve on-resonance saturation, the carrier frequency of a Gaussian pulse train was applied to the valine methyl group within the hydrophobic core region of the Aβ peptide at ∼0.75 ppm. Off-resonance saturation for the STR experiment was obtained by saturating at 30 ppm, a region in which no specific 1H resonance occurs for the samples studied here. The saturation transfer difference spectra were obtained by subtracting on-resonance and off-resonance spectra through phase cycling (19,20). The Gaussian pulse train consisted of a sequence of 40 Gaussian-shaped pulses of 50 ms each, separated by a 1 ms interpulse delay, resulting in total saturation time of ∼2 s, which was preceded by a 100 ms interscan delay. The strength of each saturating Gaussian pulse was 110.23 Hz with a 1% truncation and 1000 digitization points. A 30 ms SL pulse with strength of 2.5 kHz was applied to suppress the residual protein signal in all STD and STR experiments. For the STD experiment 128 scans and 8 dummy scans were acquired, which were reduced to 32 scans and 32 dummy scans for the more sensitive STR spectra. For each titration point two STR and four STD replica spectra were collected. All STD and STR replicas were then added to increase the S/N ratios. The errors for the STD/STR ratios were evaluated using the individual STR and STD replicas and error propagation. Before data acquisition for each protein titration point, STD experiments were preformed on a filtered Aβ (12–28) sample as well as on an aggregated Aβ (12–28) sample without protein to confirm sample stability. All 1D spectra were processed using an exponential multiplication window function before zero filling.
The 1H off-resonance nonselective relaxation rates (R35.5°,ns) experiments (ORR) were acquired at 700 MHz with an off-resonance trapezoidal SL including two adiabatic pulses of 4 ms duration and applied at the angle of 35.5° to ensure optimal NOE/ROE compensation in the spin-diffusion limit (19,20). The total SL durations were 13 and 88 ms. The strengths of the off-resonance and TOCSY SLs were 8.25 and 10 kHz, respectively. The interscan delay between the end of the acquisition and the start of the first adiabatic pulse was 2 s. The spectral widths for both dimensions were 8389.26 Hz with 256 t1 and 1024 t2 complex points, respectively. For each experiment, 16 scans and 128 dummy scans were employed. All two-dimensional (2D) replica sets were coadded to increase the S/N ratios and processed with Xwinnmr, Bruker) using a 90° phase-shifted squared sine-bell window function for both dimensions before zero filling. The 2D crosspeak intensities were measured with Sparky 3.111 (22) by Gaussian line fitting using the fit peak heights. The standard deviation of the differences in fit heights between two copies was used to estimate the error of the individual spectra. The error of the sum was scaled up proportionally to the square root of the total number of scans. For all residues, the Hα,i-HN,i crosspeaks were used for the ORR data analysis, with the exception of G25 and of the N-terminal V12. G25 was omitted from the analysis due to the overlap of its degenerate Hα protons, whereas for V12, the Hα,12-HMe,12 crosspeak was used to probe Hα relaxation rates. The nonselective ORR rates in s−1 were computed from the experimental fit heights though the equation:
| (1) |
The nonselective off-resonance relaxation rates calculated based on the previous equation are within error of the ORR rates calculated using five different SL times. The measured rates and the related errors were then normalized with respect to the maximum observed rate.
Inductively coupled plasma experiments
The ICP technique was used to detect trace metals in the buffer solution used for the NMR experiments. The ICP data were acquired using a Perkin Elmer ELAN 6100 ICP-MS set at the detection wavelength of 259.94 nm for analysis of solution-based iron contamination following a pretreatment with 1% nitric acid.
Results
To probe how Tf inhibits Aβ self-association, we initially acquired 1D NMR spectra of Aβ (12–28) in the absence and presence of Tf (Fig. 1). The self-association of Aβ (12–28) in acetate buffer has been shown to mimic in a stable and reproducible manner the early Aβ soluble oligomers, which are populated only transiently by longer Aβ constructs (1,4,5,16,17). The Aβ (12–28) peptide in acetate buffer is subject to a steady-state exchange between monomeric and oligomeric Aβ (12–28) and this dynamic self-association equilibrium is effectively monitored through NMR line-broadening (5). For instance, narrow lines are observed in the absence of salt for a filtered Aβ (12–28) sample, which is primarily monomeric (Fig. 1 a). However, when 40 mM NaCl is added to promote self-association through electrostatic screening (27), a significant line-broadening is detected for the large majority of the Aβ (12–28) signals (Fig. 1 b). The salt-induced increase in linewidth is caused by the exchange between the monomeric and oligomeric forms of Aβ (12–28) (1,4,5).
Figure 1.
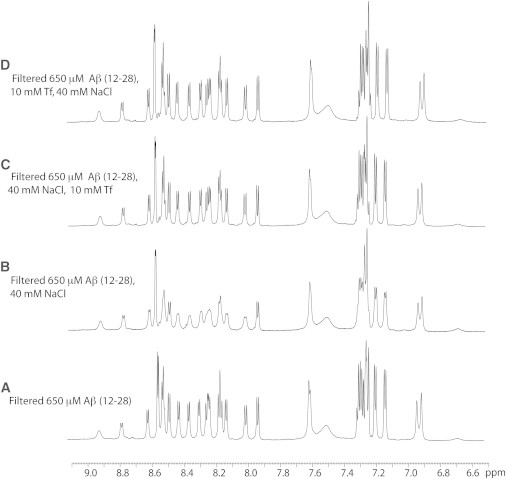
Monitoring the inhibition of Aβ (12–28) self-association by Tf using 1D NMR. 1D-WG-NMR spectra of Aβ (12–28) filtered with a 30 kDa C.O. filter before (A) and after (B) the addition of 40 mM NaCl. (C) Effect of the addition of 10 μM Tf, leading to narrow lines similar to the filtered sample A. (D) Effect of the addition of Tf before NaCl, resulting in line-narrowing comparable to C. Data were collected at 293 K and 700 MHz with a Bruker TCI cryoprobe. The horizontal axis reports the 1H chemical shift in ppm.
It is notable that no detectable self-association was observed for the filtered Aβ (12–28) sample under our experimental conditions and within the time frame of our experiments (Fig. 1 a), indicating that iron traces possibly present in our samples are not effective in promoting the aggregation of Aβ (12–28). This result is also supported by chelation experiments (vide infra) and ICP analyses, showing that the concentration of iron impurities within our samples is minimal and below 36 nM. The line-broadening observed for Aβ (12–28) in Fig. 1 b is therefore not expected to be caused by the residual free iron, but primarily by the monomer/oligomer exchange. Nevertheless, when Tf is added to the Aβ (12–28) sample in substoichiometric amounts, either before or after salt addition, a dramatic line-narrowing effect is observed (Fig. 1, c and d), which restores the Aβ (12–28) line widths to values comparable to those measured for the filtered Aβ (12–28) before the addition of salt (Fig. 1 a). No significant chemical shift changes are detected upon addition of Tf (Fig. 1).
A possible explanation for the linewidth reduction without significant chemical shift variations occurring upon Tf addition (Fig. 1) is that under our experimental conditions Tf binds selectively to the Aβ (12–28) oligomers rather than to the Aβ (12–28) monomer (Fig. 2). The resulting Aβ (12–28) oligomer/Tf complex inhibits the exchange of Aβ (12–28) polypeptide chains between the monomeric and oligomeric forms of Aβ (12–28), leading to the observed line-narrowing effect of Tf (Fig. 1, d and c; Fig. S3 in the Supporting Material). The model outlined in Fig. 2 explains also why dramatic line-narrowing is obtained even with minimal substoichiometric amounts of Tf (Fig. 1, c and d). However, additional experiments are required to further corroborate the proposed model. For instance, based on the data of Fig. 1 it is not possible to rule out the possibility that the absence of Aβ (12–28) monomer/Tf interactions is simply due to the saturation of Tf by Aβ (12–28) oligomers as opposed to the lack of any significant affinity of Tf for monomeric Aβ (12–28). To further probe this possibility, we designed a negative control experiment in which Tf was added to a sample with filtered and diluted Aβ (12–28) in the absence of salt to ensure that Aβ (12–28) is primarily monomeric without potentially Tf-saturating Aβ (12–28) oligomers (Fig. S1). No significant changes were observed for Aβ (12–28) in either line widths or chemical shifts upon the addition of Tf (Fig. S1), indicating that indeed under our experimental conditions Tf does not significantly bind monomeric Aβ (12–28) and confirming our initial hypothesis (Fig. 2).
Figure 2.
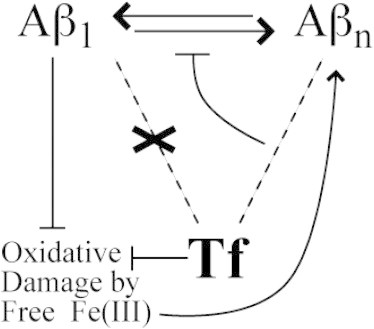
Schematic representation of the proposed direct and indirect mechanisms for the inhibition of Aβ self-association by Tf. Dashed lines indicate possible noncovalent interactions between Tf and the Aβ peptide. T-ended solid lines denote inhibition. Tf binds preferentially with Aβ oligomers, inhibiting the addition of further Aβ monomers to the preexisting Aβ oligomers (direct mechanism). Tf also binds Fe(III) inhibiting the Fe(III) inducible oxidative damage, which would otherwise promote the formation of Aβ aggregates, as indicated by the nonhorizontal arrow. This iron-mediated inhibitory mechanism is denoted here as the indirect mechanism or the iron-sequestration mechanism. Monomeric Aβ inhibits iron inducible oxidative damage (14).
The selectivity of Tf for Aβ (12–28) oligomeric species was further confirmed by a positive control experiment in which the fraction of NMR-undetectable Aβ (12–28) oligomers is effectively increased by increasing the NaCl concentration from 40 to 60 mM (Fig. S2 b). As expected, in the presence of 60 mM NaCl further line-broadening is observed (Fig. S2 b) relative to the spectra acquired with 40 mM NaCl (Fig. 1 b), confirming a higher population of Aβ (12–28) oligomers. Based on the model of Fig. 2, we would then expect that, upon addition of Tf, line-narrowing should be observed due to the inhibition of monomer/oligomer exchange, but without restoring the intensity of the NMR signal to the values measured before addition of salt, due to the sequestration of a large fraction of monomer into NMR-undetectable oligomers. Our data fully support this prediction (Fig. S2 c). When Tf is added to the Aβ (12–28) sample with 60 mM NaCl, line-narrowing is observed (Fig. S2 c), similar to Fig. 1. However, unlike Fig. 1, Tf is unable to restore the intensity of the NMR signal to the levels observed in the absence of salt (Fig. S2 a), despite the line-narrowing effect of Tf (Fig. S2 c). These observations further confirm the model of Fig. 2.
A specific implication of the proposed model (Fig. 2) is that Tf effectively screens the Aβ (12–28) oligomers from the monomeric form of Aβ (12–28). Based on the model of Fig. 2, we therefore expect that the NMR line-narrowing effect of Tf should be comparable to that obtained through physical removal of the Aβ (12–28) oligomers, for instance through filtration in the absence of salt. A stringent test of this prediction is provided by NMR experiments that quantitatively measure the linewidth change. One of these NMR methods is the 2D ORR experiment with a tilt angle of 35.5° (19,20). Fig. 3 a indicates that the residue-specific ORR rates of the filtered sample without NaCl and of the sample with both 40 mM NaCl and Tf are systematically reduced relative to those of the sample with 40 mM NaCl but without Tf. This result was expected based on the linewidth changes reported in Fig. 1. However, the quantitative ORR analysis also shows that the changes in relaxation caused by Tf addition are comparable and correlated with those caused by the physical elimination of the Aβ (12–28) oligomers through filtration (Fig. 3 b), confirming the oligomer screening function of Tf as predicted by the model shown in Fig. 2.
Figure 3.
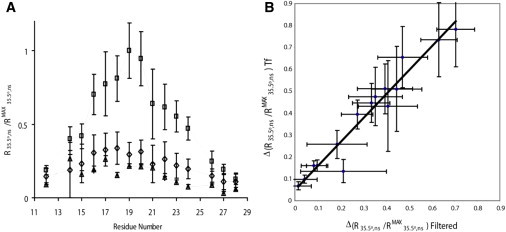
Tf addition and filtration have comparable effects on the ORR rates of Aβ (12–28). (A) Residue-specific normalized ORR rates for filtered 650 μM Aβ (12–28) (▴), 650 μM Aβ (12–28) with 40 mM NaCl (□) and 650 μM Aβ with both 40 mM NaCl and 10 μM Tf (♢). (B) Correlation between the changes in normalized ORR rates caused by Tf (vertical axis) and those caused by filtration (horizontal axis). The 2D ORR data were recorded at 293 K and at 700 MHz using a Bruker TCI cryoprobe.
Another NMR method to quantitatively monitor the Aβ (12–28) oligomer screening effect exerted by Tf is the STD NMR experiment (5,18). Fig. 4 reports the relative 1D STD changes observed for Aβ (12–28) as a function of the Tf concentration, showing a clear dose-response pattern with distinct binding and saturation regions. The plateau observed in the saturation region indicates that the interaction between the Aβ (12–28) oligomers and Tf is specific, whereas the binding region points to an IC50 ≤ 0.1 μM for the inhibition of Aβ (12–28) self-association by Tf under our experimental conditions (Fig. 4).
Figure 4.
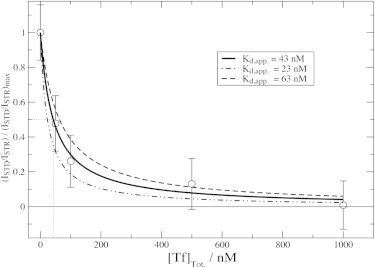
1D STD monitored isotherm for the Aβ (12–28) self-association inhibition by Tf. Tf was titrated into a solution of salt-aggregated 650 μM Aβ (12–28) and 1D STD and STR spectra were acquired at 293 K and at 600 MHz using a Bruker TBI-Z probe. Experimental data points are reported as circles, whereas the solid line was obtained through nonlinear fitting of the equation f + (1-f)∗Kd,app./(Kd,app. + [Tf]), where f is the fraction of Aβ (12–28) oligomers that are not Tf-binding competent, [Tf] is the concentration of free Tf and Kd,app. is an apparent average dissociation constant for the complexes between Tf and the other Aβ (12–28) oligomers (1). The dashed and dot-dashed lines were obtained using the same equation but by varying the Kd,app. value obtained through nonlinear curve fitting (i.e., 43 nM, assuming that the product of the total concentration of Tf-binding competent Aβ oligomers and of the ratio of the number of Tf molecules bound per Aβ oligomer versus the number of Aβ oligomers bound per Tf molecule is ≤ 5 nM. If this product is >5 nM, the Kd,app. values reported in the figure become just an upper limit to the best fit Kd,app. values). We also assumed that all Tf molecules bind to equivalent and independent binding sites (Scatchard-like model) (1). The latter assumption reflects the simplest model that fits the experimental data. The dotted line defines the IC50 for the inhibition of Aβ (12–28) self-association inhibition by Tf.
To further test the hypothesis that the inhibition of Aβ (12–28) self-association by Tf observed under our experimental conditions is not due to iron sequestration, we acquired STD spectra in the presence of a metal chelator, i.e., EDTA (Fig. 5). Fig. 5 shows that EDTA does not affect the STD magnitude of the salt aggregated Aβ (12–28) sample, indicating that under our experimental conditions iron sequestration is not a viable mechanism for inhibiting Aβ (12–28) self-association. However, when Tf is added, either before or after the addition of EDTA, the STD magnitude is decreased to values comparable to those observed for the filtered Aβ (12–28) sample in the absence of salt, which is mainly monomeric (Fig. 5). Overall, the results of Fig. 5 confirm that under our experimental conditions the mechanism for the inhibition of Aβ (12–28) self-association by Tf does not rely on iron sequestration (Fig. 2).
Figure 5.
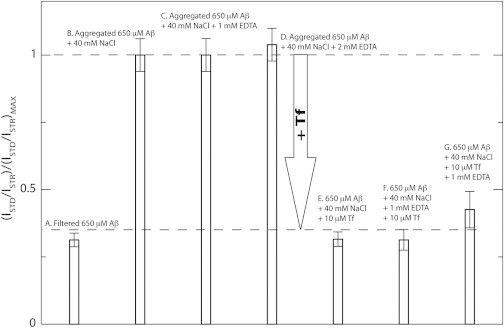
1D STD monitored chelation experiments show that Tf can inhibit Aβ (12–28) self-association even without iron sequestration. Normalized STD/STR ratios measured under multiple experimental conditions, as reported in the figure. Compounds were added in the order listed. All data were collected at 293 K and at 700 MHz using a Bruker TCI cryoprobe.
The model for the Aβ self-association inhibitory function of Tf proposed in Fig. 2 based on Aβ (12–28), also explains the effect of Tf on the longer Aβ (1–42) peptide (Fig. 6). Due to the transient nature of the STD-detectable Aβ (1–42) oligomers, the Aβ (1–42) self-association was monitored through losses in the 1D NMR signal of Aβ (1–42), which results from the formation of high molecular weight (MW) NMR-undetectable aggregates. Fig. 6 a shows the time profile for the 1D NMR signal of Aβ (1–42). In the absence of Tf, >60% of the original NMR signal is lost after 24 h (Fig. 6 a). Upon addition of 1 mM of the EDTA chelator only minimal and almost marginal changes in the Aβ (1–42) time profile are detected (Fig. 6 a), indicating that free iron sequestration is not an effective mechanism to inhibit Aβ (1–42) aggregation under our experimental conditions, likely due to the low iron content of our solutions. However, upon addition of substoichiometric amounts of Tf a significant enhancement of the 1D NMR signal is observed (Fig. 6 a), confirming that Tf is able to inhibit Aβ (1–42) self-association through a mechanism that does not necessarily rely on iron sequestration. This mechanism does not involve binding of Tf to Aβ (1–42) monomers, as no change in chemical shift for Aβ (1–42) was observed upon the Tf addition (Fig. 6, b and c). Therefore, we conclude that the Aβ (1–42) data presented in Fig. 6 fully support the model outlined in Fig. 2 and originally based on the Aβ (12–28) system.
Figure 6.
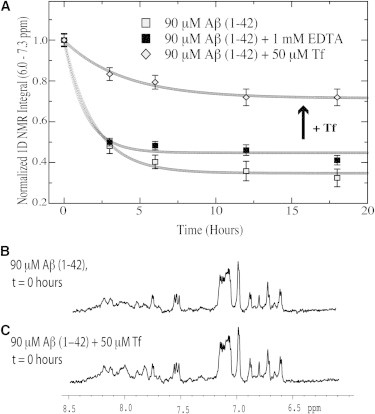
Effect of chelation and Tf addition on the aggregation profile of Aβ (1–42) as monitored by 1D NMR signal losses over time. (A) 1D NMR integral versus time for Aβ (1–42), Aβ (1–42) + 1 mM EDTA, and Aβ (1–42) + 50 μM Tf. Symbols for the actual experimental data are reported in the figure legend. The experimental data were fitted using the offset decaying exponential: a × e-bt + c, where t is in hours and the a–c parameters were obtained through nonlinear curve-fitting. The fitted curves are shown as a sequence of open circles. After ∼20 h most of the 1D NMR signal of Aβ (1–42) is lost due to the formation of high-MW NMR-undetectable aggregates. A similar pattern is observed in the presence of the EDTA chelator, however Tf considerably reduces the degree of signal loss. (B and C) 1D NMR spectra of Aβ (1–42) at the first sampled time point in the absence and presence of Tf. Tf does not cause detectable changes in the 1D NMR spectra, consistent with the absence of significant binding between Tf and monomeric Aβ (1–42). The horizontal axis reports the 1H chemical shift in ppm.
Discussion
Our data on the Aβ (12–28) peptide consistently indicate that Tf is able to inhibit Aβ self-association even without iron sequestration. This inhibitory effect is supported by multiple independent experiments, i.e., Tf-dependent changes in linewidth (Fig. 1 and Fig. S2), in ORR rates (Fig. 3), in STD/STR ratios (Fig. 4 and Fig. 5). Overall, our data are fully consistent with a model in which Tf selectively binds Aβ oligomers rather than monomers (Fig. 2). Upon binding to Tf, the Aβ oligomers are screened from the Aβ monomers hindering the growth of the Aβ oligomers into larger assemblies (Fig. 2). Under our experimental conditions, the affinity of Tf for the Aβ (12–28) oligomers appears to be in the sub-μM range, with IC50 values not greater than the plasma concentration of Tf (∼38 μM) (7). Previously, an IC50 value of 30 μM was reported for the inhibition of biotin-labeled Aβ (1–40) incorporation into immobilized Aβ (1–42) polymers by Tf (7). This IC50 value is in between the plasma and CSF concentrations of Tf and is significantly higher than the IC50 reported here (Fig. 4), but it is possible that variations in IC50 values are due to differences in experimental conditions and/or Aβ peptide used. Specifically, the data of Fig. 1 and Figs. 3–5 were acquired on the Aβ (12–28) peptide and different Aβ constructs may behave differently. However, the 1D intensity versus time profiles measured for Aβ (1–42) (Fig. 6) suggest that the model proposed for Aβ (12–28) may apply, at least at a qualitative level, also to longer Aβ peptides.
The model of Aβ self-association inhibition by Tf emerging from this study (Fig. 2) is qualitatively similar to the mechanism previously proposed for the inhibition of Aβ aggregation by another plasma protein, i.e., HSA (1,4,5). Furthermore, in the case of HSA a marked Aβ oligomer versus monomer selectivity was observed and, similar to Tf, the Aβ oligomer – HSA complexes compete with the addition of Aβ monomers to preexisting Aβ oligomers (1,4,5). It is therefore possible that the Aβ oligomer-binding model proposed in Fig. 2 represents a general Aβ self-association inhibitory strategy shared by multiple plasma proteins. In addition, other Aβ self-association inhibitory agents have been shown to act through a similar mechanism. For instance, inorganic nanoparticles such as quantum dots bind to Aβ oligomers or fibers and interfere with the conversion of monomers into fibers (28).
The Aβ oligomer-binding model is however markedly different from a previously proposed mechanism for the protective action of Tf against Aβ fibrillization (Fig. 2) (11). According to the previous model, Tf inhibits Aβ aggregation by sequestering free iron, which would otherwise induce the production of toxic Aβ aggregates either by promoting the formation of oxygen-free radicals and/or by delaying the formation of ordered and less toxic Aβ fibrils (Fig. 2) (11,15,23–26). The iron-sequestration mechanism still remains a viable strategy for the inhibition of Aβ fibrillization by Tf because Fe, together with Cu and Zn, is likely linked to pathological events in AD (29), here we show that other more direct mechanisms should be considered as well (Fig. 2). Furthermore, monomeric Aβ is known to inhibit neuronal cell death caused by Fe(II) and Fe(III) and therefore the absence of detectable interactions between Tf and Aβ monomers suggests that even in the presence of Tf the monomeric form of Aβ can still serve as an antioxidant against metal-induced oxidative damage (Fig. 2) (14). It is also possible that Tf enhances the concentration of monomeric Aβ by interfering with its binding to Aβ oligomers, thus indirectly potentiating the antioxidant effect of monomeric Aβ. Overall, we conclude that the mechanism proposed here for the inhibition of Aβ self-association by Tf, i.e., formation of Aβ oligomer:Tf complexes and consequent screening of Aβ oligomers from Aβ monomers (Fig. 2), is distinct from the previously elucidated iron-sequestration inhibitory models (Fig. 2) and should be taken into consideration when rationalizing the role of Tf in Aβ fibrillization.
Acknowledgments
The authors thank Tyler McNicholl, Elle Dunitz, Rajeevan Selvaratnam, Mostafa Elgamal, Mohammed Mazhab Jafari, Naeimeh Jafari, Soumita Das, Dr. Rahul Das, and Dr. Somenath Chowdhury for helpful discussions, along with the late Dr. Don Hughes for his help and guidance.
This work was possible through the financial support of the National Science and Engineering Research Council (NSERC).
Supporting Material
References
- 1.Milojevic J., Melacini G. Stoichiometry and affinity of the human serum albumin-Alzheimer’s Aβ peptide interactions. Biophys. J. 2011;100:183–192. doi: 10.1016/j.bpj.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Llewellyn D.J., Langa K.M., Lang I.A. Serum albumin concentration and cognitive impairment. Curr. Alzheimer Res. 2010;7:91–96. doi: 10.2174/156720510790274392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boada M., Ortiz P., Páez A. Amyloid-targeted therapeutics in Alzheimer’s disease: use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect. 2009;22:325–339. doi: 10.1358/dnp.2009.22.6.1395256. [DOI] [PubMed] [Google Scholar]
- 4.Milojevic J., Raditsis A., Melacini G. Human serum albumin inhibits Abeta fibrillization through a “monomer-competitor” mechanism. Biophys. J. 2009;97:2585–2594. doi: 10.1016/j.bpj.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milojevic J., Esposito V., Melacini G. Understanding the molecular basis for the inhibition of the Alzheimer’s Abeta-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy. J. Am. Chem. Soc. 2007;129:4282–4290. doi: 10.1021/ja067367+. [DOI] [PubMed] [Google Scholar]
- 6.Biere A.L., Ostaszewski B.L., Selkoe D.J. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 7.Bohrmann B., Tjernberg L., Nordstedt C. Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues. J. Biol. Chem. 1999;274:15990–15995. doi: 10.1074/jbc.274.23.15990. [DOI] [PubMed] [Google Scholar]
- 8.Ma B.Y., Nussinov R. Selective molecular recognition in amyloid growth and transmission and cross-species barriers. J. Mol. Biol. 2012;421:172–184. doi: 10.1016/j.jmb.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reyes Barcelo A.A., Gonzalez-Velasquez F.J., Moss M.A. Soluble aggregates of the amyloid-beta peptide are trapped by serum albumin to enhance amyloid-beta activation of endothelial cells. J Biol Eng. 2009;3:1–8. doi: 10.1186/1754-1611-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa M., Ortiz A.M., Jorquera J.I. Therapeutic albumin binding to remove amyloid-beta. J. Alzh. Disease. 2012;29:159–170. doi: 10.3233/JAD-2012-111139. [DOI] [PubMed] [Google Scholar]
- 11.Giunta S., Galeazzi R., Galeazzi L. Transferrin neutralization of amyloid beta 25–35 cytotoxicity. Clin. Chim. Acta. 2004;350:129–136. doi: 10.1016/j.cccn.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 12.Deane R., Wu Z.H., Zlokovic B.V. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35(11, Suppl 1):2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 13.Loeffler D.A., Connor J.R., LeWitt P.A. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J. Neurochem. 1995;65:710–724. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 14.Zou K., Gong J.S., Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu B., Moloney A., Crowther D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011;286:4248–4256. doi: 10.1074/jbc.M110.158980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarvet J., Damberg P., Graslund A. Reversible random coil to beta-sheet transition and the early stage of aggregation of the A beta(12–28) fragment from the alzheimer peptide. J. Am. Chem. Soc. 2000;122:4261–4268. [Google Scholar]
- 17.Mansfield S.L., Jayawickrama D.A., Larive C.K. Measurement of peptide aggregation with pulsed-field gradient nuclear magnetic resonance spectroscopy. Biochim. Biophys. Acta. 1998;1382:257–265. doi: 10.1016/s0167-4838(97)00162-3. [DOI] [PubMed] [Google Scholar]
- 18.Huang H., Milojevic J., Melacini G. Analysis and optimization of saturation transfer difference NMR experiments designed to map early self-association events in amyloidogenic peptides. J. Phys. Chem. B. 2008;112:5795–5802. doi: 10.1021/jp7118718. [DOI] [PubMed] [Google Scholar]
- 19.Esposito V., Das R., Melacini G. Mapping polypeptide self-recognition through (1)H off-resonance relaxation. J. Am. Chem. Soc. 2005;127:9358–9359. doi: 10.1021/ja051714i. [DOI] [PubMed] [Google Scholar]
- 20.Milojevic J., Esposito V., Melacini G. Analysis and parametric optimization of 1H off-resonance relaxation NMR experiments designed to map polypeptide self-recognition and other noncovalent interactions. J. Phys. Chem. B. 2006;110:20664–20670. doi: 10.1021/jp063194z. [DOI] [PubMed] [Google Scholar]
- 21.Piotto M., Saudek V., Sklenár V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 22.Sparky 3.111 Goddard, T. D. and Kneller, D. G., SPARKY 3, University of California, San Francisco.
- 23.Casadesus G., Smith M.A., Perry G. Alzheimer disease: evidence for a central pathogenic role of iron-mediated reactive oxygen species. J. Alzheimers Dis. 2004;6:165–169. doi: 10.3233/jad-2004-6208. [DOI] [PubMed] [Google Scholar]
- 24.Monji A., Utsumi H., Tashiro N. Amyloid-beta-protein (A beta) (25–35)-associated free radical generation is strongly influenced by the aggregational state of the peptides. Life Sci. 2002;70:833–841. doi: 10.1016/s0024-3205(01)01451-5. [DOI] [PubMed] [Google Scholar]
- 25.Monji A., Utsumi H., Tashiro N. The relationship between the aggregational state of the amyloid-beta peptides and free radical generation by the peptides. J. Neurochem. 2001;77:1425–1432. doi: 10.1046/j.1471-4159.2001.00392.x. [DOI] [PubMed] [Google Scholar]
- 26.Olanow C.W. A radical hypothesis for neurodegeneration. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 27.Vivekanandan S., Brender J.R., Ramamoorthy A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoo S.I., Yang M., Kotov N.A. Inhibition of amyloid peptide fibrillation by inorganic nanoparticles: functional similarities with proteins. Angew. Chem. Int. Ed. Engl. 2011;50:5110–5115. doi: 10.1002/anie.201007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeToma A.S., Salamekh S., Lim M.H. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012;41:608–621. doi: 10.1039/c1cs15112f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.