

Abstract
MicroRNAs (miRNAs) are small noncoding RNAs that play important roles in posttranscriptional regulation of gene expression. Mature miRNAs associate with the RNA interference silencing complex to repress mRNA translation and/or degrade mRNA transcripts. Mass spectrometry-based proteomics has enabled identification of several core components of the canonical miRNA processing pathway and their posttranslational modifications which are pivotal in miRNA regulatory mechanisms. The use of quantitative proteomic strategies has also emerged as a key technique for experimental identification of miRNA targets by allowing direct determination of proteins whose levels are altered because of translational suppression. This review focuses on the role of proteomics and labeling strategies to understand miRNA biology.
Keywords: Cell biology, iTRAQ, miRNA, Multiple reaction monitoring, Noncoding RNA, SILAC
1 Background
MicroRNAs (miRNAs) are small noncoding RNAs of about 22 nucleotides that regulate various cellular functions such as differentiation [1], metabolism [2], senescence [3], autophagy [4], proliferation, and apoptosis [5]. miRNAs impose this layer of posttranscriptional gene regulation on a wide spectrum of biological processes ranging from gametogenesis [6], development [7], and tissue repair [8] to aging [9]. This widespread regulation reflected in the fact that up to ∼60% of the human protein-coding genes are potentially modulated by miRNAs [10]. Given the extensive involvement of miRNA in physiology, dysregulation of miRNA expression can be associated with cancer pathobiology including oncogenesis [11], proliferation [12], epithelial-mesenchymal transition [13], metastasis [14], aberrations in metabolism [15], and angiogenesis [16], among others. To date, the online miRNA repository database, miRBase (Release 18), has documented over 2000 human mature miRNA sequences and over 1200 mouse mature miRNA sequences along with miRNAs reported in other organisms [17]. With the availability of next generation sequencing technologies, this list will undoubtedly continue to expand further [18].
In general, genes encoding miRNAs are initially transcribed as primary miRNAs (pri-miRNAs) by RNA polymerase II, and then pri-miRNAs are processed to be precursor miRNAs (pre-miRNAs) by the microprocessor complex comprising of Drosha and DGCR8 [19]. The pre-miRNAs, hairpin-like structures of ∼60–80 nucleotides, are transported to the cytosol by exportin-5 [20], where they are processed by Dicer in association with TRBP to form ∼22-nucleotide double stranded mature miRNAs (Fig. 1) [21]. In parallel, the mature miRNAs are assembled with the RNA interference silencing complex (RISC) comprising of Argonaute and TNRC6 (GW182 protein) [22]. The assembly process also involves the chaperones Hsc70/Hsp90 and ATP [23]. The mature miRNA consists of two strands – a passenger strand which is degraded and a functional guide strand which pairs with its cognate mRNA (target) mostly in the 3′ untranslated region (3′UTR) by complementary-base pairing [24]. Binding of miRNAs to their cognate target mRNAs results in translation repression and/or mRNA destabilization by either decapping target mRNAs or by deadenylation followed by mRNA degradation [25].
Figure 1.
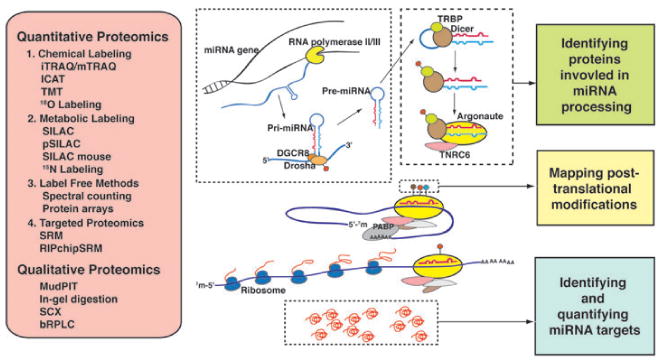
Proteomic strategies for studying the mechanism of miRNA processing and miRNA-mediated translation repression. Genes encoding miRNAs are transcribed as pri-miRNAs by RNA polymerase II and processed by the microprocessor complex consisting of Drosha and DGCR8. This complex cleaves pri-miRNAs into ∼60–80 nt hairpin structures, called pre-miRNAs, which are transported out of nucleus by exportin-5. In the cytoplasm, Dicer associates with TRBP and excises the loop portion of pre-miRNAs generating double stranded mature miRNAs. These are loaded onto RNA interference silencing complex (RISC) consisting of Argonaute and TNRC6. Translation inhibition is brought about by base-pairing of guide strand miRNA to the 3′UTR of mRNA and interfering with translation initiation. Quantitative proteomics analyses encompass different categories of strategies including chemical labeling, metabolic labeling, label-free methods and targeted proteomics. Among them, iTRAQ and SILAC are the most frequently used. Along with various techniques in qualitative proteomics, these methods can enable not only characterization of the miRNA processing pathway components and their posttranslational modifications but also discovery of miRNA targets.
The vast majority of studies on miRNA biology thus far have primarily focused on two aspects: elucidating mechanisms of miRNA-mediated posttranscriptional gene regulation and identifying targets of miRNAs. This has been made possible by the use of established methodologies such as gene expression microarrays, next generation sequencing, computational prediction of miRNA targets, and mass spectrometry-based proteomic approaches [26]. Each of these high-throughput technologies has its own advantages, thus complementing one another. In this article, we will highlight the importance and advantages of proteomics to decipher the role of miRNA in regulating biological processes.
2 Approaches for computational analysis and proteomics
2.1 Computational approaches to predict miRNA targets
A large majority of reports describing identification of miRNA targets are based on computational approaches or detection of altered mRNA levels. However, the mechanism of target recognition is still not fully understood and as a result the algorithms that are employed for target gene prediction are not accurate and often overpredict miRNA targets. Numerous web-based miRNA target prediction tools such as TargetScan (www.targetscan.org), miRanda (www.microrna.org), TarBase (diana.cslab.ece.ntua.gr), PicTar (pictar.mdcberlin.de), RNA22 (cbcsrv.watson.ibm.com/rna22.html), mirWIP (ambroslab.org), PITA (genie.weizmann.ac.il/pubs/mir07/mir07_dyn_data.html), and miRDB (mirdb.org) predict miRNA targets based on selection criteria such as sequence pairing (which involves perfect or nearly perfect complementarity between nucleotides 2 and 8 at the 5′ end of miRNA (seed sequence) to the mRNA target site), secondary structure modeling, free energy calculation, and comparative genomic sequence conservation. However, the field is greatly expanding with significant improvements in technologies resulting in the generation of a vast amount of data. Furthermore, most prediction tools overlook the potentiality of miRNA binding in 5′UTR region as well as the coding sequence, even though there is substantial evidence provided by several groups [27–29]. Thus, it should be noted that miRNA target prediction by computational methods alone is not sufficient and it becomes essential to experimentally identify and validate targets of miRNAs.
2.2 Importance of proteomics in understanding miRNA biology
One of the mechanisms by which miRNA regulates gene expression is by repressing translation without mRNA degradation [30,31]. Determining the mRNA expression levels is not ideal for identifying targets as mRNA levels do not necessarily correlate with the levels of protein expression [32–34]. In addition, the discordance between half-lives of mRNAs and proteins adds another layer of complexity in drawing conclusions about the proteome from analysis of the transcriptome. Furthermore, a recent study revealed that the efficiency of miRNA-mediated gene silencing can be downregulated by poly (ADP-ribosyl)ation of Argonaute/RISC, suggesting that the mere presence of miRNA does not necessarily reduce the protein production [35]. Therefore, the abundance of mRNA/miRNA species does not serve as the best surrogate marker for biological activity of these proteins. This problem can be alleviated by the use of mass-spectrometry-based proteomic approaches that enable studying the protein complement of cells more directly.
2.3 Mass spectrometry as a discovery tool
The dynamic nature and complexity of the proteome present enormous technological challenges. Mass spectrometry is a powerful analytical technology that enables elucidation of protein sequence as well as abundance. Recent advances in mass spectrometry have significantly enhanced the throughput of protein identification and quantification by improved sensitivity, high resolving power, reproducibility, and the dynamic range of proteomic analyses. A typical proteomic experiment involves isolation and extraction of proteins from cells or tissues, protease digestion to generate peptides generally followed by sample fractionation to reduce proteome complexity. The fractionated samples are then introduced into a mass spectrometer where the peptides are ionized and their masses measured. The peptide ions are then picked for fragmentation in a data-dependent fashion leading to generation of MS/MS spectra. Improved separation methods are essential for increasing the comprehensiveness and throughput of proteome analyses and to address the complexity of proteomes.
2.4 Proteomic strategies in miRNA research
Over the past decade, various strategies have been developed and optimized to characterize the proteome. A large majority of proteomic experiments in miRNA research have employed mass spectrometry-based quantitative proteomic approaches. Quantitative proteomics has emerged as a promising tool to identify proteins differentially expressed in various biological processes [36]. To enable protein quantitation, two main strategies are currently employed by the community in combination with mass spectrometry (Fig. 1). These are referred as “label” and “label free” strategies. Labeling techniques utilize isotopic labels as a reference for either relative or absolute quantitation. These labels can be introduced in vivo by labeling methods such as SILAC [37] or by in vitro labeling strategies such as iTRAQ [38], 18O labeling [39] or TMT tags [40], among others. Apart from labeling-based methods, a semiquantitative strategy, less frequently used, is based on differential spot analysis by two-dimensional electrophoresis [41–43].
In recent years, there has been significant progress in the development and application of technologies for targeted analysis of peptides in complex protein samples. Selected reaction monitoring (SRM) or multiple reaction monitoring is a robust tandem mass spectrometry method employed to monitor these protein-specific peptides and thus provide quantitative measurements [44,45]. SRM assays allow identification and quantitation of peptides with very low limits of detection, high reproducibility, specificity, and sensitivity. In contrast to discovery-type proteomic experiments, SRM assays allow one to perform repeat measurements with high-throughput and sensitivity. Several groups have employed labeling strategies to identify targets of miRNAs. However, to date there are only two studies (both by the same group) where SRM-based targeted studies were performed to identify and validate predicted targets in C. elegans [46, 47].
Non-MS-based proteomic approaches such as reverse phase protein microarray have also been employed to study various biological processes. These include profiling immune response to infection, protein–protein interactions, protein–nucleic acid interactions and signaling pathways and have also been utilized to understand the mechanism of miRNA regulation. In a recent study, Iliopoulos et al. carried out microRNA profiling and reverse-phase protein microarray analysis on the same patient-derived osteoarthritic cartilage to identify genes involved in osteoarthritic pathogenesis. Integrating the data from both these approaches together with the use of miRNA prediction algorithms, they created an interactome network and uniquely identified 17 miRNA-gene target pairs implicated in pathogenesis of osteoarthritis [48].
3 Application of proteomics in miRNA research
The power of MS-based proteomics has been exploited to identify several critical components of the miRNA biogenesis pathway and their posttranslational modifications. Additionally, quantitative proteomic strategies have facilitated quantifying proteomic changes secondary to the perturbation of certain miRNAs. This confers a powerful high throughput platform to predict miRNA targets that are not fully appreciated by bioinformatics tools. These aspects will be covered in the following sections.
3.1 Identification of key components in miRNA biogenesis
MS-based proteomics has enabled identification of several associating factors such as DGCR8, TRBP, and TNRC6 that are all indispensable for generating miRNAs. Gregory et al. immunoprecipitated Drosha in HEK293T cells, identifying DGCR8 as the co-eluate in the complex that decisively retained the pri-miRNA processing activity [19]. Analyzing Dicer affinity eluates through mass spectrometry, Chendrimada et al. identified TRBP, a protein necessary for the functional interaction between Dicer and Argonaute. They observed diminished generation of mature miRNA upon TRBP knockdown [21]. Using shotgun proteomic analysis, Duchaine et al. identified Dicer (DCR-1)-associated proteins in C. elegans using immunoaffinity purification approach. They identified a high confidence set of 20 interactors, many of which were previously not known to interact with DCR-1. Interaction between Dicer and the translation initiation factor, EIF2C2, has partially unveiled the mechanism of miRNA-mediated translation suppression [49]. Association between Argonaute and TNRC6 was initially revealed through multidimensional protein identification technology by Liu et al. and supported by several other groups' finding [50–53]. Identification of these core factors of miRNA processing pathways has initiated further functional and quantitative studies that will be discussed below. Wang et al. employed a quantitative proteomic approach to compare functions across different Argonaute proteins in mouse skin and human melanoma cells [54]. They employed spectral counting, a label-free quantitative method, to measure changes in the abundance of Ago1–4 in mouse skin cells. Their study showed that the abundance of Ago3/4 is negligible and the ratio between Ago1 and Ago2 is close to their associated miRNAs, an implication that miRNAs are loaded randomly onto Ago1 and Ago2.
3.2 Mapping posttranslational modifications on key components of miRNA biogenesis
The miRNA processing pathway is now known to be regulated by posttranslational modifications on key components such as Drosha, TRBP, and Argonaute. Importantly, these modifications such as phosphorylation and hydroxylation result in alteration of their activity that, in turn, influences miRNA biogenesis. To delineate the regulation of pri-miRNA processing, Tang et al. examined the mechanism by which Drosha, one of the key enzymes in the miRNA processing machinery, localizes to the nucleus. Mass spectrometric analysis revealed phosphorylation at Ser-300 or Ser-302 and with additional functional studies they proved that phosphorylation at either site is critical for nuclear localization [55]. Serine phosphorylation is also important for functions of TRBP and Argonaute. Paroo et al. have shown that Dicer complexes with phosphorylated TRBP to generate mature miRNAs in HeLa cells. They identified four serine residues (Ser-142, Ser-152, Ser-283, and Ser-286) phosphorylated by MAPK/Erk and unequivocally demonstrated that phosphorylated TRBP confers stability to the Dicer-TRBP complex [56]. Additionally, this study also revealed phosphorylation of Argonaute by MAPK-activated protein kinase 2. A study by Zeng et al. showed that phosphorylation of Ago-2 at Ser-387 mediates localization of Argonaute to processing bodies which are the site for the process of miRNA-mediated gene silencing [57]. Further, studies have shown that apart from phosphorylation at key residues, other modifications also play important roles in the functioning of Argonaute. Qi et al. studied the interactome of Argonaute with MS-based proteomics and have shown that it physically interacts with the subunits of prolyl-4-hydroxylase. They further demonstrated that hydroxylation at Pro-700 of Ago-2 is essential for its own stability and for miRNA-mediated gene silencing [58]. Overall, the regulation of the miRNA processing pathway by posttranslational modifications has been relatively less investigated and additional studies are required to reveal the intricate mechanism of miRNA processing.
3.3 Proteome analysis for suppression of miRNA processing pathways
Perturbation of miRNA processing pathway such as Dicer and Argonaute results in phenotypic changes; thus functional studies regarding these components are of prime interest. Dicer is one of the key enzymes of the miRNA processing machinery and is also known to play important roles in cell differentiation and apoptosis. Knockout of Dicer gene has been reported to cause various defects [59–62]. To investigate the functions of Dicer in vivo, our group generated SILAC mice to study the effects of inducible deletion of Dicer [63]. Our study showed abnormal lipid accumulation in small intestine. Evaluation of the proteomic changes in the small intestine revealed a critical role of Dicer in lipid transport [63]. In an episomal shRNA expression system, Drosha knockdown experiments in MCF-7 and HCT116 showed size-biased proteome modifications [42]. Similar quantitative proteomics strategies need to be employed in the future to increase our depth of understanding the components involved in the canonical and noncanonical miRNA processing pathways.
3.4 Identification of miRNA targets using quantitative proteomics
A complete delineation of the miRNA targeting mechanism entails concurrent measurement of mRNAs and their protein products. In the past few years, several groups have made significant efforts to integrate high-throughput techniques to enable identification of bona fide miRNA targets (Table 1). Most studies thus far have employed overexpression of the miRNAs of interest in a transient or stable fashion to identify potential targets. Other studies have employed anti-miRNA oligonucleotides and miRNA knockout mouse model. SILAC-based strategy has been widely used in most studies to identify miRNA targets [64–67]. Baek et al. measured the effects of addition of miRNAs such as miR-124, a brain specific miRNA, miR-1, and miR-181 on the expression levels of proteins in HeLa cells. Based on the identification of the repressed proteins, this study revealed that these miRNAs recognize seed sequences located within the 3′ UTRs. In addition, they also studied the effects of miRNA knockout to identify endogenous targets since ectopic addition of miRNAs would only provide insights into miRNA target recognition and potential targets [64]. Our group has also adopted SILAC-based strategy and knocked down expression of endogenous miR-21 to identify its potential targets [68]. This miRNA is known to play important roles in tumorigenesis. To enable identification and quantitation of proteins synthesis on a global scale, Ebner et al. devised a new strategy which is a variation of SILAC technology and designated it pulsed SILAC (pSILAC). pSILAC measures the differences in the amount of protein synthesized over a period of time depending upon the incorporation rate of heavy amino acids [66]. They applied this strategy to study the protein regulatory mechanisms by miRNAs. Kaller et al. employed pSILAC and microarray analysis to study the effects of miR-34a, an important mediator of p53-mediated tumor suppressor activities on mRNA and proteome expression [69]. Employing iTRAQ-based quantitative strategy coupled with computational prediction, Taguchi et al., identified hypoxia inducible factor-1α as a novel target of miR17–92 cluster in lung cancer cells. Overexpression of miR17–92 cluster has been shown to play important roles in lung cancer and in B-cell lymphoma development. Using an SRM-based approach, Jovanovic et al. quantified and validated predicted targets of let-7 and miR-58 in C. elegans. Their results revealed ztf-7 as a bona fide target of let-7 which was also supported by another independent study [46].
Table 1. A list of publications that have employed quantitative proteomics to identify miRNA targets.
| No. | miRNA | Type of perturbation | Cell line | Transcriptomics analysis | Quantitative proteomics | Total protein | Reference |
|---|---|---|---|---|---|---|---|
| 1 | let-7a | Stable lentiviral vector overexpression | SGC-7901 (human gastric carcinoma cells) | NA | 2DE | 10 | Zhu et al. [73] |
| 2 | let-7b | LNA-anti-let-7b knockdown | HeLa (ovarian cancer) | Microarray | pSILAC | 2700 | Selbach et al. [65] |
| 3 | miR-1 | Transient miRNA mimics overexpression | HeLa (ovarian cancer) | NA | SILAC | 504 | Vinther et al. [74] |
| 4 | miR-1 | Transient miRNA mimics overexpression | HeLa (ovarian cancer) | Microarray | pSILAC | 4962a) | Selbach et al. [65] |
| 5 | miR-1 | Transient miRNA mimics overexpression | Hela (ovarian cancer) | Microarray | SILAC | 2312 | Baek et al. [64] |
| 6 | miR-15a/16-1 | Transient miRNA mimics overexpression | MEG-01 (Leukemia) | Microarray | 2DE | 27 | Calin et al. [75] |
| 7 | miR-16 | Transient miRNA mimics overexpression | HeLa (ovarian cancer) | Microarray | pSILAC | 4962a) | Selbach et al. [65] |
| 8 | miR-17-92 | Conditional episomal miRNA vector overexpression | SHEP-TR-miR-17–92 (neuroblastoma) | NA | SILAC | NA | Mestdagh et al. [76] |
| 9 | miR-17-92 | Stable pre-miR overexpression | BEAS2B (bronchial epithelial cells) | NA | iTRAQ | 479 | Taguchi et al. [77] |
| 10 | miR-17-92 cluster | LNA-anti- miR-19a, miR-20a and miR-92–1 knockdown | SBC-3 (small-cell lung cancer cells) | NA | 2DE | 112 up-regulated proteins | Kanzaki et al. [78] |
| 11 | miR-21 | Transient anti-miRNA oligo overexpression | MCF-7 (breast cancer) | NA | iTRAQ | 1151 | Yang et al. [67] |
| 12 | miR-21 | Transient anti-miRNA oligo overexpression | MCF-7 (breast cancer) | NA | 2D-DIGE | NA | Zhu et al. [79] |
| 13 | miR-21 | Transient miRNA mimics overexpression | U266 (human myeloma cell line) | NA | SILAC | 1498 | Xiong et al. [80] |
| 14 | miR-29a | Transient miRNA mimics overexpression | DLKP-A cells (Lung carcinoma) | NA | 2D-DIGE | 75 | Muniyappa et al. [81] |
| 15 | miR-29a | Transient miRNA overexpression | HEK293T (human embryonic kidney) | NA | SILAC | 5074 | Bargaje et al. [82] |
| 16 | miR-30a | Transient miRNA mimics overexpression | HeLa (ovarian cancer) | Microarray | pSILAC | 4962a) | Selbach et al. [65] |
| 17 | miR-30e-3p | Transient miRNA mimics overexpression | DLD1 (colorectal cancer) | NA | Spectral counting | NA | Schepeler et al. [71] |
| 18 | miR-34a | Transient pre-miR mimics overexpression | HepG2 (hepatocellular carcinoma) | NA | 2D-DIGE | 116b) | Cheng et al. [43] |
| 19 | miR-34a | Episomal pri-miRNA vector overexpression | SW480 (colon cancer) | Microarray | pSILAC | 1206 | Kaller et al. [69] |
| 20 | miR-34a | Transient miRNA mimics overexpression | IMR32 (neuroblastoma) | Microarray | iCAT | 1495 | Chen et al. [83] |
| 21 | miR-34a | Transient miRNA mimics overexpression | HEK293T (human embryonic kidney) | NA | SILAC | 5688 | Bargaje et al. [82] |
| 22 | miR-122a | Stable lentiviral vector overexpression | HepG2 (Hepatocellular carcinoma) | NA | 2DE | 10 | Diao et al. [84] |
| 23 | miR-124 | Transient miRNA mimics overexpression | Hela (ovarian cancer) | Microarray | SILAC | 2120 | Baek et al. [64] |
| 24 | miR-143 | Transient miRNA mimics overexpression | MiaPaCa-2 (pancreatic cancer) | Microarray | SILAC | 1202 | Yang et al. [68] |
| 25 | miR-155 | Transient miRNA mimics overexpression | HeLa (ovarian cancer) | Microarray | pSILAC | 3299 | Selbach et al. [65] |
| 26 | miR-155 | Transient miRNA episomal vector overexpression | HEK293T (human embryonic kidney) | NA | SILAC | 3148 | Lossner et al. [85] |
| 27 | miR-155 | Transient miRNA mimics overexpression | DHL16 (diffuse large B-cell lymphoma) | NA | pSILAC | 2103 | Huang et al. [86] |
| 28 | miR-181 | Transient miRNA mimics overexpression | Hela (ovarian cancer) | Microarray | SILAC | 1774 | Baek et al. [64] |
| 29 | miR-193b | Transient pre-miRNA vector overexpression | MCF-7 (breast cancer) | Microarray | iTRAQ | 743 | Leivonen et al. [87] |
| 30 | miR-210 | Transient miRNA mimics overexpression | Human umbilical venous endothelium cells | NA | 2DE | 28 | Fasanaro et al. [88] |
| 31 | miR-223 | Congenital miRNA knockout | miR-223−/− mouse bone marrow primary culture | Microarray | SILAC | 5019 | Baek et al. [64] |
| 32 | miR-372 | Transient miRNA overexpression | CL 1–0 (lung adenocarcinoma cells) | NA | 2D-DIGE | 19 | Lai et al. [89] |
| 33 | miR-373 | Transient pre-miRNA mimics overexpression | MCF-7 (breast cancer) | NA | SILAC | 3666 | Yan et al. [90] |
| 34 | miR-516a-3p | Stable episomal miRNA vector overexpression | 44As3-miR516a-3p (gastric cancer) | NA | iTRAQ | NA | Takei et al. [91] |
2-DE: two-dimensional gel electrophoresis; 2-D DIGE: two-dimensional fluorescence difference gel electrophoresis; NA: not available.
Total number of proteins from experiments of let-7b, miR-1, miR-16, miR-30a, and miR-155.
The number of differentially expressed proteins identified by 2D gels.
In addition to identifying individual miRNA targets, the application of quantitative proteomics also reveals several signaling networks of cancer biology. Schliekelman et al. use SILAC to determine which proteins are regulated by miR-200 family to cause the epithelial-mesenchymal transition in lung cancer. Many of them are associated with TGFβ-1, a finding corroborating with the current understanding [70]. To decode the intricate network of Wnt signaling in carcinogenesis, Schepeler et al. took a two-step approach [71]. They first adopted a conditional suppression system of Wnt signaling and identified miR-145, miR-126, miR-30e-3p, and miR-139–5p as critical regulators for their ectopic expression causing growth inhibition. Subsequently, they used spectral counting to characterize proteomic changes and luciferase assays to confirm that PIK3C2A is related to aberrant Wnt signaling. Another network-revealing study about cancer biology was conducted by Cheng et al., who transfected miR-34a in a hepatocellular carcinoma cell line. In the two-dimensional gel electrophoresis analysis coupled with mass spectrometry, half of the differentially regulated proteins were found to be miR-34a targets and shown belong to p53 signaling pathway [43].
Although these studies have led to identification of many novel miRNA candidate targets, a clear distinction between direct and indirect targets is lacking. Studies have shown that identification of direct miRNA targets is possible by co immunoprecipitation of miRISCs with the associated mRNAs, which is then coupled to microarrays (RIP-Chip) or mRNA sequencing (CLIP-Seq). However, these approaches do not measure the protein expression level of the direct targets. Recently, Jovanovic et al. developed a targeted, quantitative proteomic approach called RIP-chip-SRM to identify direct targets of miRNAs. This combinatorial approach involves RIP-chip analysis of miRISC complexes isolated from wild-type and miRNA deletion mutants followed by SRM analysis to determine the abundance of the protein products of the candidate transcripts [47]. One noteworthy caveat of the immunoprecipitation-based study was recently observed by Riley et al., who showed that the association between Argonaute proteins and miRNAs can happen after cell lysis [72]. This points out the possibility of false positive results from this type of study.
3.5 Future perspective
The use of quantitative proteomic strategies to characterize targets of miRNAs has opened new avenues to study miRNA biology. More and more targets of important miRNAs that relate to cancers are being identified and helping us unravel the intricacies of cancers and potentially even develop novel anticancer approaches. Meanwhile, our knowledge of post-translational modifications on components of the miRNA processing pathway and their associated factors is also expanding, paving the road for more functional analysis of miRNA-mediated gene regulation. Much of this progress has been enabled by quantitative proteomics, and we envision that this trend will continue in miRNA research and accelerate in the years to come.
Acknowledgments
S.M.P. is a recipient of independent research fellowship from the Council of Scientific and Industrial Research (CSIR), New Delhi, India. This work was supported by an NIH roadmap grant for Technology Centers of Networks and Pathways (U54RR020839) and a contract (HHSN268201000032C) from the National Heart, Lung and Blood Institute.
Abbreviations
- miRNA
microRNA
- pre-miRNA
precursor miRNA
- pri-miRNA
primary miRNA
- pSILAC
pulsed SILAC
- RISC
RNA interference silencing complex
- SRM
selected reaction monitoring
Footnotes
The authors have declared no conflict of interest.
References
- 1.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 2.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 3.Marasa BS, Srikantan S, Masuda K, Abdelmohsen K, et al. Increased MKK4 abundance with replicative senescence is linked to the joint reduction of multiple microRNAs. Sci Signal. 2009;2:ra69. doi: 10.1126/scisignal.2000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frankel LB, Wen J, Lees M, Hoyer-Hansen M, et al. MicroRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–4641. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennecke J, Hipfner DR, Stark A, Russell RB, et al. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi K, Chuva de Sousa Lopes SM, Kaneda M, Tang F, et al. MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS One. 2008;3:e1738. doi: 10.1371/journal.pone.0001738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 8.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 9.Liu N, Landreh M, Cao K, Abe M, et al. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature. 2012;482:519–523. doi: 10.1038/nature10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, Thomson JM, Hemann MT, Hernando-Monge E, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Ma L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012 doi: 10.1007/s10555-012-9368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khew-Goodall Y, Goodall GJ. Myc-modulated miR-9 makes more metastases. Nat Cell Biol. 2010;12:209–211. doi: 10.1038/ncb0310-209. [DOI] [PubMed] [Google Scholar]
- 15.Fang R, Xiao T, Fang Z, Sun Y, et al. miR-143 regulates cancer glycolysis via targeting hexokinase 2. J Biol Chem. 2012;287:23227–23235. doi: 10.1074/jbc.M112.373084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dews M, Homayouni A, Yu D, Murphy D, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voellenkle C, van Rooij J, Guffanti A, Brini E, et al. Deep-sequencing of endothelial cells exposed to hypoxia reveals the complexity of known and novel microRNAs. RNA. 2012;18:472–484. doi: 10.1261/rna.027615.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory RI, Yan KP, Amuthan G, Chendrimada T, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 20.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwasaki S, Kobayashi M, Yoda M, Sakaguchi Y, et al. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol Cell. 2010;39:292–299. doi: 10.1016/j.molcel.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 25.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 26.Thomson DW, Bracken CP, Goodall GJ. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39:6845–6853. doi: 10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnall-Levin M, Zhao Y, Perrimon N, Berger B. Conserved microRNA targeting in Drosophila is as widespread in coding regions as in 3′UTRs. Proc Natl Acad Sci USA. 2010;107:15751–15756. doi: 10.1073/pnas.1006172107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA. 2010;16:2493–2502. doi: 10.1261/rna.2384610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson PT, Wang WX, Mao G, Wilfred BR, et al. Specific sequence determinants of miR-15/107 microRNA gene group targets. Nucleic Acids Res. 2011;39:8163–8172. doi: 10.1093/nar/gkr532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, et al. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 31.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 32.Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 33.Chen G, Gharib TG, Huang CC, Taylor JM, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1:304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 34.Griffin TJ, Gygi SP, Ideker T, Rist B, et al. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol Cell Proteomics. 2002;1:323–333. doi: 10.1074/mcp.m200001-mcp200. [DOI] [PubMed] [Google Scholar]
- 35.Leung AK, Young AG, Bhutkar A, Zheng GX, et al. Genome-wide identification ofAgo2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol. 2011;18:237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 37.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 38.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Yao X, Freas A, Ramirez J, Demirev PA, et al. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 40.Dayon L, Hainard A, Licker V, Turck N, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 41.Ding J, Guo Y, Liu S, Yan Y, et al. Embryonic stem cells derived from somatic cloned and fertilized blastocysts are post-transcriptionally indistinguishable: a MicroRNA and protein profile comparison. Proteomics. 2009;9:2711–2721. doi: 10.1002/pmic.200800824. [DOI] [PubMed] [Google Scholar]
- 42.Peric D, Labarre J, Chevalier F, Rousselet G. Impairing the microRNA biogenesis pathway induces proteome modifications characterized by size bias and enrichment in antioxidant proteins. Proteomics. 2012;12:2295–2302. doi: 10.1002/pmic.201100461. [DOI] [PubMed] [Google Scholar]
- 43.Cheng J, Zhou L, Xie QF, Xie HY, et al. The impact of miR-34a on protein output in hepatocellular carcinoma HepG2 cells. Proteomics. 2010;10:1557–1572. doi: 10.1002/pmic.200900646. [DOI] [PubMed] [Google Scholar]
- 44.Sherwood CA, Eastham A, Lee LW, Risler J, et al. Rapid optimization of MRM-MS instrument parameters by subtle alteration of precursor and product m/z targets. J Proteome Res. 2009;8:3746–3751. doi: 10.1021/pr801122b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi T, Su D, Liu T, Tang K, et al. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics. 2012;12:1074–1092. doi: 10.1002/pmic.201100436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jovanovic M, Reiter L, Clark A, Weiss M, et al. RIPchipSRM, a new combinatorial large scale approach identifies a set of translationally regulated bantam/miR58 targets in C. elegans. Genome Res. 2012;22:1360–1371. doi: 10.1101/gr.133330.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jovanovic M, Reiter L, Picotti P, Lange V, et al. A quantitative targeted proteomics approach to validate predicted microRNA targets in C. elegans. Nat Methods. 2010;7:837–842. doi: 10.1038/nmeth.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iliopoulos D, Malizos KN, Oikonomou P, Tsezou A. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS One. 2008;3:e3740. doi: 10.1371/journal.pone.0003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duchaine TF, Wohlschlegel JA, Kennedy S, Bei Y, et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124:343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 50.Ding L, Spencer A, Morita K, Han M. The developmental timing regulator AIN-1 interacts with miRISCs and may target the argonaute protein ALG-1 to cytoplasmic P bodies in C. elegans. Mol Cell. 2005;19:437–447. doi: 10.1016/j.molcel.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Landthaler M, Gaidatzis D, Rothballer A, Chen PY, et al. Molecular characterization of human Argonaute-containing ribonucleo protein complexes and their bound target mRNAs. RNA. 2008;14:2580–2596. doi: 10.1261/rna.1351608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Rivas FV, Wohlschlegel J, Yates JR, 3rd, et al. A role for the P-body component GW182 in microRNA function. Nat Cell Biol. 2005;7:1261–1266. doi: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Ding L, Cheung TH, Dong MQ, et al. Systematic identification of C. elegans miRISC proteins, miRNAs, and mRNA targets by their interactions with GW182 proteins AIN-1 and AIN-2. Mol Cell. 2007;28:598–613. doi: 10.1016/j.molcel.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang D, Zhang Z, O'Loughlin E, Lee T, et al. Quantitative functions of Argonaute proteins in mammalian development. Genes Dev. 2012;26:693–704. doi: 10.1101/gad.182758.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang X, Zhang Y, Tucker L, Ramratnam B. Phosphorylation of the RNase III enzyme Drosha at Serine300 or Serine302 is required for its nuclear localization. Nucleic Acids Res. 2010;38:6610–6619. doi: 10.1093/nar/gkq547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139:112–122. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng Y, Sankala H, Zhang X, Graves PR. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem J. 2008;413:429–436. doi: 10.1042/BJ20080599. [DOI] [PubMed] [Google Scholar]
- 58.Qi HH, Ongusaha PP, Myllyharju J, Cheng D, et al. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature. 2008;455:421–424. doi: 10.1038/nature07186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin D, Shin JY, McManus MT, Ptacek LJ, et al. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 2009;66:843–857. doi: 10.1002/ana.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koralov SB, Muljo SA, Galler GR, Krek A, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132:860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 61.Sekine S, Ogawa R, Ito R, Hiraoka N, et al. Disruption of Dicer1 induces dysregulated fetal gene expression and promotes hepatocarcinogenesis. Gastroenterology. 2009;136:2304–2315. e1–e4. doi: 10.1053/j.gastro.2009.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang TC, Saharabuddhe NA, Kim MS, Getnet D, et al. Regulation of lipid metabolism by dicer revealed through SILAC mice. J Proteome Res. 2012;11:2193–2205. doi: 10.1021/pr2009884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baek D, Villen J, Shin C, Camargo FD, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 66.Ebner OA, Selbach M. Whole cell proteome regulation by microRNAs captured in a pulsed SILAC mass spectrometry approach. Methods Mol Biol. 2011;725:315–331. doi: 10.1007/978-1-61779-046-1_20. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y, Chaerkady R, Kandasamy K, Huang TC, et al. Identifying targets of miR-143 using a SILAC-based proteomic approach. Mol Biosyst. 2010;6:1873–1882. doi: 10.1039/c004401f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Y, Chaerkady R, Beer MA, Mendell JT, et al. Identification of miR-21 targets in breast cancer cells using a quantitative proteomic approach. Proteomics. 2009;9:1374–1384. doi: 10.1002/pmic.200800551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaller M, Liffers ST, Oeljeklaus S, Kuhlmann K, et al. Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.010462. M111.010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schliekelman MJ, Gibbons DL, Faca VM, Creighton CJ, et al. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res. 2011;71:7670–7682. doi: 10.1158/0008-5472.CAN-11-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schepeler T, Holm A, Halvey P, Nordentoft I, et al. Attenuation of the beta-catenin/TCF4 complex in colorectal cancer cells induces several growth-suppressive microRNAs that target cancer promoting genes. Oncogene. 2012;31:2750–2760. doi: 10.1038/onc.2011.453. [DOI] [PubMed] [Google Scholar]
- 72.Riley KJ, Yario TA, Steitz JA. Association of Argonaute proteins and microRNAs can occur after cell lysis. RNA. 2012;18:1581–1585. doi: 10.1261/rna.034934.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu Y, Xiao X, Dong L, Liu Z. Investigation and identification of let-7a related functional proteins in gastric carcinoma by proteomics. Anal Cell Pathol (Amst) 2012;35:285–295. doi: 10.3233/ACP-2012-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vinther J, Hedegaard MM, Gardner PP, Andersen JS, et al. Identification of miRNA targets with stable isotope labeling by amino acids in cell culture. Nucleic Acids Res. 2006;34:e107. doi: 10.1093/nar/gkl590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calin GA, Cimmino A, Fabbri M, Ferracin M, et al. MiR-15a and miR-16–1 cluster functions in human leukemia. Proc Natl Acad Sci USA. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mestdagh P, Bostrom AK, Impens F, Fredlund E, et al. The miR-17–92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol Cell. 2010;40:762–773. doi: 10.1016/j.molcel.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taguchi A, Yanagisawa K, Tanaka M, Cao K, et al. Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17–92 microRNA cluster. Cancer Res. 2008;68:5540–5545. doi: 10.1158/0008-5472.CAN-07-6460. [DOI] [PubMed] [Google Scholar]
- 78.Kanzaki H, Ito S, Hanafusa H, Jitsumori Y, et al. Identification of direct targets for the miR-17–92 cluster by proteomic analysis. Proteomics. 2011;11:3531–3539. doi: 10.1002/pmic.201000501. [DOI] [PubMed] [Google Scholar]
- 79.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 80.Xiong Q, Zhong Q, Zhang J, Yang M, et al. Identification of novel miR-21 target proteins in multiple myeloma cells by quantitative proteomics. J Proteome Res. 2012;11:2078–2090. doi: 10.1021/pr201079y. [DOI] [PubMed] [Google Scholar]
- 81.Muniyappa MK, Dowling P, Henry M, Meleady P, et al. MiRNA-29a regulates the expression of numerous proteins and reduces the invasiveness and proliferation of human carcinoma cell lines. Eur J Cancer. 2009;45:3104–3118. doi: 10.1016/j.ejca.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 82.Bargaje R, Gupta S, Sarkeshik A, Park R, et al. Identification of novel targets for miR-29a using miRNA proteomics. PLoS One. 2012;7:e43243. doi: 10.1371/journal.pone.0043243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen QR, Yu LR, Tsang P, Wei JS, et al. Systematic proteome analysis identifies transcription factor YY1 as a direct target of miR-34a. J Proteome Res. 2011;10:479–487. doi: 10.1021/pr1006697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diao S, Zhang JF, Wang H, He ML, et al. Proteomic identification of microRNA-122a target proteins in hepatocellular carcinoma. Proteomics. 2010;10:3723–3731. doi: 10.1002/pmic.201000050. [DOI] [PubMed] [Google Scholar]
- 85.Lossner C, Meier J, Warnken U, Rogers MA, et al. Quantitative proteomics identify novel miR-155 target proteins. PLoS One. 2011;6:e22146. doi: 10.1371/journal.pone.0022146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang X, Shen Y, Liu M, Bi C, et al. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am J Pathol. 2012;181:26–33. doi: 10.1016/j.ajpath.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leivonen SK, Rokka A, Ostling P, Kohonen P, et al. Identification of miR-193b targets in breast cancer cells and systems biological analysis of their functional impact. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.005322. M110.005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fasanaro P, Greco S, Lorenzi M, Pescatori M, et al. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem. 2009;284:35134–35143. doi: 10.1074/jbc.M109.052779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lai JH, She TF, Juang YM, Tsay YG, et al. Comparative proteomic profiling of human lung adenocarcinoma cells (CL 1–0) expressing miR-372. Electrophoresis. 2012;33:675–688. doi: 10.1002/elps.201100329. [DOI] [PubMed] [Google Scholar]
- 90.Yan GR, Xu SH, Tan ZL, Liu L, et al. Global identification of miR-373-regulated genes in breast cancer by quantitative proteomics. Proteomics. 2011;11:912–920. doi: 10.1002/pmic.201000539. [DOI] [PubMed] [Google Scholar]
- 91.Takei Y, Takigahira M, Mihara K, Tarumi Y, et al. The metastasis-associated microRNA miR-516a-3p is a novel therapeutic target for inhibiting peritoneal dissemination of human scirrhous gastric cancer. Cancer Res. 2011;71:1442–1453. doi: 10.1158/0008-5472.CAN-10-2530. [DOI] [PubMed] [Google Scholar]