

Abstract
Heterogeneous expression of drug target proteins within tumor sites is a major mechanism of resistance to anticancer therapies. We describe a strategy to selectively inhibit, within tumor sites, the function of a critical intracellular protein, the sarcoplasmic/endoplasmic reticulum calcium adenosine triphosphatase (SERCA) pump, whose proper function is required by all cell types for viability. To achieve targeted inhibition, we took advantage of the unique expression of the carboxypeptidase prostate-specific membrane antigen (PSMA) by tumor endothelial cells within the microenvironment of solid tumors. We generated a prodrug, G202, consisting of a PSMA-specific peptide coupled to an analog of the potent SERCA pump inhibitor thapsigargin. G202 produced substantial tumor regression against a panel of human cancer xenografts in vivo at doses that were minimally toxic to the host. On the basis of these data, a phase 1 dose-escalation clinical trial has been initiated with G202 in patients with advanced cancer.
Introduction
Heterogeneous expression of therapeutic target proteins within individual tumor sites leads to the development of resistance to therapeutic inhibitors through the selection and proliferation of cancer cells in which the targeted protein is down-regulated. One strategy to overcome this heterogeneity problem is to target a protein whose continued expression is critical to the survival of all normal and tumor cell types. Proper function of the sarcoplasmic/endoplasmic reticulum calcium adenosine triphosphatase (SERCA) pump protein, which transfers Ca2+ from the cytosol of the cell to the lumen of the sarcoplasmic and endoplasmic reticulum, is necessary for normal cellular viability (1–7). The natural product thapsigargin (TG) (Fig. 1A) binds tightly to the transmembrane portion of the SERCA pump (8–10), inhibits its function (11, 12), and induces cell death in all normal and malignant cell types tested (13). Sustained TG-driven inhibition of the SERCA pump depletes endoplasmic reticulum calcium stores, which triggers the opening of plasma membrane calcium channels and the resulting rapid elevation in cytoplasmic calcium (12, 14–16). Sustained inhibition results in continued depletion of endoplasmic reticulum calcium and elevation of cytoplasmic calcium to micromolar levels (10, 14). This elevation triggers the endoplasmic reticulum stress/unfolded protein response, caspase activation, release of apoptotic factors from the mitochondria, and direct activation of calcium-dependent endonucleases that cleave cellular DNA (16–19) (Fig. 1B). Unlike cell cycle–dependent chemotherapies, low nanomolar concentrations of TG are equally effective at inducing apoptosis in both proliferating and nonproliferating cells (5, 13).
Fig. 1.
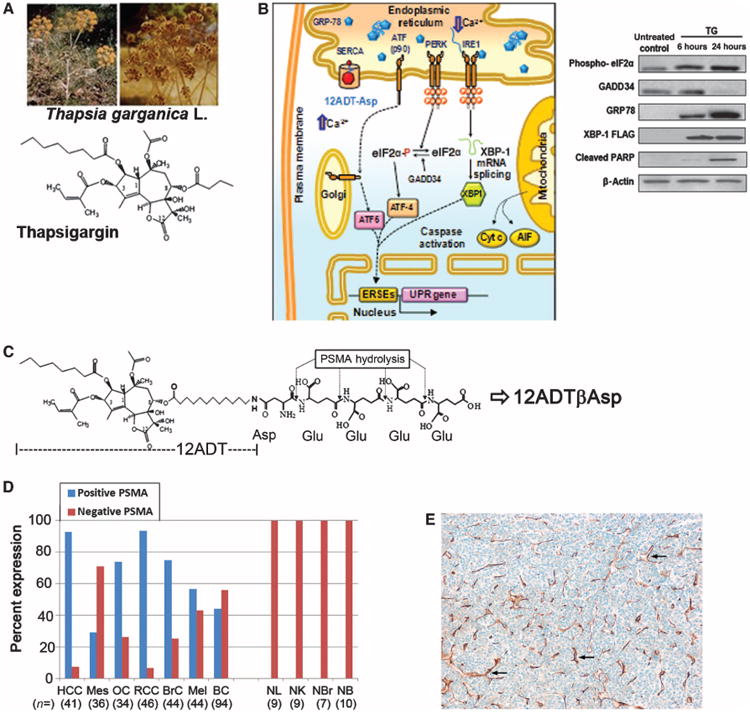
12ADT analog and prodrug structures and characterization of PSMA expression by tumor ECs. (A) Chemical structure of TG isolated from the seeds of Thapsia garganica L., which grows as a weed throughout the Mediterranean basin. (B) 12ADT-Asp (red) produces a decrease in endoplasmic reticulum calcium and an increase in cytoplasmic calcium, initiating endoplasmic reticulum stress, which results in 78-kD glucose–regulated protein (GRP78) elevation, release and processing of activating transcription factor (ATF), and production of ATF4 and X-box binding protein 1 (XBP-1), with subsequent expression of unfolded protein response (UPR) genes. Apoptotic factors cytochrome c (Cyt c) and AIF are released from the mitochondria, which activate caspases and the apoptotic program. Inset, Western (immuno) blots demonstrate TG-induced elevations in phospho–eukaryotic initiation factor 2α (eIF2α), GRP78, XBP-1, and cleaved poly(adenosine diphosphate–ribose) polymerase (PARP). (C) Schematic of sequential PSMA hydrolysis of 12ADTβAsp-GluγGluγGluγGlu (G202) to yield the cytotoxic TG analog 12ADTβAsp. (D) Tissue microarrays were stained for PSMA expression using the clone 3E6 anti-PSMA antibody. EC staining was graded on a zero- to three-point scale (table S1). Samples that contained any PSMA staining (that is, 1 to 3+) were considered to be positive. HCC, hepatocellular cancer; Mes, mesothelioma; OC, ovarian cancer; RCC, renal cell cancer; BrC, breast cancer; Mel, melanoma; BC, bladder cancer; NL, normal liver; NK, normal kidney; NBr, normal breast; NB, normal bladder; n, number of samples shown in parentheses. (E) Example of 3+ PSMA staining of ECs (brown) in a 40× section of hepatocellular carcinoma. Examples of brown EC staining indicated by arrows.
Because of its ability to kill all cell types in a proliferation-independent manner, TG is highly toxic in vivo (13). Therefore, to direct TG's potent cytotoxicity selectively to tumor cells while sparing normal tissue, we developed a protease-activated prodrug strategy in which a highly potent primary amine–containing TG analog is coupled to a protease-specific peptide carrier (13, 20). Using an iterative medicinal chemistry approach in which individual side chains of the TG molecule were selectively modified, we determined that the eighth position of TG could be modified without significantly affecting SERCA binding (21, 22). The primary amine–containing 8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin (12ADT) has an optimal-length side chain that maintains cytotoxic potency and is easily coupled to pep-tides (Fig. 1C) (22, 23).
To target 12ADT to the SERCA pump in tumor cells, we focused on the unique carboxypeptidase activity and restricted expression of the prostate-specific membrane antigen (PSMA) (24). Previous immunohistochemical studies demonstrated that PSMA is expressed by normal prostate epithelium and is even more highly expressed by a large proportion of prostate cancers, including metastatic prostate cancers (25–28). Although it was initially thought to be relatively prostate tissue–specific, PSMA expression has also been detected in tumor endothelial cells (ECs) of a variety of tumor types but is not expressed by ECs in normal tissues (29–33). Successful targeting of metastatic tumor sites using a radiolabeled humanized anti-PSMA antibody (J591) suggests that one could target PSMA expression in the vasculature of solid tumors to proteolytically activate a prodrug, which leads to the selective death of cells within the site of metastases (34, 35).
PSMA is a 100-kD type 2 cell-surface transmembrane glycoprotein whose catalytic domain projects into the extracellular space (36). Thus, release of the cytotoxic 12ADT analog by PSMA occurs extracellularly within the tumor microenvironment, which contains epithelial cancer cells supplied with nutrients by tumor ECs. This extracellular activation could overcome potential heterogeneity in PSMA expression, because both PSMA-expressing and non–PSMA-expressing cells within the tumor microenvironment could be killed by the 12ADT analog released within the peritumoral space. Thus, targeting PSMA's enzymatic activity within the tumor vasculature represents an alternative strategy that potentially could be used to treat all solid tumors.
Here, we describe a prodrug strategy that takes advantage of the unique dual exopeptidase activity of PSMA as both a pteroyl poly-γ-glutamyl carboxypeptidase (folate hydrolase) and an N-acetylated α-linked acidic dipeptidase (NAALADase) (36, 37). To target this dual enzymatic activity, we identified PSMA-specific peptide substrates that were coupled to the 12ADT analog to produce a PSMA prodrug (Fig. 1C) that selectively killed PSMA-expressing cells in vitro and produced marked regression of a variety of human tumor xenografts in mice. These preclinical studies identified a candidate PSMA-activated pro-drug that is currently being tested in clinical trials in patients with advanced solid tumors.
Results
PSMA expression by tumor ECs in human cancers
Recently, a new monoclonal antibody (mAb) to PSMA (clone 3E6, Dako) that binds to PSMA on paraffinized tissue sections better than previously available PSMA antibodies was created. The new PSMA mAb was used to perform immunohistochemical staining of paraffin-embedded sections from both cancer and normal tissues. PSMA expression was detected in the tumor-associated ECs of 66% of gastric cancers, 85% of colorectal carcinomas (32), and 100% of bladder cancers (33). Using this antibody, we stained a total of 340 human tumor samples and 32 control tissues from commercially available microarrays to more fully characterize PSMA expression by tumor ECs of a large variety of human tumors (Fig. 1, D and E). Nearly 95% of hepatocellular and renal cancers and ∼75% of ovarian and breast cancers stained positive for PSMA in the tumor vasculature. Furthermore, 57% of melanoma samples, 43% of bladder cancers (43%), and less than 30% of the mesothelioma specimens stained positive for PSMA. None of the 32 normal tissue samples that we examined demonstrated PSMA expression in the epithelium or associated normal vasculature (Fig. 1D). These results provide further evidence that a targeted therapy can be based on selective PSMA expression by tumor ECs.
Structural characterization of 12ADT-Asp analog binding to the SERCA pump
Previously, we generated a series of peptide substrates consisting of combinations of aspartates and/or glutamates coupled to the 4-([{2,4-diaminopteridin-6-yl}methyl][methyl]amino)benzoic acid (APA) portion of the drug methotrexate (38). We demonstrated that PSMA could sequentially hydrolyze various combinations of methotrexate-bound acidic amino acids, whether the peptide bonds were formed via α- or γ-linkage between amino acids, but could not cleave the ultimate aspartate or glutamate from APA. These results suggest that the appropriate TG analog for incorporation into a PSMA prodrug would be one that maintained its cytotoxic potency with an acidic amino acid such as aspartic or glutamic acid incorporated into its structure.
To determine the impact of binding to the SERCA pump produced by the incorporation of such an acidic amino acid, we solved the crystal structure of 12ADTβAsp bound to the SERCA pump (table S2) [Note that the hyphen (-) between amino acids indicates α-linkage, β indicates β-linkage, and γ indicates γ-carboxyl linkage.] The overall structure of SERCA consists of three cytoplasmic domains and 10 transmembrane helices (Fig. 2A). These structural studies demonstrated that the 12-carbon side chain in 12ADTβAsp-GluγGlu penetrated the transmembrane domain of the SERCA pump, placing the amino acid group at a cytoplasmically exposed site on the opposite side of the protein relative to the TG binding site. The Asp group was oriented in a position to produce a hydrogen bond between the α-amine of the Asp with Gln250 (2.8 Å) (Fig. 2B). In addition, a phospholipid head group from the endoplasmic reticulum membrane lipid interacts with the free amino acid of 12ADTβAsp, near the binding pocket for cyclopiazonic acid (CPA), a second type of SERCA pump inhibitor (39). Earlier structural studies demonstrated that CPA binds to the SERCA pump at a TG-independent site (39). A comparison of previous structural studies of CPA binding to SERCA demonstrated that the Asp group of 12ADTβAsp occupied a similar site within the SERCA pump, although the overlap is not as extensive as that observed in the structure for butoxycarbonyl (boc)–protected 12ADT (9) (Fig. 2C). A competition of binding sites was confirmed by intrinsic fluorescent studies demonstrating that 12ADTβAsp and CPA competitively affect the binding of each other.
Fig. 2.
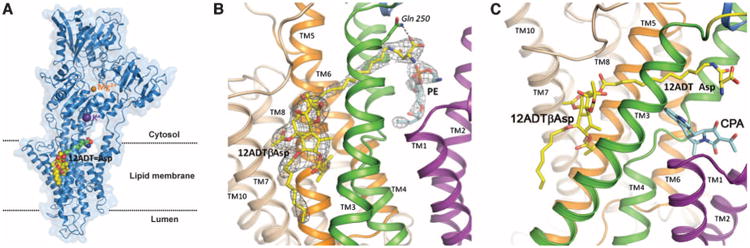
Structural analysis of 12ADTβAsp binding to SERCA pump. (A) 12ADTβAsp bound to SERCA pump (purple) demonstrating predominant binding of TG pharmacophore (yellow/red space filling) to the SERCA pump transmembrane domain with extension of the 12ADT (green) and Asp (blue/red) moieties into the space between the α helices that make up the transmembrane lipid domain. (B) The α-amine group of the Asp moiety forms a hydrogen bond (dotted line) with Gln250 of the SERCA pump (residue shown in stick representation) and a potential interaction with phosphatidylethanolamine within the membrane (blue and red in the figure). The electron density of the 12ADTβAsp is shown in gray cage (contour level 1σ). (C) The Asp moiety of 12ADTβAsp (yellow/red/blue) occupies a similar site in the SERCA pump as the known SERCA pump inhibitor CPA (aquamarine/red). (B) and (C) color code: transmembrane (TM) domains 1 and 2, purple; TM3 and 4, green; TM5 and 6, orange; TM7 to TM10, wheat. (Methods for crystal structure determination, data collection, and refinement statistics are included in the Supplementary Materials.)
In vitro characterization of a PSMA-activated prodrug
As predicted by the structural studies, 12ADTβAsp and 12ADT-Glu analogs were as potent at inhibiting the SERCA pump in broken cell microsomal studies as was TG (Fig. 3A). These TG analogs also maintained cytotoxic potencies against human cancer cell lines that were comparable to those of TG (Fig. 3A).
Fig. 3.
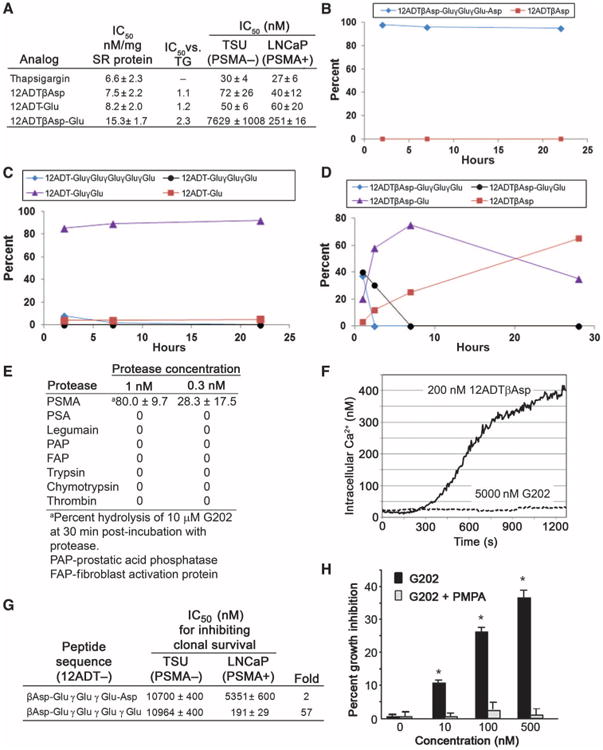
Identification and in vitro characterization of 12ADT analogs and PSMA-activated prodrugs. (A) Concentrations of TG, 12ADTβAsp, 12ADT-Glu, and 12ADTβAsp-Glu that inhibit SERCA pump function by 50% in microsomal assays, and IC50 against TSU and LNCaP cell lines in clonal survival assays conducted after 72-hour exposure to drugs. (B to D) Hydrolysis of (B) 12ADTβAsp-GluγGluγGlu-Asp, (C) 12ADT-GluγGluγGluγGluγGlu, and (D) G202 when incubated with Fc-PSMA for the indicated times. (E) Comparison of hydrolysis of G202 by PSMA and equimolar concentrations of other proteases. (F) Immediate effect of exposure to 12ADTβAsp or G202 on intracellular calcium levels in PSMA-negative TSU human bladder cancer cells loaded with Fura-2 calcium-sensitive dye. (G) Prodrug concentration that inhibits cell proliferation by 50% as determined with a clonal survival assay after exposure of either PSMA-negative TSU or PSMA-positive LNCaP cell lines to the indicated prodrugs for 72 hours. All results were performed in triplicate with average ± SE presented. (H) Growth inhibition of PSMA-positive LNCaP cells after a 48-hour exposure to increasing concentrations of G202 in the presence or absence of the PSMA inhibitor PMPA (10 μM) (*P < 0.05 for G202 + PMPA versus G202 alone by paired t test).
Structural studies of the PSMA catalytic site demonstrate that there is an ∼20 Å deep funnel that leads from the surface of PSMA to the enzyme active site (40–42). Once in the active site, glutamate residues are sequentially hydrolyzed from poly-γ-glutamated substrates without release of the shortened substrate from the catalytic site (41). Computational modeling based on these available PSMA structural studies suggested that the funnel could accommodate prodrugs containing 12ADT linked to acidic amino acids with the 12-carbon side chain being of adequate length to keep the bulky TG moiety outside of the funnel that leads to the catalytic site (42–44). We generated the dipeptide prodrug 12ADTβAsp-Glu to test the validity of the modeling. PSMA hydrolyzed >95% of the dipeptide to 12ADTβAsp but was unable to hydrolyze the Asp moiety to generate 12ADT (fig. S1), confirming that coupling 12ADT to a PSMA substrate did not compromise hydrolysis.
On the basis of our earlier results (38), peptides that contain five acidic amino acids were deemed sufficient for this prodrug strategy because they could be completely hydrolyzed by PSMA, enhance the solubility of lipophilic 12ADT, and prevent the charged nonhydrolyzed prodrug from penetrating the plasma membrane of normal cells. Because γ-linked substrates are highly unstable in plasma (38), the first prodrug we synthesized, 12ADTβAsp-GluγGluγGlu-Asp, contained α-linked amino acids in the first and last positions of the peptide (Fig. 3B). This prodrug was not only completely stable to hydrolysis in human plasma but also not hydrolyzed by PSMA (Fig. 3B). We also evaluated poly-γ-glutamated 12ADT (that is, 12ADT-GluγGluγGluγGluγGlu), and unlike poly-γ-glutamated methotrexate, this version of 12ADT was completely stable to hydrolysis in human plasma and was rapidly hydrolyzed by PSMA to the dipeptide 12ADT-Glu*Glu; however, PSMA was unable to cleave the penultimate glutamate to generate 12ADT-Glu (Fig. 3C). Because we knew that PSMA can cleave the α-linked 12ADTβAsp-Glu compound, the next prodrug we synthesized and evaluated had the sequence 12ADTβAsp-Glu, which was found to be ∼30-fold less cytotoxic to the PSMA-negative TSU bladder cancer cell line than the LNCaP line (Fig. 3A). PSMA then hydrolyzed this α-linked dipeptide to form 12ADTβAsp with kinetics that were somewhat slower than those observed for cleavage of the γ-linked intermediates (Fig. 3D). In addition, G202 was completely stable to hydrolysis in human plasma; in fact, the ability to hydrolyze G202 was relatively PSMA specific, as G202 was not appreciably hydrolyzed after incubation with a variety of other purified proteases (Fig. 3E).
To determine whether nonspecific cellular uptake could be prevented by the 5–amino acid peptide in G202, we loaded PSMA-nonproducing TSU bladder cancer cells with a calcium-sensitive dye and exposed the cells to either 12ADTβAsp (200 nM) or G202 (5 mM) (6). Exposure of the PSMA-nonproducing TSU bladder cancer cells to 12ADTβAsp produced a rapid maximal elevation in intracellular Ca2+ up to 400 nM (Fig. 3E). In contrast, exposure of the same cells to 25-fold higher concentrations of G202 resulted in no appreciable increase in intracellular calcium (Fig. 3F).
To confirm that G202 displayed selective toxicity to PSMA-producing cells, we compared the cytotoxic effects of G202 to those of the PSMA-nonhydrolyzing 12ADTβAsp-GluγGluγGlu-Asp in the PSMA-producing LNCaP human prostate cancer line and the PSMA-nonproducing TSU cell line. In this assay, the concentration that inhibited growth by 50% in cell proliferation assays (IC50) for the nonhydrolyzed prodrug was in the micromolar range against both cell lines (Fig. 3G). In contrast, the IC50 for G202 against the PSMA-nonproducing TSU cells was 57-fold higher than that for the PSMA-producing LNCaP cells (Fig. 3G). Finally, G202-induced growth inhibition of LNCaP cells could be blocked by the addition of 2-(phosphonomethyl)-pentanedioic acid (PMPA), a potent PSMA inhibitor, further confirming the PSMA requirement for activation of the prodrug in vitro (Fig. 3H).
Evaluation of G202 in vivo against human prostate cancer xenografts
Initial dose-finding studies in wild-type mice demonstrated that the maximally tolerated single intravenous dose of G202 was 112 mg/kg (2 μmol/dose), whereas up to three consecutive daily doses of 56 mg/kg (1 μmol/dose) produced mortality in <10% of treated animals. Subsequently, PSMA-producing human prostate cancer xenografts growing in intact (LNCaP and MDA-PCa2b cell lines) or castrated (CWR22R-H cell line) mice were treated with G202 [a dose of 56 mg/kg on 3 consecutive days (1 mmol × 3)] (Fig. 4, A to C). A single 3-day course of G202 produced ∼50% average regression of LNCaP xenografts over a 30-day period (Fig. 4A). Significant antitumor effects were also observed against MDA-PCa2b and CWR22R-H out to ≥30 days after a single 3-day course of G202 (Fig. 4, B and C). Overall, this 56 mg/kg × 3 dosing regimen routinely produced a maximum body weight loss of about 15% by 5 to 7 days after therapy, with weight returning to baseline levels by days 14 to 21 (Fig. 4D).
Fig. 4.
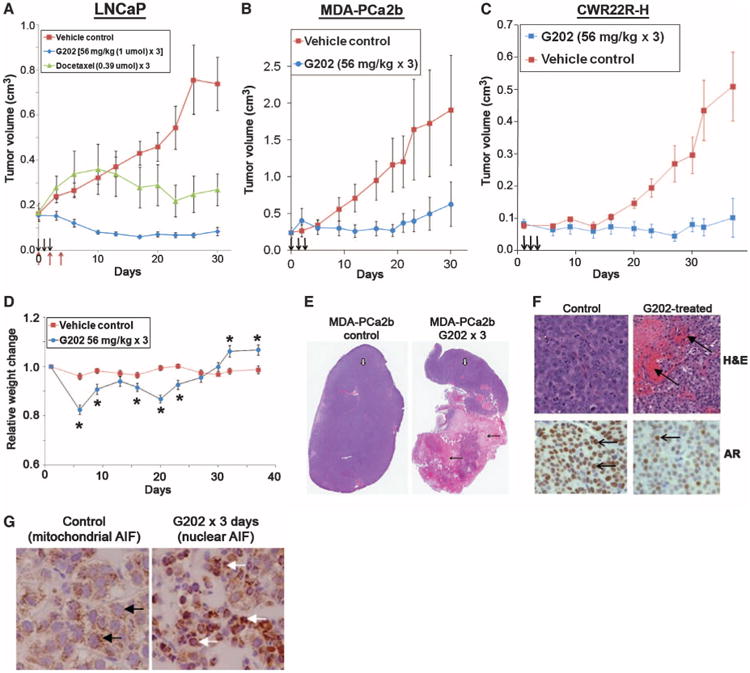
Antitumor effect of G202 in mouse xenograft models. Mice were either untreated or treated with three consecutive intravenous doses of G202 (56 mg/kg) (arrows, A to C). (A) Comparison of growth inhibition of LNCaP human prostate cancer xenografts in intact mice by G202 versus docetaxel at the indicated doses. Black arrows indicate G202 dosing on days 0,1, and 2, and red arrows indicate docetaxel dosing on days 0, 2, and 5 [P < 0.05 for tumor volume for treated versus control (untreated) mice after day 3 for G202 and after day 21 for docetaxel by Student's t test]. (B) Comparison of growth inhibition of MDA-PCa2b human prostate cancer xenografts in intact mice (P < 0.05 by Student's t test for G202-treated tumor versus control beginning after day 8). (C) Comparison of growth inhibition of CWR22R-H human prostate cancer xenografts in castrated mice (P < 0.05 by Student's t test for G202-treated tumor versus control after day 15). (D) Effect of three daily doses of G202 (56 mg/kg) on body weight of mice bearing CWR22R-H tumors (*P < 0.05 by Student's t test for animal weight in G202-treated animals versus control). (E) Histology of MDA-PCa2b tumors harvested from untreated control mice or 4 days after last dose of G202. White arrows show normal tumor morphology under 2× microscopy, whereas black arrows indicate areas of tumor necrosis in G202-treated animals. (F) Upper panels show hematoxylin and eosin (H&E) staining under 40× high power (bright-field microscopy, Zeiss Axiovert) of MDA-PCa2b tumors 4 days after treatment with G202 (56 mg/kg × 3) compared to untreated controls. Arrows indicate areas of red blood cell extravasation into tumor microenvironment in tumors treated with G202. Lower panels show intensity of AR staining in the nucleus (brown) with representative brown-stained AR-positive nuclei indicated by arrows in MDA-PCa2b tumors from untreated controls or 4 days after treatment with three daily doses of G202 (56 mg/kg). (G) AIF staining of sections of prostate cancer MDA-PCa2b xenografts from untreated control shows punctuate brown AIF staining in the mitochondria (black arrows) versus brown AIF staining in the nuclei (white arrows) after 3 days exposure to G202 (56 mg/kg).
As a comparative benchmark, we compared the effect of G202 to that of the standard chemotherapeutic agent docetaxel against the LNCaP human prostate cancer mouse xenograft model. Docetaxel was selected because it is used as standard often first-line therapy for many cancers, including prostate, breast, lung, bladder, and ovarian. We compared the maximally tolerated dose of docetaxel (0.39 μmol × 3 daily doses) to the maximally tolerated dose of G202 (1.0 μmol × 3 doses) (Fig. 4A). This dose of docetaxel was relatively toxic to the LNCaP xenograft–bearing mice and produced ∼30% average loss of body weight at 21 days after dosing (fig. S2). In contrast, although we delivered ∼2.5-fold more of the 12ADTβAsp toxin on a molar basis, the G202 prodrug produced a transient ∼15% average loss of body weight with full recovery of weight observed by day 21 (fig. S2). The G202 prodrug produced a treated–to–control tumor volume (T/C) ratio of 0.15, with 80% (seven of nine) of tumors showing >50% regression by day 21. In contrast, docetaxel treatment in this mouse model produced a T/C ratio of 0.63, with only one of eight (14%) treated animals showing >50% tumor regression out to day 30 after treatment (Fig. 4A).
Histological analysis of resected tumors revealed large areas of necrosis in the G202-treated tumors compared to controls (Fig. 4E). Within the nonnecrotic areas of the treated tumors, extensive extravasation of red blood cells was observed (Fig. 4F). Previous studies had demonstrated that 12ADTβAsp exposure produced a down-regulation of the androgen receptor (AR) in prostate cancer cells (45). Within the G202-treated AR-positive MDA-PCa2b tumors, marked down-regulation of AR expression was observed, consistent with intratumoral release of 12ADTβAsp (Fig. 5F). In addition, the uptake of 12ADTβAsp by these prostate cancer cells produced release of apoptosis-inducing factor (AIF) from the mitochondria and uptake into the nucleus as part of the apoptotic program initiated after exposure of cells to 12ADTβAsp (Fig. 4G).
Fig. 5.
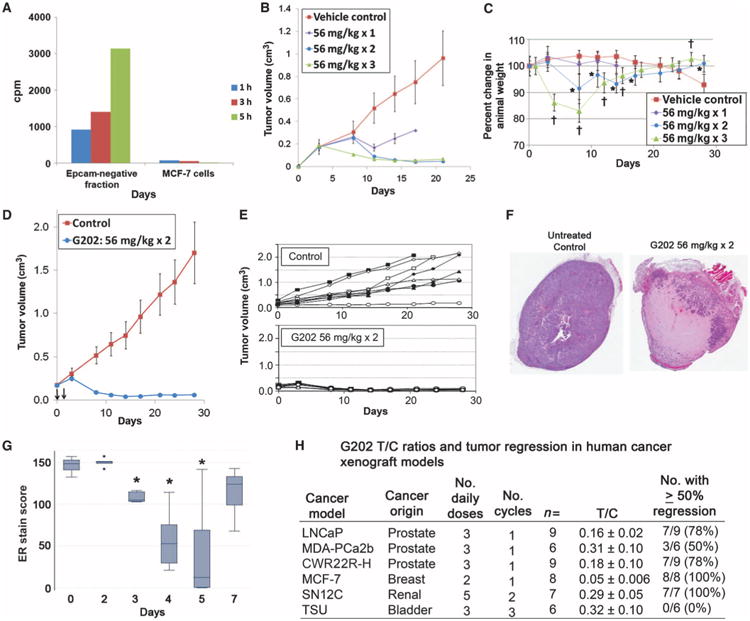
In vivo effects of G202 on MCF-7 breast cancer cells. (A) Hydrolysis of the PSMA substrate 3H-NAAG by the EPCAM-negative fraction of cells obtained from a suspension of single cells obtained from a single harvested MCF-7 xenograft compared to activity of MCF-7 cells obtained from in vitro culture. (B) Effect of one, two, or three doses of G202 (56 mg/kg) on growth of MCF-7 xenografts compared to untreated control (P < 0.05 by Student's t test for tumor treated versus control at all points after day 10 for animals treated with one or two doses of G202 and P < 0.05 at all time points after day 5 for animals treated with three doses of G202). (C) Body weight of G202-treated animals after one, two, or three doses of G202 at 56 mg/kg (P < 0.05 by Student's t test indicated by * for G202 times two doses or † for three doses versus control animal weight). (D and E) Effect of a single course of two doses of G202 (56 mg/kg) on average tumor volume of MCF-7 cells versus untreated controls over a 28-day period (P < 0.05 by Student's t test for G202-treated versus control at all points after day 3) (D) or tumor volume of individual animals (E). (F) Histology of MCF-7 xenografts harvested from untreated control or 6 days after treatment with two doses of G202 (56 mg/kg). (G) Whisker plot demonstrating the effects of two doses of G202 (56 mg/kg) on the expression of ER over a 7-day period in harvested MCF-7 xenografts. *P < 0.05 for ER stain score on indicated day versus stain score at day 0. (H) Summary of T/C ratios of human tumor models treated with indicated doses and cycles of G202.
Evaluation of G202 in vivo against other human cancer xenografts
To confirm PSMA enzymatic activity in the mouse endothelial compartment within a human tumor, we generated a single-cell suspension from a harvested MCF-7 human breast cancer xenograft. MCF-7 epithelial cells were removed using anti-human epithelial cell adhesion molecule (EPCAM)–labeled magnetic beads, and the stromal cell component (that is, all nonepithelial cells) was tested for PSMA activity using the PSMA substrate 3H-NAAG (Fig. 5A). Compared to control samples that consisted of equal numbers of MCF-7 cells growing in vitro, a large amount of PSMA substrate was hydrolyzed by the EPCAM-negative EC-containing tumor stromal cell compartment fraction from this single MCF-7 tumor xenograft. On the basis of these findings, we used MCF-7 as a nonprostate cancer model to test the antitumor activity of G202 in vivo. A dose-finding study revealed that a single dose of G202 (56 mg/kg on 1 day) produced tumor growth delay compared to the growth of control tumors (Fig. 5B). A similar degree of tumor regression was observed with two consecutive (T/C ratio of 0.05 ± 0.006) versus three consecutive (T/C ratio of 0.06 ± 0.005) doses by day 21 after initiation of therapy (Fig. 5B). Three doses, however, produced more significant weight loss than two (Fig. 5C). Furthermore, two daily doses of G202 produced sustained tumor regression out to 30 days in all treated MCF-7 xenograft–containing animals compared to untreated (control) animals (Fig. 5, D and E).
Histological analysis demonstrated a largely necrotic tumor as early as 4 to 6 days after the last dose of G202 (Fig. 5F). As was observed with the AR-positive prostate models, G202 treatment produced down-regulation of the estrogen receptor (ER) in these MCF-7 cells that reached their nadir 3 days after the last dose (Fig. 5G). Additional experiments were performed to assess the effects of G202 on human renal cell carcinoma (SN12C) and human bladder cancer (TSU) models using a more intensive treatment schedule than we used for the breast and prostate cancer xenografts (Fig. 5H). Overall, for the xenograft models tested, the T/C ratios at 3 weeks after G202 treatment ranged from 0.05 for MCF-7 to 0.32 for the TSU xenografts; in addition, G202 produced ≥50% regression of the majority of individual tumors (Fig. 5H). These results demonstrate that G202 has broad tumor regression activity in vivo for both prostate and nonprostate cancer models.
Pharmacokinetics and biodistribution of G202 in animal models
A liquid chromatography–mass spectrometry (LC-MS) method was developed to assess the amounts of G202 and PSMA cleavage products 12ADTβAsp and 12ADTβAsp-Glu in plasma and tissue. Pharmacokinetics in BALB/c mice demonstrated that G202 had a half-life of 4.9 hours after a single dose of 67 mg/kg (Fig. 6A). Less than 1% of this dose was converted in the circulation to the active 12ADTβAsp analog, consistent with the stability of G202 to nonspecific hydrolysis and a lack of PSMA in the systemic circulation.
Fig. 6.
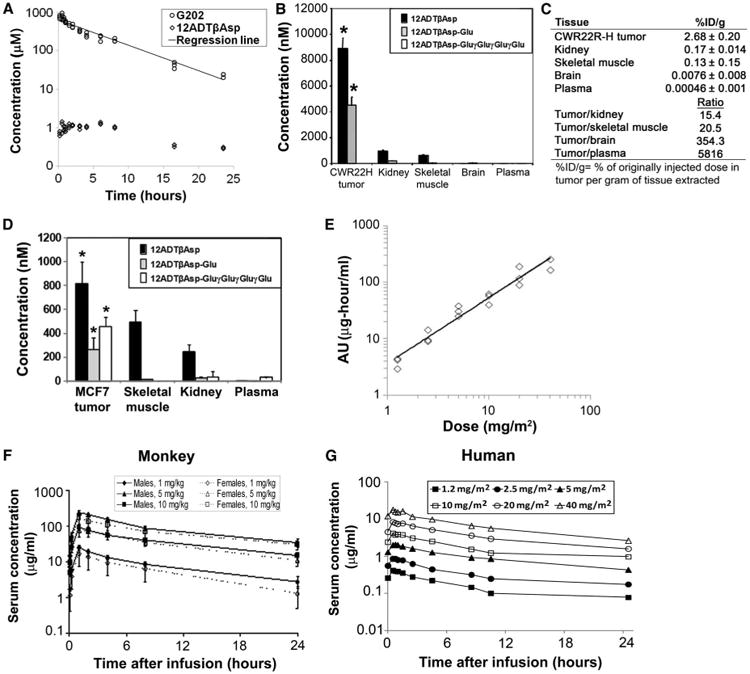
Pharmacokinetics of G202 and 12ADTβAsp in tumor xenografts, monkey, and human studies. (A) Pharmacokinetic study of a single dose of G202 (67 mg/kg) in wild-type BALB/c mice. LC-MS/MS analysis was used to determine concentrations of G202 and 12ADTβAsp in groups of three mice at the indicated time points over a 24-hour period. A semilog scale was used to calculate trend line (regression line) as indicated. (B) Tissue concentrations of 12ADTβAsp, 12ADTβAsp-Glu, and G202 in CWR22R-H-bearing castrated mice. Tissues were harvested 5 days after a single dose of G202 (56 mg/kg) administered to five tumor-bearing mice. *P < 0.05 by Student's t test for 12ADTβAsp and 12ADTβAsp-Glu levels in tumors compared to other tissues sampled. No significant difference in G202 levels in tumors versus normal tissues was observed. (C) Comparison of 12ADTβAsp concentrations in various tissues harvested from five CWR22R-H tumor–bearing mice 5 days after the final of three doses of G202 (56 mg/kg), given as a percentage of the injected dose of G202. (D) Tissue concentrations of 12ADTβAsp, 12ADTβAsp-Glu, and G202 in five MCF-7 tumor–bearing female mice. Tissues were harvested 5 days after a single dose of G202 (56 mg/kg). *P < 0.05 by Student's t test for 12ADTβAsp, 12ADTβAsp-Glu, and G202 levels in tumors compared to levels of each species in other tissues sampled. (E) AUC (ng·hour/ml) dose proportionality of G202 over doses ranging from 1.2 to 40 mg/m2 in a phase 1 clinical trial in 18 advanced cancer patients. (F) G202 serum concentrations over a 24-hour period after a single 1-hour G202 treatment of male and female monkeys over a dose range of 1 to 10 mg/kg. (G) G202 serum concentrations over a 24-hour period in 18 advanced cancer patients who received a 1-hour infusion of G202 at doses ranging from 1.2 to 40 mg/m2 in a phase 1 clinical trial.
We then performed biodistribution studies in the CWR22R-H–bearing castrated mouse xenograft model to evaluate the amounts of G202 and PSMA cleavage products in tumor and normal tissues 5 days after a single dose of G202 (56 mg/kg). These studies revealed significant accumulation of both 12ADTβAsp and 12ADTβAsp-Glu to micromolar levels in the CWR22R-H tumor tissue compared to the normal kidney, skeletal muscle, and brain (Fig. 6B). These data are consistent with lack of activation in these tissues as a result of either lack of PSMA expression (skeletal muscle) or lack of accessibility to the kidney proximal tubules and brain tissue, both of which are known to express PSMA (Fig. 6B). Overall, compared to other PSMA-expressing tissues, there was a 15-fold higher amount of 12ADTβAsp in the tumor tissue compared to normal kidney and a 354-fold higher amount of 12ADTβAsp in the tumor tissue compared to normal brain (Fig. 6C). In the MCF-7 xenograft model, the amount of 12ADTβAsp in the tumors was 1.6-fold higher than that found in skeletal muscle and 3.5-fold higher than that found in the kidney (Fig. 6D). However, 12ADTβAsp concentrations were only about 1/10 of the amounts of 12ADTβAsp observed in the CWR22R-H prostate xenografts.
Clinical development of G202
On the basis of the extensive preclinical data described above, which demonstrate the efficacy of G202 against a panel of human cancer xenografts, G202 has entered clinical development. U.S. Food and Drug Administration–required toxicological studies were performed in rats and cynomolgus monkeys [Ricerca Biosciences, LLC, Toxicology and Pharmacology (study numbers 022444, 022445, 023209, and 023210)]. The most common toxicity observed in these studies was transient reversible renal toxicity (table S3). This toxicity was thought to be an “on-target” toxicity resulting from PSMA expression in the kidney proximal tubules. In the monkey, repeated intravenous infusions of G202 (1-hour infusions for 3 consecutive days, which was repeated after a 4-week dose-free interval) were associated with nephrotoxicity at all examined doses (1,5, or 10 mg/kg). On the basis of clinical and anatomical pathology changes, toxicity was deemed to be minimal to mild at the lowest dose and reversible.
More severe renal toxicity was evident at the mid and highest doses, in a dose-responsive manner except for one male animal in the mid-dose group that experienced renal failure. In the affected animals at the mid-dose (5 mg/kg) and high-dose (10 mg/kg) groups, the renal lesions consisted of renal tubular degeneration predominantly affecting the proximal renal tubules/descending loop with an inflammatory cell component. By the end of the recovery period, the indicators of renal damage (blood urea nitrogen, creatinine, and albumin) showed clear reversibility, reflecting resolution/reversibility of the renal findings in animals at all dose levels tested. Lymphoid depletion of lymph nodes (five of six) and spleen (five of six) was evident in monkeys at a dose of G202 of 10 mg/kg along with thymic involution for all animals at the day 33 necropsy. Although not statistically significant because of the small number of animals, splenic/body weight and thymus/body weight ratios decreased by 44% and 67%, respectively, in three of three male monkeys at a dose of 10 mg/kg compared to the vehicle-treated animals. The splenic ratio increased by 25% in three of three female monkeys, whereas the thymic ratio decreased by 35% at a dose of 10 mg/kg. Lymphoid lesions were characterized by mild to moderate lymphoid depletion of follicles (germinal centers) in the lymph nodes, mild to moderate diffuse lymphoid depletion of the spleen, and increased involution of the thymus. However, although PSMA is also expressed in the brain, in the six monkeys treated with three consecutive doses of G202 (10 mg/kg), the brains were found to be within normal limits on pathological analysis. A similar lack of toxicity to the brain was observed in the rat pathological studies. Overall, the nonseverely toxic dose of G202 administered by this regimen in the monkey was considered to be more than 1 mg/kg per infusion but less than 5 mg/kg per infusion, and this dose range was used to identify a starting dose of 1.5 mg/m2 for human trials (Investigational New Drug 105,830). This phase 1 dose-escalation study (identifier # NCT01056029) is testing the safety and efficacy of three consecutive daily doses of G202 given in monthly cycles in patients with metastatic cancer. Pharmacokinetics from the first six dose cohorts demonstrated that systemic exposure area under the curve (AUC) to G202 increased slightly greater than dose proportionally; on average, for a doubling in dose, AUC was predicted to increase 2.23-fold (Fig. 6E). The terminal half-life of 15.1 hours was ∼1.5-fold greater than the 9.7-hour half-life observed in the monkey toxicological studies (Fig. 6, F and G, and fig. S3). To date, 29 patients in eight dose cohorts ranging from 1.2 to 88 mg/m2 have been treated in this ongoing trial.
Discussion
The goal of these studies was to develop a prodrug strategy to selectively deliver an analog of the highly potent, non–cell type–specific cytotoxin TG to human tumors while minimizing toxicity to normal tissues. G202, the lead PSMA-activated prodrug identified here, was efficiently hydrolyzed by PSMA and was ∼60-fold more toxic in vitro to PSMA-positive cells compared to PSMA-negative cells. Whereas TG has no therapeutic index and a lethal dose for 100% of the test population (LD100) equals to 0.2 mg/kg in BALB/c mice, two to three doses of G202 (5.6 to 56 mg/kg) produced tumor regression and growth inhibition in vivo in six separate tumor xenograft models with T/C ratios ranging from 0.05 to 0.32. Thus, the PSMA-activated prodrug approach allowed for the systemic delivery of >150-fold more TG molar equivalents compared to TG alone without significant host toxicity.
The choice of PSMA as the activating enzyme was based on its unique enzymatic activity as both a NAALADase and a folate hydrolase, which could allow for the identification and incorporation of specific peptide substrates into a prodrug strategy that are not hydrolyzed by other proteases present in nonprostatic tissues (Fig. 3E). In addition, besides almost universal expression by malignant prostate epithelial cells, PSMA is selectively expressed by tumor ECs within a variety of tumor types but is not expressed by normal ECs or by most normal tissues. The results from our staining of 340 human cancers in a tumor tissue array format are consistent with earlier results in which immunohistochemical staining was used to document PSMA expression in tumor ECs.
Our findings document that PSMA is more highly expressed by certain tumor types, with >60% of samples from hepatocellular, ovarian, renal, and breast cancers demonstrating PSMA expression, whereas a lower percentage of PSMA-expressing samples were observed for melanoma, bladder cancer, and mesothelioma. Consistent with earlier studies, we detected no PSMA expression by ECs from 32 normal tissue samples. However, other studies have documented PSMA expression in the proximal tubule of the kidney and in the brain (28, 46). In this regard, we detected 12ADTβAsp in the kidney in biodistribution studies in the mouse, although 12ADTβAsp concentrations in tumors were 3- to 15-fold higher than that in normal kidney tissue. In addition, transient reversible renal toxicity was observed in animal toxicological studies in the rat and monkey (table S3). However, pathological analyses of the brains of rats and monkeys treated with three consecutive doses of G202 (10 mg/kg) showed no evidence of G202-related toxicity. This lack of toxicity most likely stems from the inability of the highly charged prodrug to cross the blood-brain barrier, as evidenced by the low amounts of G202 detected in the brain of mice in the biodistribution studies. Finally, unlike traditional cytotoxic chemotherapies, G202 caused no bone marrow toxicity in mouse, rat, or monkey toxicology studies. These results suggest that G202 could be used clinically without a requirement for blood-product support.
Besides PSMA targeting, a further advantage of this approach lies in the use of a TG analog as the targeted cytotoxin. The 12ADTβAsp analog is highly lipophilic and rapidly partitions into cell membranes after release from the peptide carrier, thus minimizing systemic exposure that can result from leakage of an active toxin from a tumor. Unlike the heterogeneous expression of most current anticancer therapeutic targets, the SERCA pump is expressed homogeneously by all cell types and its continued expression is critically important for cell viability. Thus, resistance to 12ADTβAsp via down-regulation of SERCA expression is unlikely to occur. We validated this hypothesis in experiments in which cancer cells were repeatedly exposed to a cytotoxic dose of TG in an attempt to reproduce in vitro the type of exposure that would be achieved in vivo with repeat doses of therapeutic amounts of a TG analog. Here, no change in clonal survival was observed after 11 cycles of repeat exposure and clonal expansion (fig. S4). In addition, unlike other commonly used chemotherapeutic agents, which are typically cell proliferation–dependent cytotoxins, TG and its analogs can kill both rapidly proliferating and nonproliferating cells with equal potency. The ability to kill nonproliferating cells makes a TG-based approach a particularly suitable choice of agent for the treatment of prostate cancer, because we have demonstrated that within sites of metastatic prostate cancer >95% of the cancer cells are in a proliferatively quiescent G0 state (13, 47, 48). Furthermore, like human prostate cancer cells, ECs and fibroblasts within the tumor stromal compartment also have a low rate of cell proliferation (49–51). These observations suggest that TG also represents a favorable choice for a tumor EC–targeted approach.
Finally, a series of mechanistic studies have demonstrated that TG inhibition of the SERCA pump has catastrophic consequences for the cell. Function of the SERCA pump is critical to the maintenance of intracellular calcium homeostasis after calcium-mediated signaling events and to keep endoplasmic reticulum calcium at the high levels required for the maturation of nascent proteins. Perturbation of the endoplasmic reticulum environment by TG results in elevation of cytoplasmic calcium and depletion of endoplasmic reticulum calcium, thus activating the endoplasmic reticulum stress response. Sustained inhibition of the SERCA pump by TG produces an elevation in cytoplasmic calcium to micromolar concentrations that results in the subsequent activation of the apoptotic caspase cascade, release of pro-apoptotic factors from the mitochondria, and direct stimulation of Ca2+-activated endonucleases, with the end result being the induction of apoptosis in all cell lines tested (6, 16). Although these mechanistic studies have highlighted the profound intracellular sequelae that follow TG inhibition of the SERCA pump in all cell types, they also underscore the requirement for a targeting strategy if TG analogs are to be considered as potential therapeutic agents for cancer.
The preclinical results presented here demonstrate that the PSMA-activated tumor EC–targeted prodrug strategy produces a sufficient therapeutic index and tumor targeting to support clinical development of this approach. Results from our dose-finding phase 1 study and from the PSMA tumor tissue array staining will guide the design of future studies that test the effectiveness of this unique targeted approach in the treatment of human cancer.
Materials and Methods
Materials
TG was ethanol-extracted from the harvested seeds of Thapsia garganica L. obtained from Thapsibiza Inc., and boc-protected 12ADT was synthesized as previously described (22). PSMA prodrugs for in vitro studies were produced by coupling boc-protected 12ADT to peptides (California Peptide Research Inc.) with previously described methods (13). Purified G202 for animal studies was provided by GenSpera Inc. PSMA fusion protein (Fc-PSMA) (Lexigen Pharmaceuticals) (38) enzymatic activity was confirmed by 3H-NAAG hydrolysis, and purity was confirmed by Coomassie staining (38). PMPA (52) was provided by Guilford Pharmaceuticals. All other reagents, unless otherwise specified, were from Sigma.
Cell lines
The human cancer cell lines LNCaP (prostate), TSU (bladder), SN12C (renal), and MCF-7 (breast) were obtained from the American Type Culture Collection. The MDA-PCa2b human prostate cancer line was provided by N. Navone (M.D. Anderson Cancer Center). The CWR22R-H xenograft was derived from the androgen-sensitive CWR22 xenograft model (provided by T. Pretlow, Case Western Reserve University). This line was generated from a CWR22 tumor that relapsed in a castrate host. The generation and characterization of this CWR22R-H xenograft have been described previously (53).
Immunohistochemical staining
Tumor tissue microarrays were purchased from Imgenex with the exception of the bladder cancer array that was purchased from Pantomics Inc. Arrays were stained for PSMA according to the method of Milowsky et al. with a PSMA antibody (clone 3E6; Dako) diluted 1:20 in antibody diluent (Dako) (35). PSMA staining was scored according to previously published methods (54). AIF staining was performed with anti-AIF antibody from Santa Cruz Biotechnology (13) as previously described. AR staining of prostate cancer xenografts was performed with rabbit polyclonal anti-AR antibody (Santa Cruz Biotechnology), and ER staining of MCF-7 xenografts was performed with ERα, mAb (Ventana) according to previously published methods (45, 55).
PSMA prodrug hydrolysis
Prodrugs were incubated with 10 μM Fc-PSMA in tris buffer, containing 0.01 mM CoCl2. Aliquots were removed at various time points and analyzed by high-performance LC with 0.1% trifluoroacetic acid (TFA) in water as solvent A and 0.1% TFA in acetonitrile as solvent B. A gradient of 40% B to 100% B over 25 min was used to analyze cleavage products. Each hydrolysis product was identified by co-elution and confirmed by MS.
Calcium measurements and SERCA pump assays
Determination of intracellular free calcium levels was performed with the ratiometric method in a cuvette assay with 7.5 μM fura-2-acetoxymethyl ester (Molecular Probes)–loaded TSU cells as previously described (15). SERCA pump assays were performed with microsomes from rabbit skeletal muscle as previously described (13, 56). The SERCA activity was measured with a coupled enzyme assay as the rate of adenosine 5′-triphosphate hydrolysis as previously described (13, 57).
Crystal structure determination
Procedures for solubilization and crystallization of the 12ADTβAsp complex of rabbit SERCA1a, as well as for crystal stabilization and mounting, were essentially as described previously for other TG derivatives and are described in detail in the Supplementary Materials (Methods and table S2) (10, 39, 58, 59).
Cytotoxicity assays
To determine cytotoxic activity of prodrugs, clonal survival assays were performed with PSMA-negative TSU human bladder cancer and PSMA-positive LNCaP human prostate cancer cells as previously described (13).
In vivo studies
Maximum tolerated dose (single intravenous dose) was determined and used to perform pharmacokinetic studies. Single-dose pharmacokinetics were assessed by noncompartmental analysis. The AUC from time zero to infinity (AUC0–∞) was calculated with the linear trapezoidal method. The terminal half-life (t1/2) was determined from the terminal slope (Ke) on a log-linear plot of concentration versus time. Biodistribution studies were performed on tumor-bearing mice by harvesting tissues (liver, kidney, skeletal muscle, brain, and tumor) from mice (n = 3) at varying time points after single or multiple intravenous injections of G202. (Analytical methods for processing, extracting, and analyzing plasma and tissue drug concentrations are included as supplemental information.)
In vivo efficacy studies
LNCaP, MDA-PCa2b, or SN12C (2 × 106) cells in 100 μl of Matrigel (Collaborative Research) were inoculated into the flank of 6-week-old male nude mice (Harlan Sprague Dawley). MCF-7 cells (2 × 106) were injected into the flanks of female nude mice pretreated with subcutaneous estrogen pellet according to the previously described method (60). CWR22R-H xenografts were generated by subcutaneous inoculation of 10 mg of minced CWR22R-H tumor in Matrigel as previously described (11). Statistical analysis of differences in tumor volumes and weights between G202 and vehicle controls was performed with Student's t test, and P values of <0.05 were reported in the text All animal studies were performed according to the protocols approved by the Johns Hopkins Animal Care and Use Committee.
G202 clinical development
The synthesis of Good Manufacturing Practice–grade G202 was performed by GenSpera Inc. Good Laboratory Practice toxicology studies in rat and cynomolgus monkey were performed by Ricerca Biosciences, LLC, Toxicology and Pharmacology (study numbers 022444, 022445, 023209, and 023210). An Institutional Review Board-approved phase 1 open-label dose-escalation trial in patients with advanced cancer is being conducted at Johns Hopkins (Baltimore, MD), the University of Wisconsin (Madison, WI), and the Cancer Therapy and Research Center (San Antonio, TX) and sponsored by GenSpera Inc.
Statistical analysis
Student's paired t test analyses were performed to ascertain statistical significance. All tests were two-sided, and P values of <0.05 were considered statistically significant. All error bars in the figures represent ±SEM. The analyses were performed with statistical software SAS version 9.3.
Supplementary Material
Table S1. Tumor endothelial cell staining scores for individual tumor types.
Table S2. Crystal structure data collection and refinement statistics.
Table S3. Effect of G202 on laboratory values in cynomolgus monkeys.
Fig. S1. PSMA hydrolysis of the 12ADT|3Asp-Glu dipeptide.
Fig. S2. Effect of G202 or docetaxel on body weight of LNCaP human prostate cancer xenograft-bearing mice.
Fig. S3. Serum concentrations of G202 over a 24-hour period in 18 individual patients who received G202 at doses ranging from 1.2 to 40 mg/m2 in a phase 1 clinical trial.
Fig. S4. Clonal survival of PC-3 human prostate cancer cells after repeated exposure to 500 nM TG.
Acknowledgments
We thank J. Hicks, A. Gady, R. Ricklis, A. Solomon, L. Anthony, and R. Becker for excellent technical assistance and D. Hart for technical assistance with preparation of figures.
Funding: Grant funding provided by the Department of Defense Prostate Cancer Research Program, the Prostate Cancer Foundation and Mr. David Koch, the Danish Cancer Society, the Danish Research Council for Strategic Research, the Danish National Research Foundation, the Danish Medical Research Council, the Aarhus University Research Foundation, and the National Cancer Institute Specialized Program of Research Excellence.
Footnotes
Author contributions: S.R.D., J.T.I., I.D., C.O., and S.B.C. designed the research; S.R.D., A.M.M., D.M.R., W.N.B., S.D., I.D., C.O., B.G., A.M.D., G.W., M.A.C., J.V.M., P.N., S.B.C., and J.T.I. performed the research; B.G. and A.M.D. contributed analytic tools; S.R.D., I.D., C.O., C.A.D., P.N., S.B.C., and J.T.I. analyzed the data; S.D. prepared cell lines for xenograft studies; I.D. and C.O. acquired and analyzed crystallographic data; and S.R.D., P.N., and J.T.I. wrote the paper.
Competing interests: S.R.D., S.B.C., and J.T.I. are consultants for GenSpera Inc. and have received equity and financial compensation. This relationship for S.R.D. and J.T.I. has been disclosed and is under the management of the Johns Hopkins University School of Medicine Conflict of Interest Committee. C.A.D. is an employee of GenSpera Inc.
References and Notes
- 1.Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT. The role of calcium, pH, and cell proliferation in the programmed (apoptotic) death of androgen-independent prostatic cancer cells induced by thapsigargin. Cancer Res. 1994;54:6167–6175. [PubMed] [Google Scholar]
- 2.Furuya Y, Krajewski S, Epstein JI, Reed JC, Isaacs JT. Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clin Cancer Res. 1996;2:389–398. [PubMed] [Google Scholar]
- 3.Tombal B, Denmeade SR, Gillis JM, Isaacs JT. A supramicromolar elevation of intracellular free calcium ([Ca2+]i) is consistently required to induce the execution phase of apoptosis. Cell Death Differ. 2002;9:561–573. doi: 10.1038/sj.cdd.4400999. [DOI] [PubMed] [Google Scholar]
- 4.Denmeade SR, Lin XS, Tombal B, Isaacs JT. Inhibition of caspase activity does not prevent the signaling phase of apoptosis in prostate cancer cells. Prostate. 1999;39:269–279. doi: 10.1002/(sici)1097-0045(19990601)39:4<269::aid-pros7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 5.Lin XS, Denmeade SR, Cisek L, Isaacs JT. Mechanism and role of growth arrest in programmed (apoptotic) death of prostatic cancer cells induced by thapsigargin. Prostate. 1997;33:201–207. doi: 10.1002/(sici)1097-0045(19971101)33:3<201::aid-pros9>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 6.Tombal B, Weeraratna AT, Denmeade SR, Isaacs JT. Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate. 2000;43:303–317. doi: 10.1002/1097-0045(20000601)43:4<303::aid-pros10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4:14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 8.Toyoshima C, Nomura H, Sugita Y. Crystal structures of Ca2+-ATPase in various physiological states. Ann NY Acad Sci. 2003;986:1–8. doi: 10.1111/j.1749-6632.2003.tb07131.x. [DOI] [PubMed] [Google Scholar]
- 9.Søhoel H, Jensen AM, Møller JV, Nissen P, Denmeade SR, Isaacs JT, Olsen CE, Christensen SB. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg Med Chem. 2006;14:2810–2815. doi: 10.1016/j.bmc.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Winther AM, Liu H, Sonntag Y, Olesen C, le Maire M, Soehoel H, Olsen CE, Christensen SB, Nissen P, Møller JV. Critical roles of hydrophobicity and orientation of side chains for in-activation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J Biol Chem. 2010;285:28883–28892. doi: 10.1074/jbc.M110.136242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen U, Christensen SB, Sandberg F. Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm Suec. 1978;15:133–140. [PubMed] [Google Scholar]
- 12.Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, Christensen SB, Isaacs JT. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003;95:990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- 14.Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 15.Tombal B, Denmeade SR, Isaacs JT. Assessment and validation of a microinjection method for kinetic analysis of [Ca2+]i in individual cells undergoing apoptosis. Cell Calcium. 1999;25:19–28. doi: 10.1054/ceca.1998.0005. [DOI] [PubMed] [Google Scholar]
- 16.Deniaud A, Sharaf el dein O, Malillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 17.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 18.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 19.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denmeade SR, Isaacs JT. Enzymatic activation of prodrugs by prostate-specific antigen: Targeted therapy for metastatic prostate cancer. Cancer J Sci Am. 1998;4(Suppl. 1):S15–S21. [PubMed] [Google Scholar]
- 21.Christensen SB, Andersen A, Kromann H, Treiman M, Tombal B, Denmeade S, Isaacs JT. Thapsigargin analogues for targeting programmed death of androgen-independent prostate cancer cells. Bioorg Med Chem. 1999;7:1273–1280. doi: 10.1016/s0968-0896(99)00074-7. [DOI] [PubMed] [Google Scholar]
- 22.Jakobsen CM, Denmeade SR, Isaacs JT, Gady A, Olsen CE, Christensen SB. Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J Med Chem. 2001;44:4696–4703. doi: 10.1021/jm010985a. [DOI] [PubMed] [Google Scholar]
- 23.Singh P, Mhaka AM, Christensen SB, Gray JJ, Denmeade SR, Isaacs JT. Applying linear interaction energy method for rational design of noncompetitive allosteric inhibitors of the sarco- and endoplasmic reticulum calcium-ATPase. J Med Chem. 2005;48:3005–3014. doi: 10.1021/jm049319a. [DOI] [PubMed] [Google Scholar]
- 24.Horoszewicz JS, Kawinski E, Murphy GP. Monoclonal antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic cancer patients. Anticancer Res. 1987;7:927–935. [PubMed] [Google Scholar]
- 25.Israeli RS, Powell CT, Corr JG, Fair WR, Heston WDW. Expression of the prostate-specific membrane antigen. Cancer Res. 1994;54:1807–1811. [PubMed] [Google Scholar]
- 26.Kawakami M, Nakayama J. Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res. 1997;57:2321–2324. [PubMed] [Google Scholar]
- 27.Minner S, Wittmer C, Graefen M, Salomon G, Steuber T, Haese A, Huland H, Bokemeyer C, Yekebas E, Dierlamm J, Balabanov S, Kilic E, Wilczak W, Simon R, Sauter G, Schlomm T. High level PSMA expression is associated with early PSA recurrence in surgically treated prostate cancer. Prostate. 2011;71:281–288. doi: 10.1002/pros.21241. [DOI] [PubMed] [Google Scholar]
- 28.Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–85. [PubMed] [Google Scholar]
- 29.Liu H, Moy P, Kim S, Xia Y, Rajasekaran A, Navarro V, Knudsen B, Bander NH. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997;57:3629–3634. [PubMed] [Google Scholar]
- 30.Chang SS, O'Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin Cancer Res. 1999;5:2674–2681. [PubMed] [Google Scholar]
- 31.Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59:3192–3198. [PubMed] [Google Scholar]
- 32.Haffner MC, Kronberger IE, Ross JS, Sheehan CE, Zitt M, Mühlmann G, Ofner D, Zelger B, Ensinger C, Yang XJ, Geley S, Margreiter R, Bander NH. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum Pathol. 2009;40:1754–1761. doi: 10.1016/j.humpath.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Samplaski MK, Heston W, Elson P, Magi-Galluzzi C, Hansel DE. Folate hydrolase (prostate-specific antigen) 1 expression in bladder cancer subtypes and associated tumor neovasculature. Mod Pathol. 2011;24:1521–1529. doi: 10.1038/modpathol.2011.112. [DOI] [PubMed] [Google Scholar]
- 34.Bander NH, Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Oncol. 2005;23:4591–4601. doi: 10.1200/JCO.2005.05.160. [DOI] [PubMed] [Google Scholar]
- 35.Milowsky MI, Nanus DM, Kostakoglu L, Sheehan CE, Vallabhajosula S, Goldsmith SJ, Ross JS, Bander NH. Vascular targeted therapy with anti–prostate-specific membrane antigen monoclonal antibody J591 in advanced solid tumors. J Clin Oncol. 2007;25:540–547. doi: 10.1200/JCO.2006.07.8097. [DOI] [PubMed] [Google Scholar]
- 36.Pinto JT, Suffoleto BP, Berzin TM, Qiao CH, Lin S, Tong WP, May F, Mukherjee B, Heston WDW. Prostate-specific membrane antigen: A novel folate hydrolase in human prostatic carcinoma cells. Clin Cancer Res. 1996;2:1445–1451. [PubMed] [Google Scholar]
- 37.Carter RE, Feldman AR, Coyle JT. Prostate-specific membrane antigen is a hydrolase with substrate and pharmacologic characteristics of a neuropeptidase. Proc Natl Acad Sci USA. 1996;93:749–753. doi: 10.1073/pnas.93.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mhaka A, Gady AM, Rosen DM, Lo KM, Gillies SD, Denmeade SR. Use of methotrexate-based peptide substrates to characterize the substrate specificity of prostate-specific membrane antigen (PSMA) Cancer Biol Ther. 2004;3:551–558. doi: 10.4161/cbt.3.6.846. [DOI] [PubMed] [Google Scholar]
- 39.Laursen M, Bublitz M, Moncoq K, Olesen C, Møller JV, Young HS, Nissen P, Morth JP. Cyclopiazonic acid is complexed to a divalent metal ion when bound to the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 2009;284:13513–13518. doi: 10.1074/jbc.C900031200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci USA. 2005;102:5981–5986. doi: 10.1073/pnas.0502101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mesters JR, Barinka C, Li W, Tsukamoto T, Majer P, Slusher BS, Konvalinka J, Hilgenfeld R. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006;25:1375–1384. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barinka C, Byun Y, Dusich CL, Banerjee SR, Chen Y, Castanares M, Kozikowski AP, Mease RC, Pomper MG, Lubkowski J. Interactions between human glutamate carboxypeptidase II and urea-based inhibitors: Structural characterization. J Med Chem. 2008;51:7737–7743. doi: 10.1021/jm800765e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aggarwal S, Singh P, Topaloglu O, Isaacs JT, Denmeade SR. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006;66:9171–9177. doi: 10.1158/0008-5472.CAN-06-1520. [DOI] [PubMed] [Google Scholar]
- 44.Kularatne SA, Zhou Z, Yang J, Post CB, Low PS. Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted 99mTc-radioimaging agents. Mol Pharm. 2009;6:790–800. doi: 10.1021/mp9000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vander Griend DJ, Antony L, Dalrymple SL, Xu Y, Christensen SB, Denmeade SR, Isaacs JT. Amino acid containing thapsigargin analogues deplete androgen receptor protein via synthesis inhibition and induce the death of prostate cancer cells. Mol Cancer Ther. 2009;8:1340–1349. doi: 10.1158/1535-7163.MCT-08-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cunha AC, Weigle B, Kiessling A, Bachmann M, Rieber EP. Tissue-specificity of prostate specific antigens: Comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett. 2006;236:229–238. doi: 10.1016/j.canlet.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 47.Berges RR, Vukanovic J, Epstein JI, CarMichel M, Cisek L, Johnson DE, Veltri RW, Walsh PC, Isaacs JT. Implication of cell kinetic changes during the progression of human prostatic cancer. Clin Cancer Res. 1995;1:473–480. [PMC free article] [PubMed] [Google Scholar]
- 48.Pinski J, Parikh A, Bova GS, Isaacs JT. Therapeutic implications of enhanced G0/G1 checkpoint control induced by coculture of prostate cancer cells with osteoblasts. Cancer Res. 2001;61:6372–6376. [PubMed] [Google Scholar]
- 49.Fox SB, Gatter KC, Bicknell R, Going JJ, Stanton P, Cooke TG, Harris AL. Relationship of endothelial cell proliferation to tumor vascularity in human breast cancer. Cancer Res. 1993;53:4161–4163. [PubMed] [Google Scholar]
- 50.Di X, Nishizaki T, Harada K, Kajiwara K, Nakayama H, Ito H. Proliferative potentials of glioma cells and vascular components determined with monoclonal antibody MIB-1. J Exp Clin Cancer Res. 1997;16:153–157. [PubMed] [Google Scholar]
- 51.Hasebe T, Sasaki S, Sugitoh M, Ono M, Saitoh N, Ochiai A. Proliferative activities of tumor stromal cells play important roles in tumor thickness and progression of T3 ulcerative-type colorectal cancer. Virchows Arch. 2003;442:569–576. doi: 10.1007/s00428-003-0811-5. [DOI] [PubMed] [Google Scholar]
- 52.Jackson PF, Cole DC, Slusher BS, Stetz SL, Ross LE, Donzanti BA, Trainor DA. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated α-linked acidic dipeptidase. J Med Chem. 1996;39:619–622. doi: 10.1021/jm950801q. [DOI] [PubMed] [Google Scholar]
- 53.Dalrymple SL, Becker RE, Isaacs JT. The quinoline-3-carboxamide anti-angiogenic agent, tasquinimod, enhances the anti-prostate cancer efficacy of androgen ablation and taxotere without effecting serum PSA directly in human xenografts. Prostate. 2007;67:790–797. doi: 10.1002/pros.20573. [DOI] [PubMed] [Google Scholar]
- 54.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, Isaacs WB, Luo J, De Marzo AM. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu JM, Fackler MJ, Halushka MK, Molavi DW, Taylor ME, Teo WW, Griffin C, Fetting J, Davidson NE, De Marzo AM, Hicks JL, Chitale D, Ladanyi M, Sukumar S, Argani P. Heterogeneity of breast cancer metastases: Comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin Cancer Res. 2008;14:1938–1946. doi: 10.1158/1078-0432.CCR-07-4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kosk-Kosicka D. Measurement of Ca2+-ATPase activity (in PMCA and SERCA1) Methods Mol Biol. 1999;114:343–354. doi: 10.1385/1-59259-949-4:343. [DOI] [PubMed] [Google Scholar]
- 57.Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- 58.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 59.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 60.Kyprianou N, English HF, Davidson NE, Isaacs JT. Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation. Cancer Res. 1991;51:162–166. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Tumor endothelial cell staining scores for individual tumor types.
Table S2. Crystal structure data collection and refinement statistics.
Table S3. Effect of G202 on laboratory values in cynomolgus monkeys.
Fig. S1. PSMA hydrolysis of the 12ADT|3Asp-Glu dipeptide.
Fig. S2. Effect of G202 or docetaxel on body weight of LNCaP human prostate cancer xenograft-bearing mice.
Fig. S3. Serum concentrations of G202 over a 24-hour period in 18 individual patients who received G202 at doses ranging from 1.2 to 40 mg/m2 in a phase 1 clinical trial.
Fig. S4. Clonal survival of PC-3 human prostate cancer cells after repeated exposure to 500 nM TG.