

Abstract
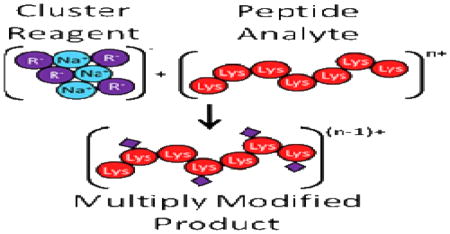
Multiple gas phase ion/ion covalent modifications of peptide and protein ions are demonstrated here using cluster-type reagent anions of N-hydroxysulfosuccinimide acetate (sulfo-NHS acetate) and 2-formyl-benzenesulfonic acid (FBMSA). These reagents are used here to selectively modify unprotonated primary amine functionalities of peptides and proteins. Multiple reactive reagent molecules can be present in a single cluster ion, which allows for multiple covalent modifications to be achieved in a single ion/ion encounter and at the ‘cost’ of only a single analyte charge. Multiple derivatizations are demonstrated when the number of available reactive sites on the analyte cation exceeds the number of reagent molecules in the anionic cluster (e.g., data shown here for reactions between the polypeptide [K10+3H]3+ and the reagent cluster [5R5Na-Na]−). This type of gas phase ion chemistry is also applicable to whole protein ions. Here, ubiquitin was successfully modified using an FBMSA cluster anions which, upon collisional activation, produced fragment ions with various numbers of modifications. Data for the pentamer cluster are included here as illustrative of the results obtained for the clusters comprised of 2–6 reagent molecules.
INTRODUCTION
Mass spectrometry (MS) and tandem mass spectrometry (MSn) approaches continue to play vital roles in the identification and structural characterization of peptides and proteins[1]. Solution phase chemical derivatization approaches are routinely employed to aid in these efforts in a number of ways [2,3]. Some recent uses include improving ionization characteristics [4], allowing accurate relative quantification [5,6], and facilitating structural characterization [7,8]. For example, N-hydroxysuccinimide (NHS) ester derivatives have been used to characterize the relative reactivities of primary amine-containing amino acid residues (viz. the ε-amino side chain of lysine and the N-terminus) in denatured and native-state ubiquitin within the context of a top-down Fourier transform ion cyclotron resonance (FTICR) experiment [9]. Derivatization techniques have also been employed for cross-linking purposes [10,11,12] and to attach chromophores to bioanalytes to facilitate photodissociation experiments [13]. While these techniques are normally performed in an off-line fashion, it has recently been demonstrated that reactive desorption electrospray ionization (DESI) can be used to perform rapid derivatization as part of the ionization process [14,15,16,17,18]. Each of these techniques has the potential to be useful in an array of proteomics applications.
Recently, it has been shown that these types of bioconjugation techniques, commonly conducted in solution, can also be performed in the gas phase via ion/ion reactions [19]. Some of the inherent properties of these gas phase reactions make gas phase bioconjugation an attractive option, relative to solution phase bioconjugation, in some protein analysis scenarios. For example, attractive aspects of the gas phase approach include short reaction times (i.e., on the order of 100 milliseconds), facile comparison between modified and unmodified analytes, a high degree of control over reactant identities via precursor mass selection, the avoidance of highly complex reaction mixtures often generated in solution phase approaches, and the ability to control the number of modifications by varying the reaction time and/or the identity of the reagent ion. Recent demonstrations of gas phase ion/ion bioconjugation include the use of NHS ester derivatives in cross-linking experiments [20,21], the use of NHS ester derivatives in covalent labeling studies [22,23,24], the use of 4-formyl-1,3-benzenedisulfonic acid (FBDSA) to tag and enhance the primary sequence coverage attained from collision induced dissociation (CID) experiments [25,26,27,28], and the use of N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC) in covalent labeling studies [29].
NHS ester and FBDSA reagents have chiefly been used to selectively target primary amine functionalities for modification via ion/ion reactions, though it has recently been shown that the guanidinium functionality of arginine can also undergo covalent modification by NHS ester derivatives under certain conditions [24]. A necessary condition for both of these gas-phase reaction types is the formation of a long-lived ion/ion reaction complex (i.e., composed of ionic forms of the analyte and reagent, at least one of which must be multiply charged). NHS ester modification then proceeds via nucleophilic attack of the carbonyl carbon of the NHS ester by an unprotonated primary amine on the peptide or protein, resulting in spontaneous or collision-induced signature neutral loss of either NHS or sulfo-NHS (depending on the reagent). FBDSA modification similarly proceeds via nucleophilic attack of the aldehyde by an unprotonated primary amine that results in signature water loss, either spontaneously or as a result of CID, and the formation of an imine bond (i.e., a Schiff base formation).
The extent to which the gas phase reactions can be performed on a given system (i.e., the maximum number of modifications that can be made to any one analyte) is limited by the number of available reaction sites in the analyte (e.g., neutral primary amines) and the initial charge state of the analyte. If the number of reactive primary amines is greater than one, a situation common with many protein ions, for example, the number of available reaction sites in the analyte is then larger than the number of covalent modifications that can be formed in any single ion/ion transformation with a single reagent ion with only one reactive site. In some experiments, it may be desirable to perform multiple modifications (e.g., to attach multiple chromophores that, upon laser irradiation, would facilitate dissociation). Ultimately, the number of ion/ion reactions that can be performed for any multiply charged analyte ion (e.g., of the general form [M+nH]n+) is one less than the total number of analyte charges (or simply n−1), neglecting the possibility of charge inversion. As such, while sequential ion/ion reactions could be used to effect multiple modifications, the system would ultimately be limited by the initial charge state of the analyte ion. It is therefore desirable to develop a method by which multiple gas phase derivatizations can be performed in a single ion/ion reaction (i.e., in parallel and at the cost of only a single analyte charge).
Cluster ions provide the possibility of effecting multiple reactions in a single ion/ion encounter. Historically, cluster ion types have seen use as primary ion sources in secondary ion mass spectrometry (SIMS) [30,31]. Additionally, the clustering of ions is of fundamental importance in ion/molecule reactions [32,33,34] and in ion-mobility spectrometry (IMS) [35,36]. However, cluster ions as they relate to the field of proteomics, especially with respect to electrospray ionization (ESI), are typically minimized to avoid compromising mass measurements. These types of clusters are typically composed of an analyte ion and any number of neutral solvent molecules and/or salt adducts. Most commercial electrospray sources employ interfaces designed to reduce or eliminate these clustering effects [37,38]. However, a cluster ion composed of multiple reagent molecules (and a single charge-bearing ion) could be used to result in multiple reactions within the context of a single ion/ion counter. Previously, multiple proton transfer reactions have been performed in a single ion/ion encounter using dendrimer reagent ions [39,40,41,42,43,44,45,46]. During the long-lived ion/ion complex, multiple proton transfers can occur, indicating that performing multiple reactions within a single ion/ion complex is indeed possible. Proton hydrate clusters have also been used previously in ion/ion reactions as “soft” proton transfer reagents [47].
Here, we demonstrate the modification of multiple primary amine functionalities in a single ion/ion encounter using reagent ion clusters. Specifically, sulfo-NHS acetate and 2-formyl-benzenesulfonic acid (termed FBMSA for our purposes so as to explicitly distinguish this reagent from the aforementioned FBDSA) reagent anion clusters are used to modify peptide and protein analyte cations.
EXPERIMENTAL
Materials
FBMSA and bovine ubiquitin were purchased from Sigma-Aldrich (St. Louis, MO). Sulfo-NHS acetate was purchased from PierceNet (Thermo Fisher Scientific Inc., Rockford, IL). FBMSA, which also leads to gas phase Schiff base chemistry, is used here because the cluster reagents ions are more easily generated than with FBDSA. The peptide KKKKKKKKKK (K10) was purchased from Pepnome Limited (Jida Zhuhai City, Guangdong Province, China). Methanol was purchased from Mallinckrodt (Phillipsburg, NJ). Ubiquitin and peptide stock solutions were prepared in a 50/50 (v/v) solution of water/methanol at an initial concentration of ~1 mg/mL and then diluted up to 100-fold prior to use. FBMSA and sulfo-NHS acetate solutions were prepared in water at concentrations of ~1 mg/mL. The high concentration of the reagent solutions aided in the formation of the cluster ions upon nESI.
Mass Spectrometry
All experiments were performed on a QTRAP 4000 hybrid triple quadrupole/linear ion trap mass spectrometer (AB Sciex, Concord, ON Canada) modified for ion/ion reactions as described previously48. Briefly, cation (multiply charged protein or peptide analyte) and anion (singly charged reagent cluster) populations are sequentially injected into the q2 reaction cell using alternatively pulsed nanoelectrospray ionization (nESI) [49,50]. The precursor ions are each isolated prior to injection into the q2 cell using the Q1 mass filter. Following a defined mutual storage ion/ion reaction period (typically 500 to 1500 milliseconds), the product ions are transmitted from q2 to the Q3 linear ion trap where subsequent MSn and mass analysis via mass-selective axial ejection are performed [51].
RESULTS
Recent demonstrations of sulfo-NHS ester and FBDSA gas phase reactivities have been limited to derivatizations using a single reagent. While multiple derivatizations could be performed sequentially (i.e., by increasing the reaction time or by increasing the number density of the reagent ions with respect to the analyte ions), each ion/ion reaction takes place at the cost of a charge. The number of possible modifications before reaching neutralization is therefore highly dependent on the initial charge state of the analyte (assuming an excess of reactive sites on the analyte). A method to circumvent this restriction is to alter the reagent ion type to an ion which contains multiple reactive reagent molecules, but which is still singly charged. For sulfo-NHS acetate and FBMSA, these clusters take the form of [nRnNa-Na]−, where n represents the number of reagent molecules present and R is the reagent anion (in Figure 1, R is used to denote sulfo-NHS acetate (Fig. 1a) and R is used to denote FBMSA (Fig. 1b). The sodium cations act as counter ions within the cluster, stabilizing the sulfonate moieties. Upon nanoelectrospray ionization, one counter ion is lost, forming singly deprotonated reagent clusters containing various numbers of reagent molecules.
Figure 1.

nESI generated clusters of (a) sulfo-NHS acetate (see structure inset), denoted in this text as R, and (b) FBMSA (see structure inset), denoted in this text as R. The ‘Na’ subscripts indicate the reagent counter ion, which is equal to the number of reagent molecules present (one of which is subsequently lost upon ionization to generate the anionic cluster). The unlabeled ions represent partially fragmented clusters and/or clusters with a slightly different chemical makeup.
Sulfo-NHS Acetate
Mass selection of the desired reagent cluster can be performed in the Q1 mass filter prior to the mutual storage ion/ion reaction with the analyte. This process is first demonstrated with sulfo-NHS acetate (Figure 2). The ion/ion reaction between [K10+3H]3+ and [2R2Na-Na]− mainly produces a charged reduced complex, [M+3H+2R2Na-Na]2+, which is stabilized by an electrostatic interaction between the anionic sulfonate and a protonated site on the peptide (Figure 2a). Proton transfer, a competing reaction pathway that leads to [M+2H]2+, is minimal in this instance. The neutral loss of a sulfo-NHS acetate molecule (with a sodium counter ion) from the ion/ion complex is also present. Unmodified peptide fragment ions (b82+ and b92+) arise from CID resulting from energetic ion transfer conditions between the q2 reaction cell and Q3 linear ion trap. Subsequent isolation and ion trap CID of the charge-reduced ion/ion complex results in losses of one and two of sulfo-NHS molecules, neutral losses which have been identified as signatures for covalent bond formation with a primary amine functionality (Figure 2b). Although there is a distribution of sulfo-NHS losses containing either a proton or sodium counter ion, there are only up to two acetylation modifications observed, as would be expected based on the initial reagent dimer cluster. Subsequent CID of the first sulfo-NHS loss reveals only one further signature sulfo-NHS loss and confirms this observation (Figure 2c).
Figure 2.

(a) An ion/ion reaction between the triply protonated K10 peptide, [M+3H]3+, and dimer sulfo-NHS acetate reagent cluster, [2R2Na-Na]−, results mainly in complex formation. (b) Ion trap CID, denoted by the lightning bolt, of the ion/ion complex, [M+3H+2R2Na-Na]2+, results in up to 2 sulfo-NHS signature neutral losses. (c) Subsequent CID of the first sulfo-NHS loss reveals a second sulfo-NHS loss, a neutral loss of an intact sulfo-NHS acetate reagent molecule, and several sequence ions containing 2 covalent modifications (denoted by asterisks). The ‘Na’ and ‘H’ subscripts denote the counter ion associated with the indicated neutral loss.
The K10 peptide analyte very likely contains more reactive primary amines than were derivatized in the experiment depicted by Figure 2. K10 has 11 primary amines (10 ε-amino side chains and the N-terminus), of which 3 are protonated upon ionization, leaving up to 8 available for covalent modification (though intramolecular charge solvation by multiple amino groups may reduce the reactivities of some of the potential reactive sites). Therefore, employing reagent anion clusters that contain additional sulfo-NHS acetate molecules (R>2) should allow for further gas phase derivatizations of the peptide beyond the number possible via sequential ion/ion reactions (i.e., given the initial charge state of the peptide is 3+, only 2 sequential ion/ion derivatizations could be performed before neutralizing the analyte). CID of ion/ion complexes formed using sulfo-NHS acetate reagent clusters composed of 3 and 4 sulfo-NHS acetate molecules is shown in Figures 3a and 3c, respectively. CID of the ion/ion complex formed using the trimer reagent cluster, [3R3Na-Na]−, results in a similar distribution to that observed in Figure 2b, except now up to 3 signature sulfo-NHS losses (with both proton, and sodium counter ions) are observed (Figure 3a). The sodium counter ions originate from the reagent cluster while the proton counter ions originate from the peptide. CID of the second sulfo-NHS loss confirms that only 3 covalent modifications are possible with this trimer reagent cluster (Figure 3b). CID of the ion/ion complex formed using the tetramer reagent cluster, [4R4Na-Na]−, behaves in much the same way as the dimer and trimer sulfo-NHS acetate clusters (Figure 3c). Up to 4 signature sulfo-NHS losses are observed, as confirmed by CID of the second sulfo-NHS loss (Figure 3d). Loss of an intact neutral sulfo-NHS acetate reagent molecule with either a proton or sodium counter ion (−RH/Na) represents a minor competing pathway.
Figure 3.

(a) CID of the ion/ion complex formed using the trimer reagent cluster, [M+3H+3R3Na-Na]2+, results in up to 3 sulfo-NHS signature neutral losses. (b) Subsequent CID of the second sulfo-NHS loss confirms only a third signature loss and concomitant analyte derivatization is possible with the trimer reagent. (c) CID of the ion/ion complex formed using the tetramer reagent cluster, [M+3H+4R4Na-Na]2+, results in up to 4 sulfo-NHS signature neutral losses. (d) Subsequent CID of the second sulfo-NHS loss confirms that only up to four modifications are possible with the tetramer reagent cluster.
An interesting feature of the spectra shown in Figures 2 and 3 is the competition in the neutral loss channels between proton and sodium counter ions. CID of the ion/ion complexes shows that proton counter ion loss (i.e., −sulfo-NHSH) is initially favored over sodium counter ion loss (i.e., −sulfo-NHSNa) for the first neutral loss (Figures 2b, 3a, and 3c). After the first loss, sequential fragmentation and statistical availability of the counter ions cloud further interpretation. However, the relative abundances of the proton and sodium counter ion losses produced via CID of the ion/ion complex stand in contrast to neutral losses observed in the ion/ion reaction (Figure 2a). Based on the reported relative proton and sodium ion affinities of lysine [52] and sulfate [53,54] functionalities, the loss of sulfo-NHSH is thermodynamically favored over loss of sulfo-NHSNa by about 6 kcal/mol (Table 1), which is consistent with the CID result. The favored loss of RNa in the ion/ion reaction spectrum (Figure 2a) is not consistent with expectation based on thermodynamic grounds. This observation is presumed to be due to the much shorter lifetimes of the complexes that spontaneously lose RNa as a result of the ion/ion reaction. The long-lived (> 100 ms) complexes that survive long enough to be isolated and undergo CID have far more time to undergo cation exchange as well as covalent reactions.
Table 1.
Estimated proton and sodium affinities of sulfate and lysine. For the sulfate moiety, the proton affinity of hydrogen sulfate53 and the sodium affinity of methyl sulfate54 were used.
| Functional Group | Proton Affinity (kcal/mol) | Sodium Affinity (kcal/mol) |
|---|---|---|
| Sulfate | 320 | 127 |
| Lysine | 238 | 51 |
| Δ(sulfate-lysine) | 82 | 76 |
FBMSA
Multiple gas phase derivatizations are also possible using FBMSA-containing reagent clusters. FBMSA covalent derivatization proceeds via a similar nucleophilic attack mechanism as does FBDSA, with a neutral H2O loss representing the signature for covalent imide bond formation. Di-, tri-, tetra-, and pentamer FBMSA anionic clusters (R=2–5) react with triply protonated K10 to give as many signature water losses from the ion/ion complexes as there are reagent molecules present in the initial FBMSA cluster (Figure 4a–d). Similar to sulfo-NHS acetate, proton transfer (i.e., [M+2H]2+) and intact FBMSA reagent neutral loss with a sodium counter ion [e.g., −(H2O−RNa) in Figure 4a] compete with covalent bond formation during the ion/ion reaction. As would be expected, the summed number of intact neutral FMBSA losses (R) and signature water losses never exceeds the number of reagent molecules in the initial cluster. For example, with the pentamer FBMSA cluster (Figure 4d), neutral losses of 1 water and 4 intact FBMSA molecules [− (H2O+4R4Na)], 2 waters and 3 intact FBMSA molecules [− (2H2O+3R3Na)], 4 waters and 1 intact FBMSA molecule [− (4H2O+RNa)], and 5 waters ([M+3H+5R5Na-Na-5H2O]2+) are all present. In contrast to sulfo-NHS acetate, however, no supplemental activation step (i.e., CID) is necessary to drive off the neutral product of the reaction (i.e., H2O) in the case of FBMSA. Water loss appears to occur spontaneously from the ion/ion complex (e.g., [M+3H+2R2Na-Na-2H2O]2+ in Figure 4a), though some degree of ion activation due to energetic transfer conditions between the q2 reaction cell and the Q3 linear ion trap cannot be precluded. In any case, no intact ion/ion reaction complex (i.e., [M+3H+nRnNa-Na]2+) for any of the experiments associated with Figure 4 survives to detection. The minimal need for activation of the initial complex formed with FBMSA clusters, in contrast to the case for the sulfo-NHS ester clusters, is likely due, at least in part, to the fact that the neutral product of the reaction (i.e., water) does not interact strongly with the peptide, in contrast with the relatively strong electrostatic interaction of the sulfo-NHS group with the peptide in the latter case.
Figure 4.

Ion/ion reactions between [K10+3H]3+ and FBMSA reagent clusters (a) [2R2Na-Na]−, (b) [3R3Na-Na]−, (c) [4R4Na-Na]−, and (d) [5R5Na-Na]−.
The effectiveness of reagent cluster anions for the multiple modification of an intact protein ion was explored with cations of ubiquitin. Ubiquitin, a ~8.5 kDa protein, has a total of 13 basic sites, including 4 arginines, 7 lysines, 1 histidine, and the N-terminus. The 7+ charge state of ubiquitin was subjected to separate ion/ion reaction experiments with deprotonated FBMSA (i.e., [R-H]−) and [nRnNa-Na]− clusters, where n=2–6. In each case, the maximum number of water losses from the ion/ion reaction complex was found to be equal to the number of FBMSA molecules in the reagent anion. The results for the ion/ion reaction between the 7+ charge state of ubiquitin and the pentamer FBMSA cluster ([5R5Na-Na]−) are shown in Figure 5 as an illustrative case. Firgure 5a is the post-ion/ion reaction spectrum resulting from the reaction of [M+7H]7+ with [5R5Na-Na]−, which shows residual unreacted [M+7H]7+ and the ion/ion reaction complex with various numbers of water losses, as highlighted in the insert. The ions in the insert of 5a were subsequently isolated and swept through a resonance excitation voltage designed to sequentially activate each of the ions in the insert of Figure 5a, the results being shown in Figure 5b. Activation of the ion/ion reaction complex ions of Figure 5a results in a mixture of products that imply varying extents of covalent reaction. For example, the loss of five water molecules is the dominant product among the water losses from the initially formed complex. While water loss is common from protonated peptide and protein ions, the fact that five water losses is the dominant product implies the formation of a [M+7H+5⋄5Na-Na]6+ species, where the ⋄ is used to signify a covalent modification. Likewise, for all other reagents of the form [nRnNa-Na]− the loss of n water molecules was observed as the dominant number of water molecule loss from the initial ion/ion reaction complex (data not shown). However, the possibility for some of the water loss associated with the ion/ion reaction complexes arising from water loss from elsewhere in the protein cannot be precluded. Major products in Figure 5b were also observed for the loss of a single intact RH group along with loss of 4 water molecules and loss of two RH molecules in conjunction with loss of 3 water molecules. The latter products suggest that one and two, respectively, of the reagent cluster components did not undergo a covalent reaction with the protein while the remainder of the cluster components (i.e., 4 and 3, respectively) engaged in a covalent reaction.
Figure 5.

(a) Post-ion/ion reaction spectrum of ubiquitin [M+7H]7+ with FBMSA [5R5Na-Na]−. (b) Product ions generated by a rapid sequential collisional activation step applied to the isolated population of ions shown in the insert of (a).
Figure 6 compares the ion trap CID spectra of the ubiquitin [M+6H]6+ ion (Figure 6a), the ubiquitin [M+6H+⋄]6+ ion generated by ion/ion reaction of [M+7H]7+ and [R-Na]− (Figure 6b), and ubiquitin [M+7H+5R5Na-Na-5H2O]6+ generated in the experiments described for Figure 5 (Figure 6c). The latter ion is also indicated as [M+7H+5⋄5Na-Na]6+, under the assumption that all five water losses are associated with covalent reactions. We note that a peak for loss of RNa is apparent in Figure 6c, which indicates that at least some of the water loss is not associated with covalent bond formation. However, little or no evidence for losses of more than one intact reagent molecules is observed, which suggests that most of the observed water loss is due to covalent bond formation associated with the Schiff base reaction. Fragment ions with 1, 2, 3, and 4 covalent derivatizations are all readily apparent in the product ion spectrum of Figure 6c, which show that at least some of the protein ions underwent four covalent modifications. As some of the modified ions are likely to be complementary products (e.g., the (y24⋄+Na)2+/(b524⋄+3Na)4+ ions), the covalent modification of 5 sites in some of the protein cannot be precluded. We note that in no cases are any fragment ions observed to contain more modifications than there are lysine residues in the fragment, which is consistent with previous observations that neutral primary amines are the major reactive sites of peptides with FBMSA anions. Collectively, the water and RH loss products and the CID data of the modified ubiquitin ions are all consistent with multiple modifications of a whole protein ion in a single ion/ion encounter with a reagent cluster anion.
Figure 6.

CID of (a) the 6+ charge state of unmodified ubiquitin, (b) singly modified ubiquitin, and (c) ubiquitin modified using a pentamer FBMSA cluster. Hollow diamonds (◇) are used to denote the dehydrated product ion and correspond to covalently modified peaks.
CONCLUSIONS
Multiple gas phase covalent modifications of peptide and protein cations have been demonstrated in a single ion/ion encounter using sulfo-NHS acetate and FBMSA reagent cluster anions. Employing clusters as reagent ion types allows for the presence of multiple reactive reagent molecules in a single ion. Additionally, the nature of the cluster is such that there is one fewer sodium counter ion than reagent molecule present, resulting in a net cluster charge of 1-, irrespective of the number of reagent molecules present in the cluster. This allows for multiple gas phase modifications at the ‘cost’ of only a single analyte charge and in a single ion/ion encounter. With FBMSA reagent clusters the covalent modification process requires little or no supplemental activation, whereas with sulfo-NHS acetate reagent clusters the resulting charge-reduced ion/ion complex requires supplemental activation (CID) in order to remove the relatively strongly bound neutral product(s) of the reaction (i.e., sulfo-NHS molecules). This chemistry has been demonstrated to occur when the number of available reactive sites on the analyte cation exceeds the number of reagent molecules in the anionic cluster (e.g., data shown here for reactions between [K10+3H]3+ and [5R5Na-Na]−). The number of modifications observed does not exceed the number of reagents present in the initial cluster or the number of unprotonated primary amines in the analyte, whichever is less. Ubiquitin cations have been covalently modified using FBMSA clusters of the form, [nRnNa-Na]− for n=2–6. Ion trap CID product ions that have lost multiple water molecules, which is consistent with multiple covalent modifications, such as the ([M+7H+5⋄5Na-Na]6+) product ion, for example, produced fragment ions with various numbers of covalent modification. The results shown illustrate the use of reagent cluster ions as means for effecting multiple selective covalent modifications of a multiply charged analyte ion.
Acknowledgments
This work was sponsored by AB Sciex and by the National Institutes of Health under Grant GM 45372. B.M.P. acknowledges receipt of a Purdue University Bilsland Dissertation Fellowship.
References
- 1.Biemann K. Mass Spectrometry of Peptides and Proteins. Annu Rev Biochem. 1992;61:977–1010. doi: 10.1146/annurev.bi.61.070192.004553. [DOI] [PubMed] [Google Scholar]
- 2.Halket JM, Zaikin VG. Derivatization in mass spectrometry – 1. Silylation. Eur J Mass Spectrom. 2003;9:1–21. doi: 10.1255/ejms.527. [DOI] [PubMed] [Google Scholar]
- 3.Hermanson GT. Bioconjugate techniques. 2. Academic Press; Amsterdam: 1998. [Google Scholar]
- 4.Yang WC, Mirzaei H, Liu XP, Regnier RE. Enhancement of amino acid detection and quantification by electrospray ionization mass spectrometry. Anal Chem. 2006;78:4702–4708. doi: 10.1021/ac0600510. [DOI] [PubMed] [Google Scholar]
- 5.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 6.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Beardsley RL, Sharon LA, Reilly JP. Peptide de novo sequencing facilitated by a dual-labeling strategy. Anal Chem. 2005;77:6300–6309. doi: 10.1021/ac050540k. [DOI] [PubMed] [Google Scholar]
- 8.Madsen JA, Brodbelt JS. Simplifying fragmentation patterns of multiply charged peptides by N-terminal derivatization and electron transfer collision activated dissociation. Anal Chem. 2009;81:3645–3653. doi: 10.1021/ac9000942. [DOI] [PubMed] [Google Scholar]
- 9.Novak P, Kruppa GH, Young MM, Schoeniger J. A top-down method for the determination of residue-specific solvent accessibility in proteins. J Mass Spectrom. 2004;39:322–328. doi: 10.1002/jms.587. [DOI] [PubMed] [Google Scholar]
- 10.Tubb MR, Silva RAGD, Fang J, Tso P, Davidson WS. Lipids and lipoproteins: metabolism, regulation, and signaling. J Biol Chem. 2008;283:17314–17323. doi: 10.1074/jbc.M800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh P, Panchaud A, Goodlett DR. Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal Chem. 2010;82:2636–2642. doi: 10.1021/ac1000724. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowsk-Willis JC, Nilges M, Cramer P, Rappsilber J. Architecture of the RNA polymerase II–TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasicek L, Brodbelt JS. Enhancement of ultraviolet photodissociation efficiencies through attachment of aromatic chromophores. Anal Chem. 2010;82:9441–9446. doi: 10.1021/ac102126s. [DOI] [PubMed] [Google Scholar]
- 14.Venter A, Sojka PE, Cooks RG. Droplet dynamics and ionization mechanisms in desorption electrospray ionization mass spectrometry. Anal Chem. 2006;78:8549–8555. doi: 10.1021/ac0615807. [DOI] [PubMed] [Google Scholar]
- 15.Girod M, Moyano E, Campbell DI, Cooks RG. Accelerated bimolecular reactions in microdroplets studied by desorption electrospray ionization mass spectrometry. Chem Sci. 2011;2:501–510. [Google Scholar]
- 16.Miao Z, Chen H. Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS) J Am Soc Mass Spectrom. 2009;20:10–19. doi: 10.1016/j.jasms.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 17.Perry RH, Splendore M, Chien A, Davis NK, Zare RN. Detecting Reaction Intermediates in Liquids on the Millisecond Time Scale Using Desorption Electrospray Ionization. Angew Chem Int Ed. 2011;50:250–254. doi: 10.1002/anie.201004861. [DOI] [PubMed] [Google Scholar]
- 18.Badu-Tawiah AK, Li A, Jjunju FPM, Cooks RG. Peptide cross-linking at ambient surfaces by reactions of nanosprayed molecular cations. Angew Chem Int Ed. 2012;51:9417–9421. doi: 10.1002/anie.201205044. [DOI] [PubMed] [Google Scholar]
- 19.Prentice BM, McLuckey SA. Gas-phase ion/ion reactions of peptides and proteins: acid/base, redox, and covalent chemistries. Chem Comm. 2013;49:935–942. doi: 10.1039/c2cc36577d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mentinova M, McLuckey SA. Intra- and inter-molecular cross-linking of peptide ions in the gas-phase: reagents and conditions. J Am Soc Mass Spectrom. 2011;22:912–921. doi: 10.1007/s13361-011-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb IK, Mentinova M, McLuckey SA. Gas-phase intramolecular protein crosslinking via ion/ion reactions: ubiquitin and a homobifunctional sulfo-NHS ester. J Am Soc Mass Spectrom. doi: 10.1007/s13361-013-0590-4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mentinova M, McLuckey SA. Covalent modification of gaseous peptide ions with N-hydroxysuccinimide ester reagent ions. J Am Chem Soc. 2010;132:18248–18257. doi: 10.1021/ja107286p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mentinova M, Barefoot NZ, McLuckey SA. Solution versus gas-phase modification of peptide cations with NHS-ester reagents. J Am Soc Mass Spectrom. 2011;23:282–289. doi: 10.1007/s13361-011-0291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGee WM, Mentinova M, McLuckey SA. Gas-phase conjugation to arginine residues in polypeptide ions via N-hydroxysuccinimide ester-based reagent ions. J Am Chem Soc. 2012;134:11412–11414. doi: 10.1021/ja304778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han H, McLuckey SA. Selective covalent bond formation in polypeptide ions via gas-phase ion/ion reaction chemistry. J Am Chem Soc. 2009;131:12884–12885. doi: 10.1021/ja904812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassell KM, Stutzman JR, McLuckey SA. Gas-phase bioconjugation of peptides via ion/ion charge inversion: Schiff base formation on the conversion of cations to anions. Anal Chem. 2010;82:1594–1597. doi: 10.1021/ac902732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stutzman JR, Hassel KM, McLuckey SA. Dissociation behavior of tryptic and intramolecular disulfide-linked peptide ions modified in the gas phase via ion/ion reactions. Int J Mass Spectrom. 2012;312:195–200. doi: 10.1016/j.ijms.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stutzman JR, McLuckey SA. Ion/ion reactions of MALDI-derived peptide ions: increased sequence coverage via covalent and electrostatic modification upon charge inversion. Anal Chem. 2012;84:10679–10685. doi: 10.1021/ac302374p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prentice BM, Gilbert JD, Stutzman JR, Forrest WP, McLuckey SA. Gas-phase reactivity of carboxylic acid functional groups with carbodiimides. J Am Soc Mass Spectrom. 2013;24:30–37. doi: 10.1007/s13361-012-0506-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gillen G, Roberson S. Preliminary evaluation of an SF5+ polyatomic primary ion beam for analysis of organic thin films by seconday ion mass spectrometry. Rapid Commun Mass Spectrom. 1998;12:1303–1312. doi: 10.1002/(SICI)1097-0231(19981015)12:19<1303::AID-RCM330>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 31.Mahoney CM. Cluster secondary ion mass spectrometry of polymers and related materials. Mass Spectrom Rev. 2010;29:247–293. doi: 10.1002/mas.20233. [DOI] [PubMed] [Google Scholar]
- 32.McLuckey SA, Van Berkel GJ, Glish GL. Reactions of dimethylamine with multiply charged ions of cytochrome c. J Am Chem Soc. 1990;112:5668–5670. [Google Scholar]
- 33.McLuckey SA, Glish GL, Van Berkel GJ. Charge determination of product ions formed from collision-induced dissociation of multiply protonated molecules via ion/molecule reactions. Anal Chem. 1991;63:1971–1978. doi: 10.1021/ac00018a014. [DOI] [PubMed] [Google Scholar]
- 34.Brutschy B. Ion-molecule reactions within molecular clusters. Chem Rev. 1992;92:1567–1587. [Google Scholar]
- 35.Spangler GE. Fundamental considerations for the application of miniature ion mobility spectrometry to field analytical applications. Field Anal Chem Technol. 2000;4:255–267. [Google Scholar]
- 36.Krylova N, Krylov E, Eiceman GA, Stone JA. Effect of moisture on the field dependence of mobility for gas-phase ions of organophosphorus compounds at atmospheric pressure with field asymmetric ion mobility spectrometry. J Phys Chem A. 2003;107:3648–3654. doi: 10.1021/jp0221136. [DOI] [PubMed] [Google Scholar]
- 37.Bruins AP, Covey TR, Henion JD. Ion spray interface for combined liquid chromatography/atmospheric pressure ionization mass spectrometry. Anal Chem. 1987;59:2642–2646. [Google Scholar]
- 38.Chowdhury SK, Katta V, Chait BT. An electrospray-ionization mass spectrometer with new features. Rapid Commun Mass Spectrom. 1990;4:81–87. doi: 10.1002/rcm.1290040305. [DOI] [PubMed] [Google Scholar]
- 39.He M, McLuckey SA. Two ion/ion charge inversion steps to form a doubly protonated peptide from a singly protonated peptide in the gas phase. J Am Chem Soc. 2003;125:7756–7757. doi: 10.1021/ja0354521. [DOI] [PubMed] [Google Scholar]
- 40.He M, McLuckey SA. Increasing the negative charge of a macroanion in the gas phase via sequential charge inversion reactions. Anal Chem. 76:41889–4192. doi: 10.1021/ac496087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He M, Emory JF, McLuckey SA. Reagent anions for charge inversion of polypeptide/protein cations in the gas phase. Anal Chem. 2005;77:3173–3182. doi: 10.1021/ac0482312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gunawardena HP, Emory JF, McLuckey SA. Phosphopeptide anion characterization via sequential charge inversion and electron-transfer dissociation. Anal Chem. 2006;78:3788–3793. doi: 10.1021/ac060164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emory JF, McLuckey SA. Charge inversion of polypeptide anions using protein and dendrimer cations as charge inversion reagents. Int J Mass Spectrom. 2008;276:102–109. [Google Scholar]
- 44.Emory JF, McLuckey SA. The role of amino acid composition in the charge inversion of deprotonated peptides via gas-phase ion/ion reactions. J Am Soc Mas Spectrom. 2009;20:180–187. doi: 10.1016/j.jasms.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 45.Hassell K, LeBlanc Y, McLuckey SA. Conversion of multiple analyte cation types to a single analyte anion type via ion/ion charge inversion. Analyst. 2009;134:2262–2266. doi: 10.1039/b914304a. [DOI] [PubMed] [Google Scholar]
- 46.Hassell K, LeBlanc Y, McLuckey SA. Chemical noise reduction via mass spectrometry and ion/ion charge inversion: amino acids. Anal Chem. 2011;83:3252–3255. doi: 10.1021/ac200439k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bower JJ, Hodges BDM, Saad OM, Leary JA, McLuckey SA. Proton hydrates as soft ion/ion proton transfer reagents for multiply deprotonated biomolecules. Int J Mass Spectrom. 2008;276:153–159. [Google Scholar]
- 48.Xia Y, Wu J, Londry FA, Hager JW, McLuckey SA. Mutual storage mode ion/ion reactions in a hybrid linear ion trap. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 49.Liang X, Xia Y, McLuckey SA. Alternately pulsed nano-electrospray ionization/atmospheric pressure chemical ionization for ion/ion reactions in an electrodynamic ion trap. Anal Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang X, Han H, Xia Y, McLuckey SA. A pulsed triple ionization source for sequential ion/ion reactions in an electrodynamic ion trap. J Am Soc Mass Spectrom. 2007;18:369–376. doi: 10.1016/j.jasms.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Londry FA, Hager JW. Mass selective axial ion ejection from a linear quadrupole ion trap. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 52.Kish MM, Ohanessian H, Wesdemiotis C. The Na+ affinities of α-amino acids: side-chain substituent effects. Int J Mass Spectrom. 2003;227:509–524. [Google Scholar]
- 53.House JE, Jr, Kemper KA. Proton affinities of sulfate and bisulfate ions. J Therm Anal. 1987;32:1855–1858. [Google Scholar]
- 54.Remko M, Van Duijnen PTh, von der Lieth C. Structure and stability of Li(I) and Na(I)- carboxylate, sulfate and phosphate complexes. J Mol Struct THEOCHEM. 2007;814:119–125. [Google Scholar]