

Abstract
A new potent antiinfective and antiparasitic 2,3-dihydro-1H-indolizinium chloride, (1), was isolated from Prosopis glandulosa Torr. var. glandulosa. Three additional new (2–4) and one known (5), indolizidines were also isolated, and the dihydrochloride salts of 1–3 (compounds 6, 7 and 8) were prepared. Structures were determined by 1D and 2D NMR and mass spectra. Compound 1 showed potent in vitro antifungal activity against Cryptococcus neoformans and Aspergillus fumigatus (IC50 values = 0.4 and 3.0 μg/mL, respectively), and antibacterial activity against methicillin-resistant Staphylococcus aureus and Mycobacterium intracellulare (IC50 values of 0.35 and 0.9 μg/mL, respectively). The remarkable in vitro fungicidal activity of 1–4 against C. neoformans (MFCs = 0.63→1.25 μg/mL) and 2, 3, and 5 against A. fumigatus (MFCs = 0.63→2.5 μg/mL) were similar to amphotericin B, but >2–4-fold more potent than 6–8. Prosopilosidine (1) showed potent in vivo activity at 0.0625 mg/Kg/day/ip for 5 days in a murine model of cryptococcosis by eliminating ~76% of C. neoformans infection from brain tissue compared to ~83% with amphotericin B at 1.5 mg/Kg/day. Compounds 1 and 4 exhibited potent activity and high selectivity index (SI) values against chloroquine sensitive (D6) and chloroquine resistant (W2) strains of Plasmodium falciparum with IC50 values of 39 and 95 ng/mL, and 42 and 120 ng/mL, respectively; (chloroquine, IC50 = 17 and 140 ng/mL). Prosopilosine (1) also showed in vivo antimalarial activity with an ED50 value of ~2 mg/Kg/day/ip against Plasmodium berghei-infected mice after 3 days of treatment.
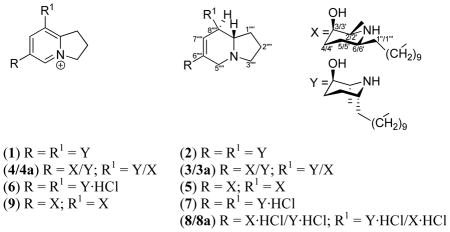
Plants of the genus Prosopis are trees or shrubs distributed in arid and semiarid tropical and subtropical regions. P. glandulosa Torrey var. glandulosa (Leguminosae), a medium-sized tree, is one of the two varieties of honey mesquite available in North America.1,2 Generally, mesquite is a popular adjuvant for preparing smoked cuisine in the southern part of the U.S. Many tribes of the southwestern United States and Mexico have long utilized the medicinal values of this plant, including treatment of eye infections, open wounds, dermatological ailments and stomach problems.3,4 Decoctions of leaves and pods are generally used to make eye washes to treat pink eye.5 An ethanolic extract of P. glandulosa from Pakistan yielded triterpenes, flavonoids, glycosides and the indolizidine alkaloid juliprosopine.6–10 Among the indolizidines reported from Prosopis species to date,6–11 the stereochemistry of juliprosine and juliprosopine were established by chemical synthesis.12 The piperidinyl indolizidines, such as juliprosopine, and their analogs exhibited in vitro antibmicrobial, antidermatophytic, pesticidal, and amebicidal activities.11,13–20 In addition, their toxicity,20–22 DNA binding activity23, inhibitory effects on β-glucosidase enzymes23 and plant growth inhibitory activities were also reported.10,24 This paper describes the isolation of the new potent antifungal and antimalarial dihydroindolizinium chloride prosopilosidine (1) from honey mesquite, together with three new analogs prosopilosine (2), isoprosopilosine (3) and isoprosopilosidine (4), and the known juliprosopine (5).25,26 P. glandulosa Torrey var. glandulosa, has not previously been subjected to chemical or biological investigations. Prosopilosidine (1) possesses the pharmacophore 2,3-dihydro-1H-indolizinium, a quaternary salt substituted with two units of 10-(5R-hydroxy-6R-methylpiperidin-2R-yl)decyl at C-6‴ and C-8‴ positions. In this paper, we report the isolation, structure elucidation, and in vitro and in vivo antiinfective, antimalarial and cytoxic evaluation of these compounds.
Results and Discussion
An EtOH extract of P. glandulosa var. glandulosa leaves showed weak in vitro antiinfective and antiparasitic activities, but the presence of alkaloids warranted bioassay-guided fractionation. The bioactivity of the EtOH extract was significantly increased in the alkaloid-enriched CH2Cl2 fraction. Column chromatography followed by centrifugal preparative TLC of the CH2Cl2 fraction resulted in the isolation of compounds 1–4, juliprosopine (5)25,26 and tryptamine,23 in the yields of 0.07%, 0.015%, 0.08%, 0.03%, 0.04% and 0.11%, respectively.
Prosopilosidine (1) was analyzed for C40H72N3O2 by electro-spray isonization high-resolution mass spectroscopy (ESIHRMS). The UV spectrum of 1 demonstrated absorption bands at λmax 198 and 230 nm, typical of indolizinium chromophore.8 The NMR spectra of 1 revealed a 2,3-dihydro-1H-indolizinium nucleus8,9 consisting of two trisubstituted and a tetrasubstituted olefin [δH-5′8.64 and δH-7″″ 8.18, each 1H, s; δC 137.3, 144.3 (2 × d); δC 139.1, 141.3, 155.0 (3 × s)], and three methylenes at δC-1″″ 31.6, δC-2″″ 20.9 and δC-3″″ 59.4. In addition, NMR spectra displayed oxymethine, methine adjacent to nitrogen, methylene, and methyl signals, each accounting for 2 carbons and assigned to two piperidinyl rings, as well as ten methylenes for two decanyl moieties. Comparison of the NMR spectra of 1 and (2S,3R,6S)-juliprosine (9)8,12 suggested close similarities for the dihydroindolizidinium ring and attached decanyl residues, but showed notable differences for the piperidinyl rings (δC 51.5, 68.4, 26.7, 28.2, 49.0, 11.9 and 29.3; C-2/2′-C-7/7′, and C-1″/1‴; vs. δC 57.2, 67.6, 31.4, 25.6, 55.8, 18.1, and 36.0/36.1 for 9), thereby suggesting that 1 was a diastereoisomer of 9. Strong shielding of 13C chemical shift values for C-2/2′, C-6/6′, C-7/7′ and C-1″/1‴ of the piperidine ring and attached methylene carbons indicated that the relative configurations of C-2/2′ and C-6/6′ were different (ie. R and R, respectively, vide infra), compared to 9 (ie. 2/2′S, 6/6′S). The COSY, HMBC and NOESY correlations, together with 13C assignments of C-2/2′-C-7/7′ confirmed the presence of two 2α-methyl-3β-hydroxy-6α-decanyl-piperidinyls with the same configuration for both rings (2/2′R, 3/3′R, 6/6′R). The HMBC spectrum of 1 demonstrated 3J- correlations between H-5″″ and C-7″″, C-8a″″, C-3″″, and C-10″; H-7″″ and C-8a″″, C-10″, and C-10″″; H-3″″ and C-8a″″ and C-1″″; H-1″″ and C-8″″; H-2″″ and C-8a″″, and 2J-correlations between H-2″″ and C-3″″, and C-1″″, confirming that 1 possessed a 6,8-dialkylated dihydroindolizinium ring. Finally, a 2D NMR NOESY experiment on 1 showed correlation between H-3/3′, H-7/7′and H-1″/1‴, suggesting that these protons were cis and on the α-face of the molecule, thereby the C-3/3′ OH group was on the β-face (3R) of the molecule. Thus, the structure of 1 was deduced as shown, and it was named prosopilosidine.
Prosopilosine (2) was isolated as gum and analyzed for C40H75N3O2 by ESIHRMS. The 1H and 13C NMR spectra of 2 (Table 1) were similar to those observed for 1, except for differences associated with the presence of a dehydroindolizidine carbon skeleton,25,26 consisting of four triplets (δC 33.4, 21.8, 54.7, 55.5), three doublets (δC 123.8, 42.8, 65.8) and a singlet (δC 136.1), instead of a 2,3-dihydro-1H-indolizinium nucleus8,9 in 1 (Table 1). Comparison of NMR spectra of 2 with spectra of other dehydroindolizidines such as juliprosopine (5) and related analogs6,9 led to the conclusion that 2 contained two identical units of 10-(2-methyl-3-hydroxy-6-alkylpiperidinyl)decane at C-6″″ and C-8″″ positions of the dehydroindolizidine nucleus, suggesting that 2 was a diastereoisomer of 5. A 2D NMR HMBC experiment of 2 (Figure 1) showed correlations between H-7″″ and C-5″″, C-8″″, C-8a″″ and C-10″; H-10″ and C-5″″, C-6″″, and C-7″″; Heq-3″″ and C-8a″″; Hax-3″″ and C-2″″, and Heq-5″″ and C-10″ and C-8a″″, confirming a 6,8-dialkylated 6,7-dehydroindolizidine nucleus. The configuration of 6,7-dehydroindolizidine nucleus of 2 was assigned as trans-(8R, 8aS) due to its close similarities of 1H NMR data and 13C chemical shift values for C-1″″, C-8″″ and C-8a″″ (δC 33.4, 42.8 and 65.8, respectively) with those of 5.12 These chemical shift values complies well with the literature values for a trans (8R, 8aS)-configuration, while the cis- configuration was reported as δC-1′27.9, δC-8′36.9 and δC-8a′63.6.12 The COSY and HMBC correlations, together with 13C assignments of C-2/2′ - C-7/7′ confirmed the presence of two 2α-methyl-3β-hydroxy-6α-piperidinyl rings. The shielding of 13C chemical shift values for C-2/2′, C-6/6′, C-7/7′ and C-1″/1‴ (δC 50.5, 50.1, 15.3, and 28.3 vs. δC 57.2, 55.8, 18.4, and 37.0 for 5) indicated that the relative configuration of C-2/2′ and C-6/6′ was R and R, respectively, compared to 2/2′S, 6/6′S of 5. A NOESY experiment on 2 (Figure 1) further showed correlation between H-3/3′, H-7/7′and H-1″/1‴, like 1, suggesting these protons were cis and on the α-face of the molecule; thus, the C-3/3′ OH group was on the β-face (3R) of the molecule.
Table 1.
1H and 13C NMR Data† (J values in Hz, in parenthesis) for 1, 2 and 5
| H/C | Prosopilosidine (1) | Prosopilosine (2) | Juliprosopine (5)+ | |||
|---|---|---|---|---|---|---|
|
| ||||||
| δH | δC | δH | δC | δH | δC | |
| 2, 2′ | 3.19 dq (6) | 51.5 d | 3.13 q (7.8) | 50.5 d | 2.75 q (6.5) | 57.2 d |
| 3, 3′ | 3.75 ddd (4.8) | 68.4 d | 3.66 br s | 68.9 d | 3.48 br.s | 67.8 d |
| 4, 4′ | 1.70 m | 26.7 t | 1.72 m | 27.9 t | 1.84 m | 26.1 t |
| 1.59 m | 1.62 m | 1.46 m | ||||
| 5, 5′ | 1.82 m | 28.2 t | 1.49 m | 26.9 t | 1.40 m | 25.8 t |
| 1.21 m | 1.22 m | 1.20 m* | ||||
| 6, 6′ | 2.80 m* | 49.0 d | 2.85 m | 50.1 d | 2.56 q (8) | 55.8 d |
| 7, 7′ | 1.14 d (7) | 11.9 q | 1.12 d (6.5) | 15.3 q | 1.07 d (6.4) | 18.4 q |
| 1″, 1‴ | 1.37 m | 29.3 t | 1.40 m | 28.3 t | 1.40–1.20 m* | 37.0 t |
| 2″, 2‴ | 30.7, 29.8, | 27.0 t | 32.2 t | |||
| 3″-8″ | 1.35–1.27 m* | 29.6, 29.5, | 1.39–1.27 m* | 30.5, 30.1, | 1.30–1.20 m* | 29.3, 29.5, |
| 29.4, 29.2, | 30.0, 29.8, | 29.6, 29.8, | ||||
| 3‴-8‴ | 29.1, 29.0, | 29.8, 29.7* | 30.1 (5 × t) | |||
| 9″, 9‴ | 26.3 (9 × t) | 1.43 m | (6 × t) | 26.6 t | ||
| 10″ | 2.80 m* | 32.1 t | 2.00 t (7.5) | 35.4 t | 2.00 t (7.4) | 35.1 t |
| 10‴ | 2.80 m* | 31.7 t | 1.39–1.27 m* | 29.8* | 1.40–1.20 m* | 28.0 t |
| 1″″ | 2.80 m* | 31.6 t | 1.52 m | 33.4 t | 1.40–1.20 m* | 33.1 t |
| 2″″ | 2.50 m | 20.9 t | 2.05 m | 21.8 t | 2.04 m | 21.5 t |
| 1.80 m | 1.76 m | |||||
| 3″″ | 4.88 m* | 59.4 t | 3.13 dd (18, 7.8) | 54.7 t | 3.11 ddd (17.6, 7.8) | 54.5 t |
| 2.10 dd (18, 9) | 2.08 dd (17.6, 9) | |||||
| 5″″ | 8.64 s | 137.3 d | 3.29 d (15) | 55.5 t | 3.26 d (15.2) | 55.3 t |
| 2.64 d (15) | 2.58 d (15.2) | |||||
| 6″″ | - | 139.1 s | - | 136.1 s | - | 136.3 s |
| 7″″ | 8.18 s | 144.3 d | 5.37 s | 123.8 d | 5.33 s | 123.9 d |
| 8″″ | - | 141.3 s | 2.00 m | 42.8 d | 2.00 m | 42.6 d |
| 8a″″ | - | 155.0 s | 1.82 m | 65.8 d | 1.74 m | 65.6 d |
Figure 1.

2D NMR HMBC (solid lines) and NOESY (dashed lines) correlations of 2
The structure of isoprosopilosine (3), C40H75N3O2, also showed the presence of two 2-methyl-3-hydroxy-6-piperidinyl rings, and a (8R, 8aS)-dehydroindolizidine nucleus, as observed for 2 and 5. Its NMR data (Table 2) were similar to those of 2, except for the presence of a 2β-methyl-3β-hydroxy-6β-piperidinyl ring (δC 56.0, 68.0, 32.5, 26.5, 57.4 and 18.8, C-2′-C-7′, respectively), instead of 2α-methyl-3β-hydroxy-6α-piperidinyl rings for 2. COSY and HMBC correlations led to the 13C assignments for C-2/2′-C-7/7′ which indicated two different configurations for the alkylpiperidine rings. This was substantiated by comparing the 13C data of the 2β-methyl-3β-hydroxy-6β-piperidine ring of isoprosopilosine and juliprosopine (5),25, 26 suggesting two alternative structures [3 (R = X, R1 = Y) or 3a (R = Y, R1 = X)] for isoprosopilosine. This compound contained two different decanyl- substituted piperidine rings, where the configurations of the C-2 methyl and C-6 methylene groups were different compared to 2 or 5. The 13C NMR signals for the piperidine ring carbons C-2/2′ - C-7/7′ of 2 (2R, 3R, 6R) and 5 (2S, 3R, 6S) were assigned to δ 50.5, 68.9, 27.9, 26.9, 50.1 and 15.3, and δC 57.2, 67.8, 26.1, 25.8, 55.8 and 18.4, respectively, and were in agreement with 3 or 3a. Based on above discussion, the configurations of two piperidine rings of isoprosopilosine were determined to be 2S, 3R, 6S and 2R, 3R, 6R.
Table 2.
1H and 13C NMR Data† (J values in Hz, in parenthesis) for (3/3a) and (4/4a)
| H/C | Isoprosopilosine (3/3a) | Isoprosopilosidine (4/4a) | ||
|---|---|---|---|---|
|
| ||||
| δH | δC | δH | δC | |
| 2, 2′ | 3.12 dq | 49.9 d | 3.18 dq (6) | 51.6 d |
| 2.77 dq (6) | 56.0 d | 2.78 dq (6) | 55.0 d | |
| 3, 3′ | 3.62 m | 69.1 d | 3.78 ddd (5, 5, 5) | 69.0 d |
| 3.50 br. s | 68.0 d | 3.61 br.s | 66.9 d | |
| 4, 4′ | 1.63 m | 27.0 t | 1.72 m, 1.62 m | 26.9 t |
| 1.51 m, 1.86 m | 32.5 t | 1.93 m, 1.65 m | 31.6 t | |
| 5, 5′ | 1.43 m, 1.63 m | 28.1 t | 1.69 m | 29.1 t |
| 1.23 m | 26.5 t | 1.53 m | 25.2 t | |
| 6, 6′ | 2.84 m | 50.4 d | 2.84 m | 48.4 d |
| 2.58 m | 57.4 d | 2.58 m | 56.5 d | |
| 7, 7′ | 1.08 d (6) | 16.0 q | 1.14 d (7) | 16.0 q |
| 1.09 d (6) | 18.8 q | 1.14 d (7) | 18.8 q | |
| 1″, 1‴ | 1.72 m | 37.3 t | 1.72 m | 37.3 t |
| 1.32 m | 28.4 t | 1.32 m | 28.4 t | |
| 2″, 2‴ | 30.4, 30.2, | 30.7, 29.9, | ||
| 3″-8″ | 30.0, 29.9, | 29.6, 29.4, | ||
| 3‴-8‴ | 1.41–1.26 m* | 29.9, 29.8, | 1.48–1.25 m* | 29.2, 26.4, |
| 9″, 9‴ | 29.6, 26.2 (8 × t) | 25.9 (7 × t) | ||
| 10″ | 2.00 t (7.5) | 35.4 t | 2.83 m* | 32.1 t |
| 10‴ | 1.41–1.26 m* | 29.8* | 2.83 m* | 31.7 t |
| 1″″ | 1.45 m | 33.4 t | 2.83 m* | 30.7 t |
| 2″″ | 1.81 m | 21.8 t | 2.54 m | 21.1 t |
| 3″″ | 3.11 dd (18, 7.8) | 54.7 t | 4.87 m | 59.4 t |
| 2.09 dd (18, 9.5) | ||||
| 5″″ | 3.29 d (15.5) | 55.5 t | 8.64 s | 137.3 d |
| 2.61 d (15.5) | ||||
| 6″″ | - | 136.2 s | - | 141.3 s |
| 7″″ | 5.36 s | 123.8 d | 8.17 s | 144.3 d |
| 8″″ | 2.00 m | 42.9 d | - | 139.2 s |
| 8a″″ | 1.72–1.81 m | 65.7 d | - | 155.0 s |
All spectra recorded in CD3OD at 500 MHz (1H) and 125 MHz (13C).
Overlapped signals.
The NMR spectral data (Table 2) of isoprosopilosidine (4), C40H72N3O2, were similar to those observed for 1, except for the presence of one 2β-methyl-3β-hydroxy-6β-piperidinyl ring (δC 55.0, 66.9, 31.6, 25.2, 56.5 and 18.8; C-2′-C-7′, respectively), instead of one of the 2α-methyl-3β-hydroxy-6α-piperidinyl rings for 1. This was substantiated by comparing the 13C NMR data for the 2β-methyl-3β-hydroxy-6β-piperidine ring with 5, suggesting two alternative structures, 4 (R = X, R1 = Y) or 4a (R = Y, R1 = X), due to two different substituted piperidine rings, one with 2S, 3R, 6S and the other with 2R, 3R, 6R configurations. The 13C NMR signals for the piperidine ring carbons (C-2/2′ - C-7/7′) of isoprosopilosidine, with 2R, 3R, 6R and 2S, 3R, 6S configurations, respectively, were assigned to δ 51.6, 69.0, 26.9, 29.1, 48.4 and 16.0, and δC 55.0, 66.9, 31.6, 25.2, 56.5 and 18.4. These values were in agreement with those observed for 3 or 3a, and thus isoprosopilosidine (4 or 4a) was established as a diastereoisomer of 1.
Prosopilosidine (1), its analogs 2–5 and salts 6–8 were tested for in vitro antibacterial, antifungal, antimalarial, antileishmanial and cytotoxic activities. Compound 1 showed potent in vitro antifungal activity against Cryptococcus neoformans, and antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) and Mycobacterium intracellulare with IC50/MIC values of 0.4/0.63, 0.35/1.25 and 0.9/2.5 μg/mL, and 2 against C. neoformans and Aspergillus fumigatus (IC50/MIC = 0.8/1.25 and 0.45/0.63 μg/mL, respectively) (Table 3). The minimum fungicidal concentration (MFC) of 1 against C. neoformans was equipotent to amphotericin B, but the selectivity index (SI, ratio of IC50 vs. VERO to IC50 C. neoformans) value was >3-fold more than amphotericin B. Compound 2 was also fungicidal to C. neoformans and A. fumigatus with MFCs of 1.25 and 0.63 μg/mL, respectively, more potent than amphotericin B. On the other hand, juliprosopine (5) exhibited strong activity against A. fumigatus (IC50/MIC/MFC = 0.9/1.25/1.25 μg/mL), while the dihydrochloride salts 6–8 showed moderate activity against C. neoformans, MRSA, and M. intracellulare. Finally, the in vitro antifungal and antibacterial activity was not decreased in the presence of 5% human serum (data not shown), indicating that these compounds could be active in vivo as well.
Table 3.
Antimicrobial Activity of P. glandulosa Extracts and Compounds 1–8
| Extract/Compound | IC50/MIC/MFC or MBC (μg/mL)
|
||||
|---|---|---|---|---|---|
| C. albicans | C. neoformans | MRSA | M. intracellulare A. fumigatus | ||
| P. glandulosa (EtOH extract) | - / - / - | 20 / - / - | 200 / - / - | - / - / - | - / - / - |
| P. glandulosa (Alkaloid Fr.) | 80 / - / - | <8 / - / - | <8 / - / - | <8 / - / - | <8 / - / - |
| 1 | 15 / - / - | 0.4 / 0.63 / 1.25 | 0.35/1.25/5.0 | 0.9 / 2.5 / 20.0 | 3.0 / 5.0 / - |
| 2 | 15 / - / - | 0.8 / 1.25 / 1.25 | 1.5 / 2.5 / 10 | 2.0 / 5.0 / 20.0 | 0.45 / 0.63 / 0.63 |
| 3 | - / - / - | 0. 4/ 0.63 / 0.63 | 1.0 / 2.5 / - | 1.5 / 5.0 / - | 0.1 / 0.16 / 2.5 |
| 4 | - / - / - | 0.55 / 1.25 / 1.25 | 0.6 / 1.25 / 20.0 | 1.5 / 2.5 / - | 6.5 / 10.0 / 10.0 |
| 5 | - / - / - | 1.5 / 5.0 / 5.0 | 3.5 / 5.0 / - | 6.0 / 20.0 / 20.0 | 0.9 / 1.25 / 1.25 |
| 6 | 15.0 / 20.0 / - | 1.5 / 2.5 / 2.5 | 1.5 / 2.5 / 5.0 | 3.5 / 10.0 / - | - / - / - |
| 7 | - / - / - | 2.0 / 5.0 / 5.0 | 3.0 / 5.0 / 5.0 | 9.5 / 20.0 / - | - / - / - |
| 8 | 7.5 / 10.0 / 10.0 | 0.8 5/ 1.25 / 2.5 | 3.0 / 5.0 / 5.0 | 10.0 / - / - | 6.5 / 10.0 / - |
| Amphotericin B | 0.35 / 0.63 / 1.25 | 0.45 / 1.25 / 2.5 | NT | NT | 0.40 / 0.63 / 2.5 |
| Ciprofloxacin | NT | NT | 0.1 / 0.3 / - | 0.25 / 0.63 / - | NT |
IC50 is the concentration that affords 50% inhibition of growth; MIC (Minimum Inhibitory Concentration) is the lowest test concentration that allows no detectable growth; MFC/MBC (Minimum Fungicidal/Bactericidal Concentration) is the lowest test concentration that kills the organism.
- = Not active at the highest test concentration; NT = Not tested.
In a separate in vivo study, experimentally infected mice were treated with compound 1. At 0.0625 mg/Kg/day, administered intraperitonially for 5 days, it eliminated ~76% of C. neoformans infection from brain tissue compared to ~83% with amphotericin B at 1.5 mg/Kg/day. These results showed that 1 is effective against C. neoformans infection in vivo, and also indicated that the compound passes through the blood brain barrier in sufficient amount to kill the organisms in the brain tissue. The potent in vivo anticryptococcal activity of 1 (0.0625 mg/Kg/day/ip) in a mouse model experiment showed the antifungal dose-response effect where 1 is active at doses much lower than its maximum tolerated dose (2.5 mg/Kg/day/ip). This makes it a strong lead candidate for development of a drug for cryptococcosis, cryptococcal meningitis and cryptococcus mediated opportunistic infections in AIDS patients. A full report of the in vivo study of 1 together with the toxicological evaluations will be published elsewhere.
Among the compounds (1–8) tested for in vitro antimalarial activity (Table 4), the dihydroindolizinium salts (1 and 4) exhibited the most potent activity and high SI against P. falciparum. They showed IC50 values of 39 and 95 ng/mL, and 42 and 120 ng/mL, respectively, against chloroquine sensitive (D6) and chloroquine resistant (W2) strains, which were similar to the standard antimalarial drug chloroquine (IC50 = 17 and 140 ng/mL). However, the dehydroindolizidine bases prosopilosine (2) and isoprosopilosine (3) were less potent than 1 and 4 (IC50 = 120 and 230 ng/mL against D6 strain, and 83 and 150 ng/mL against W2 strain), but more toxic against mammalian kidney fibroblast (VERO) cells (IC50 = 5600 and 1800 ng/mL for 2 and 3 vs. >23800 ng/mL for 1 and 4, respectively). Therefore, 1 and 4 exhibited higher SI values against P. falciparum D6 and W2 strains (SI >610, 567 and SI >250 198, respectively) than the corresponding analogs 2, 3 and 5 (SI 47, 24, 23 and SI 22, 12, 13 respectively), suggesting that dihydroindolizinium quaternary alkaloids 1 and 4 are better candidates than the tertiary bases, 2, 3 and 5 for further in vivo antimalarial studies. The dihydrochloride salts 6, 7 and 8, prepared from compounds 1, 2 and 3, respectively, retained weak activities compared to their corresponding bases. Based on investigations of the in vitro antimalarial activity profile, cytotoxicity (vide infra) and mammalian toxicity of 1–5 (Table 6), compound 1 was selected for preliminary in vivo antimalarial screening at two doses, 1 and 2 mg/Kg, in a rodent model for malaria. Compound 1 showed an ED50 value of ~2 mg/Kg against P. berghei infected mice, thereby exhibiting ~48% suppression of parasitemia after 3 days of treatment (Table 5). Compound 1 also caused 40.5% suppression in parasitemia at 1 mg/Kg/day dose for 3 days, and no significant toxicity was observed due to treatment with 1 at these doses. Higher doses were not tested since the estimated maximum tolerated dose of compound 1 was 2.5 mg/Kg.
Table 4.
Antiparasitic Activities of P. glandulosa Extracts and Compound 1–8
| Extract/compound | P. falciparum (ng/mL) | VERO L. donovani | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| D6a | W2b | (ng/mL) | (μg/mL) | ||||
|
|
|||||||
| IC50 | SIc | IC50 | SIc | IC50 | IC50 | IC90 | |
| P. glandulosa (EtOH extr.) | 13000 | >3.7 | 10000 | >4.8 | NC | 44 | 90 |
| P. glandulosa (Alkaloid Fr.) | 420 | 64 | 630 | 43 | 27000 | 18 | 35 |
| 1 | 39 | >610 | 95 | >250 | >23800 | 0.7 | 1.9 |
| 2 | 120 | 47 | 230 | 24 | 5600 | 0.65 | 2.0 |
| 3 | 83 | 22 | 150 | 12 | 1800 | 0.26 | 1.2 |
| 4 | 42 | >567 | 120 | >198 | >23800 | 0.8 | 2.5 |
| 5 | 220 | 23 | 380 | 13 | 5000 | 0.75 | 2.4 |
| 6 | 430 | >11.1 | 270 | >17.6 | NCd | 2.8 | 8.0 |
| 7 | 600 | >7.9 | 560 | >8.5 | NCd | 3.1 | 7.0 |
| 8 | 380 | >12.5 | 290 | >16.4 | NCd | 0.7 | 1.2 |
| Chloroquine | 17 | 140 | NC | NT | NT | ||
| Artemisinin | 16 | 17 | NC | NT | NT | ||
| Pentamidine | NT | NT | NT | 0.5 | 2.0 | ||
| Amphotericin B | NT | NT | NT | 0.1 | 0.4 | ||
Chloroquine-sensitive clone.
Chloroquine-resistant clone.
Selectivity index = IC50 VERO cells/IC50 P. falciparum.
NC = Not cytotoxic (up to the maximum dose tested; 4760 ng/mL for pure compounds and 47600 ng/mL for crude extracts). NT= Not tested.
Table 6.
Cytotoxic Activities of Compounds 1–5
| Compound | IC50 (μg/mL)
|
|||||
|---|---|---|---|---|---|---|
| SK-MELa | KBb | BT-549c | SK-OV-3d | VEROe | LLC-PK1f | |
| 1 | 4.5 | 7.5 | 8.75 | 7.5 | NA | NA |
| 2 | <1.1 | 6.0 | 6.0 | 2.0 | 5.7 | 1.9 |
| 3 | <1.1 | 2.2 | 2.5 | <1.1 | 1.8 | 1.8 |
| 4 | 3.3 | 7.0 | 6.75 | 4.5 | NA | 21.3 |
| 5 | 2.4 | 6.2 | 6.0 | 4.7 | 5.0 | 1.8 |
| Artemisinin | NT | NT | NT | NT | >4.76 | NT |
| Amphotericin B | 5.0 | >25 | 5.5 | >25 | 7.5 | 0.6 |
| Doxorubicin | <0.55 | <0.55 | <0.55 | <0.55 | 3.2 | <0.55 |
SK-MEL: Human malignant melanoma.
KB: Human epidermoid carcinoma.
BT-549: Human ductal carcinoma.
SK-OV-3: Human ovary carcinoma.
VERO: Monkey kidney fibroblast.
LLC-PK1: Pig kidney epithelial.
Table 5.
In vivo Antimalarial Activity (Suppressive/Curative; P. berghei Mouse model) of Compound 1
| Compound/Drug | Animals per group | Dose, mg/kg x days | Suppression in parasitemia (% activity)a
|
Cureb | |
|---|---|---|---|---|---|
| Day 10 | Day 17 | ||||
| 1 | 5 | 1 × 3 | 40.5 | nil | 0/5 |
| 5 | 2 × 3 | 48.4 | nil | 0/5 | |
| β-Arteether | 5 | 10 × 3 | 100 | 100 | 5/5 |
% suppression in parasitemia = 100 – [Parasitemia in treated group/parasitemia in untreated group × 100].
Number of mice without parasitemia till day 28/Total number of mice in group.
Compounds 1–5 also demonstrated potent in vitro activity against Leishmania donavani promastogotes (IC50 0.26–0.80 μg/mL) and were as potent as the standard control pentamidine (Table 4). Finally, all of the indolizidine compounds were tested for cytotoxic activity against selected human cancer cell lines, namely SK-MEL, KB, BT-549 and SK-OV-3 (Table 6). Both 1 and 4 were weakly active towards all of these cancer cell lines, and were inactive against mammalian VERO (monkey kidney fibroblast and LLC-PK1 (pig kidney epithelial) cells (IC50 21.3–25 μg/mL). However, compounds 2, 3 and 5 were toxic to VERO (IC50 5.7, 1.8, 5.0 μg/mL) and LLC-PK1 (1.9 and 1.8 μg/mL) cells.
This appears to be the first report of compounds 1–4 from a natural source. In contrast, the only other diastereoisomer of prosopilosidine (1), juliprosine (9), was reported without biological activity, while several dehydroindolizidine tertiary bases, natural and / or synthetic, showed potential therapeutic value. Compound 1 has favorable drug-like properties since its calculated log Po/w value is 4.4, which is an indication of a lipophilic property, lies within the value set forth by “Lipinski’s rule of 5”.27 Additionally, compound 1 was >4-fold less toxic than amphotericin B in regard to hemolytic activity [IC50 (RBC lysis) of 1 was 16 μg/mL, vs. 3.9 μg/mL for amphotericin B]. The in vivo antimalarial assay of prosopilosidine (1) in a mouse model showed an ED50 value of ~2 mg/Kg/day against P. berghei. Indolizidines have not been previously reported as antimalarial agents, against either P. falciparum strain in vitro or P. berghei infected mice in vivo, or any other species, suggesting indolizidine to be a new antimalarial pharmacophore. Potent in vitro antimalarial activity of 1 against both chloroquine susceptible and resistant strains of P. falciparum, low toxicity against mammalian cells and significant activity in vivo in the mouse malaria model at low doses indicate 1 to be a promising new antimalarial lead for further optimization.
Experimental Section
General Experimental Procedures
Optical rotations were measured using an AUTOPOL® IV instrument at ambient temperature; UV spectra were obtained in MeOH using a Hewlett Packard 8453 UV/VIS Spectrometer; IR spectra were obtained using a Bruker Tensor 27 instrument; The NMR spectra were acquired on a Bruker Avance DRX-500 instrument at 500 MHz (1H), 125 (13C) in CDCl3 or CD3OD using the residual solvent as internal standard. Multiplicity determinations (DEPT) and 2D NMR spectra (COSY, HMQC, HMBC) were obtained using standard Bruker pulse programs. HRMS were obtained by direct injection using a Bruker Bioapex-FTMS with Electro-Spray Ionization (ESI). TLC was carried out on aluminum oxide IB-F plates (Baker-flex®) using CH2Cl2–MeOH-NH3·H2O (8:2:0.1), as solvent. For flash column chromatography, basic alumina (Brockman activity I, 60–325 mesh, Fisher Scientific) was used with CH2Cl2–MeOH-NH3·H2O (9:1:0.1 → 8:2:0.1) mixtures as solvents. Centrifugal preparative TLC (CPTLC, using a Chromatotron, Harrison Research Inc., model 8924, tagged with a fraction collector) was carried out on 1 or 2 mm alumina (coated with F254 indicator) rotors, using CH2Cl2-MeOH-NH3·H2O (95:5:0.1) as eluant. Samples were dried using a Savant Speed Vac Plus SC210A concentrator. The compounds were visualized by observing under UV light at 254 or 365 nm, followed by spraying, separately, with Dragendorff’s and 1% vanillin-H2SO4 spray reagents.
Plant Material
Leaves, flowers, stems and aerial parts of P. glandulosa were collected from Nevada (voucher # PRGAGL 2884) in June 1998 and May 2004 (and 2006) by Mr. Elray Nixon, Las Vegas, Nevada. Voucher specimens are deposited at the Herbarium of NCNPR, University of Mississippi.
Extraction and Isolation
The powdered air-dried leaves (485 g) were extracted by percolation with 95% EtOH (2 L × 3) for 48 h and the combined extract was evaporated to dryness (70 g). The dried EtOH extract (50 g) was dissolved in aqueous 0.1 N HCl (500 mL, pH 1), and defatted by partitioning successively with n-hexane, followed by CH2Cl2 (each 100 mL × 3). The aqueous acidic layer was then basified with 0.1 N NH4OH to pH 11 (based on pKa 10.2 of tryptamine), followed by partitioning successively with CH2Cl2 (each 100 mL × 3), and the combined CH2Cl2 fractions afforded tryptamine.23 The aqueous basic layer was then made transparent by adding MeOH (final volume 500 mL) and the pH was adjusted to 12 (based on pKa values 11 of indolizidines 1–5) by adding conc. NH4OH, followed by successively with CH2Cl2 and EtOAc (each 100 mL × 3). The CH2Cl2 and EtOAc fractions were filtered separately over anhydrous Na2SO4, and then evaporated under vacuum to dryness (yields 2.35 g and 1.49 g of mixtures 1–5, respectively). This alkaloid enriched CH2Cl2 fraction demonstrated strong in vitro activities against P. falciparum D6 and W2 strains (Table 1). The CH2Cl2 fraction (2.17 g) was subjected to flash chromatography over alumina (Al2O3, 80 g), eluted with CH2Cl2 and then with increasing concentrations of CH3OH (1% → 10%) in CH2Cl2-NH3·H2O mixtures, and 20 mL fractions were collected. Fractions were pooled by TLC, combined, and then evaporated under reduced pressure (total 1.64 g from six combined fractions, ie. A-F). Further purification of fractions A (43 mg), B (62 mg), C (146 mg), D (111 mg), E (43 mg) and F (185 mg) was achieved by repeated CPTLC, using 1 or 2 mm alumina rotors with CH2Cl2-MeOH-NH3·H2O (99:1:0.1 → 95:5:0.1) as solvent system, which afforded 1 (28 mg), 2 (28 mg), 3 (35 mg), 4 (26 mg) and 5 (13 mg), and tryptamine23 (39 mg). Using this procedure, bulk quantities of 1–5 (ie. 80–100 mg) were isolated from leaves collected in 2005 and 2006. Compound 5 {gum; [α]D28 +1° (c 0.4, MeOH); HRESIMS m/z 630.6061 [M+H]+ (calcd. for C40H76N3O2, 630.5937)} was identified as juliprosopine by comparison of its physical and NMR data with those reported previously.25,26
Prosopilosidine (1), (6,8-bis-[(2R,5R,6R)-5-hydroxy-6-methylpiperidin-2-yl]decyl-2,3-dihydro-1H-indolizinium chloride): colorless gum; [α]D28 +6.0 (c 0.3, MeOH); UV (MeOH) λmax, nm: 204, 220, 276; IR (film) νmax, cm-1: 3309 (OH, NH), 2924, 2853, 1635, 1076; 1H and 13C NMR data, see Table 1; HRESIMS m/z 626.5635 [M]+ (calcd. for C40H72N3O2, 626.5624).
Prosopilosine (2), (6,8-bis-[(2R,5R,6R)-5-hydroxy-6-methylpiperidin-2-yl]decyl-[8R,8aS]-6,7-dehydroindolizidine): colorless gum; [α]D28 +9.4 (c 0.32, MeOH); UV (MeOH) λmax, nm: 204, 224; IR (film) νmax, cm−1: 3284 (OH, NH), 2925, 2853, 1660, 1072; 1H and 13C NMR data, Table 1; HRESIMS m/z 630.5966 [M+H]+ (calcd. for C40H76N3O2, 630.5937).
Isoprosopilosine (3): colorless gum; [α]D28 +5.3 (c 0.45, MeOH); UV (MeOH) λmax, nm: 199, 226; IR (film) νmax, cm−1: 3336 (OH, NH), 2924, 2852, 1661, 1088; 1H and 13C NMR spectral data see Table 2; HRESIMS m/z 630.5968 [M+H]+ (calcd. for C40H76N3O2, 630.5937).
Isoprosopilosidine (4): colorless gum; [α]D28 +5.6 (c 0.25, MeOH,); UV (MeOH) λmax, nm: 204, 220, 276; IR (film) νmax, cm−1: 3285 (OH, NH), 2924, 2853, 1624, 1083; 1H and 13C NMR spectral data see Table 2; HRESIMS m/z 626.5636 [M]+ (calcd. for C40H72N3O2, 626.5624).
Preparation of Dihydrochloride Salts of Indolizidines (6–8)
Excess HCl in Et2O (2 M, 0.5 mL) was added to compounds 1, 2 and 3 (each 5–6 mg), and dissolved in 2 mL of CH2Cl2. The reaction mixtures were kept overnight under nitrogen, and the solutions were dried to give dihydrochloride salts 6, 7 and 8, as yellow gums in yields of 8 mg, 5.5 mg and 6 mg, respectively. The Rf values for 6–8 were found to be 0.16, 0.36, 0.62, respectively (TLC; Alumina, solvent: CH2Cl2: MeOH: NH3.H2O; 8.5:1.5:0.1), vesus Rf values of 0.08, 0.43 and 0.49 for 1–3 (TLC; Alumina, solvent: CH2Cl2: MeOH: NH3.H2O; 9:1:0.1).
Prosopilosidine dihydrochloride (6)
[α]D28 −0.7 (c 0.8, MeOH); IR (film) νmax, cm−1: 3474 (OH, NH), 3431 (OH, NH), 3146, 3071, 2928, 2855, 1710, 1292, 1076.
Prosopilosine dihydrochloride (7)
[α]D28 −0.9° (c 0.6, MeOH); IR (film) νmax, cm−1: 3531 (OH, NH), 3487 (OH, NH), 3159, 3088, 2927, 2855, 1708, 1295, 1076; 1H NMR (CD3OD) δH 5.64 (1H, s, H-7″″), 3.87 (2H, m, H-3, 3′), 3.49 (2H, q, J= 7.5 Hz, H-2, 2′), 3.37 (2H, m, H-6, 6′), 3.15 (3H, m, H-3″″, 5″″), 2.49 (1H, m, H-3″″), 2.14 (5H, m, H-8″″, 2″″, 10″), 1.88 (2H, m, H-2″″, 8a″″), 1.78 (2H, m, H-4, 4′), 1.68, 1.58, 1.53–1.26 (m), 1.32 (6H, d, J = 6 Hz, H-7, 7′).
Isoprosopilosine dihydrochloride (8)
[α]D28 −1.7 (c 1.0, MeOH); IR (film) νmax, cm−1: 3473 (OH, NH), 3425 (OH, NH), 3149, 3073, 2928, 2855, 1708, 1295, 1076. However, 1H NMR signals of 6 and 8 were broad compared to those of 7 in CD3OD.
Antimicrobial Assay
All organisms were obtained from the American Type Culture Collection (Manassas, VA) and include the fungi Candida albicans ATCC 90028, Cryptococcus neoformans ATCC 90113, and Aspergillus fumigatus ATCC 90906 and the bacteria methicillin-resistant Staphylococcus aureus ATCC 43300 (MRS), Escherichia coli ATCC 35218, Pseudomonas aeruginosa ATCC 27853, and Mycobacterium intracellulare ATCC 23068. For all organisms excluding M. intracellulare and A. fumigatus, susceptibility testing was performed using a modified version of the CLSI (formerly NCCLS) methods28,30, and optical density was used to monitor growth. Media supplemented with 5% Alamar Blue™ (BioSource International, Camarillo, CA) was utilized for growth detection of M. intracellulare31–32 and A. fumigatus29. Samples were serially-diluted in 20% DMSO/saline and transferred in duplicate to 96 well flat bottom microplates. Microbial inocula were prepared by correcting the OD630 of cell/spore suspensions in incubation broth (RPMI @ pH 4.5 for C. albicans, Sabouraud Dextrose for C. neoformans, cation-adjusted Mueller-Hinton @ pH 7.3 for MRS, and 5% Alamar Blue™ (BioSource International, Camarillo, CA) in Middlebrook 7H9 broth with OADC enrichment, pH = 7.3 for M. intracellulare and 5% Alamar Blue in RPMI @ pH 7.3 for A. fumigatus to afford final target inocula (1·104, 1·105, 5·105, 2·106, and 3·104 CFU/mL, respectively). Drug controls [Ciprofloxacin (ICN Biomedicals, Ohio) for bacteria and Amphotericin B (ICN Biomedicals, Ohio) for fungi] were included in each assay. All organisms were read at either 630 nm using the Biotek Powerwave XS plate reader (Bio-Tek Instruments, Vermont) or 544ex/590em, (M. intracellulare, A. fumigatus) using the Polarstar Galaxy Plate Reader (BMG LabTechnologies, Germany) prior to and after incubation. Minimum fungicidal or bactericidal concentrations were determined by removing 5 μ-L from each clear well, transferring to agar and incubating until growth was seen. The MFC/MBC is defined as the lowest test concentration that kills the organism (allows no growth on agar).
Antimalarial/Parasite LDH Assay
The in vitro antimalarial activity was measured by a colorimetric assay that determines the parasitic lactate dehydrogenase (pLDH) activity. 33,34,45 The assay was performed in 96-well microplate and included two P. falciparum clones [Sierra Leone D6 (chloroquine-sensitive) and Indochina W2 (chloroquine-resistant)]. For the assay, a suspension of red blood cells infected with P. falciparum (D6 or W2) strains (200 μL, with 2% parasitemia and 2% hematocrit in RPMI - 1640 medium supplemented with 10% human serum and 60 μg/mL amikacin) was added to the wells of a 96- well plate containing 10 μL of test samples at various concentrations. The plate was flushed with a gas mixture of 90% N2, 5% O2, and 5% CO2, in a modular incubation chamber (Billups-Rothenberg, 4464 M] and incubated at 37 °C, for 72 h. Plasmodial LDH activity was determined by using Malstat™ reagent (Flow Inc., Portland, OR) as described earlier.34,35 The IC50 values were computed from the dose response curves generated by plotting percent growth against test concentrations. DMSO (0.25%), artemisinin and chloroquine were included in each assay as vehicle and drug controls, respectively. The selectivity indices (SI) were determined by measuring the cytotoxicity of samples towards mammalian cells (VERO; monkey kidney fibroblast).
In vivo Antimalarial Assay36
The in vivo antimalarial activity of the compounds was determined in mice infected with P. berghei (NK-65 strain) according to the Peter’s 4-day suppressive test. Male mice (Swiss Webster strain) weighing 18–20 g were intraperitoneally inoculated with 2·107 parasitized red blood cells obtained from a highly infected donor mouse. Mice were divided into different groups with 5 mice in each group. Test compounds were prepared in DMSO:PEG:Water (1:3:6) and administered intraperitoneally to the mice about 2 h after the infection (Day 0). The test compounds were administered to the mice once a day for 3 consecutive days (Days 0–2). A control group was treated with an equal volume of vehicle while another control group was treated with the standard antimalarial compound, β-arteether. The mice were closely observed after every dose for any apparent signs of toxicity. Blood smears were prepared on different days (until day 28 post infection) by clipping the tail end, stained with Giemsa and observed under a microscope for determination of the parasitemia. Mice without parasitemia until day 28 post infection were considered as cured.
Antileishmanial Assay
Antileishmanial activity of the compounds was tested in vitro against a culture of L. donovani promastigotes, grown in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS, Gibco Chem. Co.) at 26 °C. A 3 day-old culture was diluted to 5·105 promastigotes/mL. Drug dilutions (50 – 3.1 μg/mL) were prepared directly in cell suspension in 96-well plates. Plates were incubated at 26 °C for 48 hours, and growth of leishmania promastigotes was determined by Alamar Blue assay.37 Standard fluorescence was measured on a Fluostar Galaxy plate reader (BMG LabTechnologies) at excitation wavelength of 544 nm and emission wavelength of 590 nm. Pentamidine and amphotericin B were used as the standard antileishmanial agents. Percent growth was calculated and plotted versus test concentration for computing the IC50 and IC90 values.
Cytotoxicity Assay
The in vitro cytotoxic activity was determined against four human cancer cell lines, SK-MEL, KB, BT-549 and SK-OV-3, as well as noncancerous cell lines, VERO and LLC-PK1 (Table 6), obtained from the American Type Culture Collection (ATCC, Rockville, MD). The assay was performed in 96-well tissue culture-treated microplates. Cells (25,000 cells/well) were seeded to the wells of the plate and incubated for 24 hours. Samples were added and plates were again incubated for 48 hours. The number of viable cells was determined using Neutral Red according to a modification of the procedure of Borenfreund et al.38 IC50 values were determined from logarithmic graphs of growth inhibition vs. concentration. Doxorubicin was used as a positive control, while DMSO was used as the negative (vehicle) control.
Acknowledgments
The authors sincerely thank Dr. Alice M. Clark, Vice-Chancellor for Research and sponsored programs, UM, for her valuable advise and suggestions on antifungal activity of compounds during new drugs for OI infection meetings, and Dr. Vaishali Joshi, Dr. Troy Smillie, Dr. D. Chuck Dunbar, Ms. Sharon Sanders, Mr. John Trott, Ms. Marsha Wright, Dr. Anupam Pradhan, Ms. Lavanya Madgula and Mr. Mohammed A. Hammad, NCNPR, for assistance in plant identification, plant acquisition and biological work. This work was supported in part by the United States Department of Agriculture, Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009, and NIH, NIAID, Division of AIDS, Grant No. AI 27094.
References and Notes
- 1.Burkhart A. A Monograph of the Genus Prosopis. J Arnold Arboretum. 1976;57:450–525. [Google Scholar]
- 2.Hilu YW, Boyd S, Felker P. Morphological diversity and taxonomy of California mesquites (Prosopis, Lepminosae) Madrono. 1982;29:237–254. [Google Scholar]
- 3.Davidow J. Infusions of Healing: A Treasury of Mexican-American Herbal Remedies. Simon and Schuster Inc; 1999. pp. 1–149. [Google Scholar]
- 4.Kay MA. Healing with Plants in the American and Mexican West. The University of Arizona Press; Tuscon: 1996. pp. 221–224. [Google Scholar]
- 5.Ali AAN, Al-Rahwi K, Lindequist U. Afr J Trad, Complementary & Alt Med. 2004;1:72–76. [Google Scholar]
- 6.Ahmad VU, Qazi S. J Chem Soc Pak. 1985;7:347–350. [Google Scholar]
- 7.Ahmad VU, Basha A, Haque W. Zeitschrift fuer Naturforschung, Teil B: Anorganische Chemie, Organische Chemie. 1978;33B:347–348. [Google Scholar]
- 8.Daetwyler P, Ott-Longoni R, Schoepp E, Hesse M. Helv Chim Acta. 1981;64:1959–1963. [Google Scholar]
- 9.Ahmad VU, Sultana A, Qazi S. Alkaloids from the leaves of Prosopis juliflora. J Nat Prod. 1989;52:497–501. [Google Scholar]
- 10.Nakano H, Nakajima E, Hiradate S, Fujii Y, Yamada K, Shigemori H, Hasegawa K. Phytochem. 2004;65:587–591. doi: 10.1016/j.phytochem.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad VU, Ali MS, Bano N, Bano S, Fatima A, Fatima I, Fizza K, Farooqui TA, Khan MA. Nat Prod Chem. Vol. 3. Springer-Verlag; Berlin: 1988. pp. 355–358. [Google Scholar]
- 12.Snider BB, Neubert BJ. Org Lett. 2005;7:2715–2718. doi: 10.1021/ol050931l. [DOI] [PubMed] [Google Scholar]
- 13.Aqeel A, Khursheed AK, Viqaruddin A, Sabiha Q. Arzneimittel-Forschung. 1989;39:652–655. [PubMed] [Google Scholar]
- 14.Ahmad A, Ahmad V, Khalid SM, Ansari FA, Khan KA. Philippine J Sci. 1997;126:175–182. [Google Scholar]
- 15.Ahmad A, Khan KA, Ahmad V. Pak J of Zoology. 1996;28:365–367. [Google Scholar]
- 16.Ahmad A, Khan KA, Ahmad VU, Qazi S. Planta Med. 1986;4:285–288. [PubMed] [Google Scholar]
- 17.Ahmad A, Kazmi SU, Khan KA, Ahmad VU, Qazi S. JPMA J Pak Med Asson. 1988;38:20–24. [PubMed] [Google Scholar]
- 18.Khursheed AK, Arshad HF, Viqaruddin A, Sabiha Q, Sheikh AR, Tahir SH. Arzneimittel-Forschung. 1986;36:17–19. [PubMed] [Google Scholar]
- 19.Ahmad A, Khursheed AK, Sabiha Q, Viqaruddin A. Fitoterapia. 1989;60:86–89. [Google Scholar]
- 20.Tabassum R, Naqvi SNH, Ahmad VU, Rani S, Jahan M, Azmi MA. Proceedings of Pakistan Congress of Zoology. 1994;14:326–333. [Google Scholar]
- 21.Naqvi SNH, Yasmin N, Mahmood RA, Nizam S, Ahmad VU, Qazi S. Zeitschriftfuer Angewandte Zoologie. 1994;80:155–164. [Google Scholar]
- 22.Aqeel A, Khursheed AK, Viqaruddin A. Toxicological studies of the antimicrobial alkaloid juliflorine. Arzneimittel-Forschung. 1991;41:151–154. [PubMed] [Google Scholar]
- 23.Tapia A, Egly Feresin G, Bustos D, Astudillo L, Theoduloz C, Schmeda-Hirschmann G. J of Ethnopharmacol. 2000;71:241–246. doi: 10.1016/s0378-8741(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 24.Nakano H, Nakajima E, Fujii Y, Shigemori H, Hasegawa K. Plant Growth Regulation. 2004;44:207–210. [Google Scholar]
- 25.Usmanghani K, Saqib QN, Ahmad VU. J Chem Soc Pak. 1982;4:285–287. [Google Scholar]
- 26.Ott-Longoni R, Viswanathan N, Hesse M. Helv Chim Acta. 1980;6:2119–2129. [Google Scholar]
- 27.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 28.NCCLS. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard. National Committee for Clinical Laboratory Standards. (2) 2002;22:1–51. [Google Scholar]
- 29.NCCLS. Reference method for broth dilution antifungal susceptibility testing of conidium forming filamentous fungi. Proposed standard M38-P. National Committee for Clinical Laboratory Standards. 1998;18:1–39. [Google Scholar]
- 30.NCCLS. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. NCCLS Document M7-A5. National Committee for Clinical Laboratory Standards. 2000;20:1–58. [Google Scholar]
- 31.NCCLS. Susceptibility testing of mycobacteria, nocardia, and other aerobic actinomycetes. Tentative standard M24-T2. National Committee for Clinical Laboratory Standards. (2) 2000;20:1–81. [PubMed] [Google Scholar]
- 32.Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. J Clinical Microbiol. 1998:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dou J, McChesney JD, Sindelar RD, Goins DK, Walker LA. J Nat Prod. 1996;59:73–76. doi: 10.1021/np960013j. [DOI] [PubMed] [Google Scholar]
- 34.Makler MT, Hinrichs DJ. Am J Trop Med & Hygiene. 1993;48:205–210. doi: 10.4269/ajtmh.1993.48.205. [DOI] [PubMed] [Google Scholar]
- 35.Makler MT, Ries JM, Williams JA, Bancroft JE, Piper RC, Gibbins BL, Hinrichs DJ. Am J Trop Med & Hygiene. 1993;48:739–741. doi: 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
- 36.Peters W, Portus JH, Robinson BL. Annals of Trop Med Parasitol. 1975;69:155–171. [PubMed] [Google Scholar]
- 37.Mikus J, Steverding D. Parasitol Int. 2000;48:265–269. doi: 10.1016/s1383-5769(99)00020-3. [DOI] [PubMed] [Google Scholar]
- 38.Borenfreund E, Babich H, Martin-Alguacil N. In Vitro Cellular & Developmental Biology: J Tissue Culture Asson. 1990;26:1030–1034. doi: 10.1007/BF02624436. [DOI] [PubMed] [Google Scholar]