

Abstract
Effective drug delivery to tumors requires both transport through the vasculature and tumor interstitium. Previously, it was shown that gold nanorod (GNR) mediated plasmonic photothermal therapy (PPTT) is capable of increasing the overall accumulation of N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers in prostate tumors. In the present study, it is demonstrated that PPTT is also capable of increasing the distribution of these conjugates in tumors. Gadolinium labeled HPMA copolymers were administered to mice bearing prostate tumors immediately before treatment of the right tumor with PPTT. The left tumor served as internal, untreated control. Magnetic resonance imaging (MRI) of both tumors showed that PPTT was capable of improving the tumor mass penetration of HPMA copolymers. Thermal enhancement of delivery, roughly 1.5-fold, to both the tumor center and periphery was observed. Confocal microscopy of fluorescently labeled copolymers corroborates these findings in that PPTT is capable of delivering more HPMA copolymers to the tumor’s center and periphery. These results further demonstrate that PPTT is a useful tool to improve the delivery of polymer-drug conjugates.
Keywords: HPMA copolymers, gold nanorods, photothermal therapy, hyperthermia, MRI, drug delivery
1. Introduction
The conjugation of hydrophobic anticancer drugs to water-soluble polymers represents an effective way of solubilizing them in blood plasma, prolonging blood circulation half-life, targeting biodistribution to tumors and overcoming multidrug resistance [1]. In this way, drugs can be retained in the blood and specifically delivered to the cancerous tissue with dramatically reduced accumulation in healthy organs. While the advantages of targeted delivery using polymer-drug conjugates are well known, clinical translation has been slow. There are many reasons why this is the case, including poor drug release kinetics and carrier biocompatibility. However, the major barrier to obtaining favorable clinical outcome remains limited tumor and cancer cell delivery [2].
There are many available techniques to improve the delivery of polymer-drug conjugates. The most obvious and widely used method involves tailoring the size of the conjugates so that the therapeutic takes advantage of the increased vascular permeability of tumors to macromolecules. Coined the ‘Enhanced Permeability and Retention’ (EPR) effect, large intercellular and transcellular openings between endothelial cells that line the tumor vasculature allow macromolecules up to roughly 1 μm in size to partition from the blood and enter the tumor interstitial space with limited lymphatic drainage [3–4]. Another common approach involves the conjugation of biorecognizable motifs such as peptides or antibodies for cancer cell receptor-mediated targeting [5]. Such active targeting then enables drug carriers to specifically bind to cancer cells which express the targeted receptor and trigger internalization and drug release. Finally, a number of other pharmacologic based methods for improving delivery have been shown including treatment with angiotensin to raise the patient’s blood pressure [6–8], application of nitroglycerin [9] or heme oxygenase-1 [10], pre-treatment with vascular disrupting [11–12] or anti-angiogenic agents [13–14], as well as direct injection of extracellular matrix enzymes to reduce the interstitial density [15]. Each of these tools provides greater selectivity of nanocarrier delivery to tumors.
Another technique which is shown to improve the delivery of nanocarriers involves treating the tumors with hyperthermia. Recent findings, for example, have shown that hyperthermia can increase the rate of both endo- and phagocytosis which may then potentiate macromolecular uptake and intracellular delivery [16–17]. At the vascular level, when tumors are heated up to 43°C, tumor blood flow can increase roughly two-fold [18]. This change in blood flow then increases the overall availability of macromolecules to extravasate. The resulting increased vascular pressure and heat-induced cytoskeletal injury then causes endothelial cell damage [19–21]. This causes further expansion of the intercellular openings and therefore increased vascular permeability to macromolecules [22–23].
The observed changes in tumor vascular dynamics with heating have been leveraged to improve the delivery of various nanomedicines. In particular, tumor hyperthermia has been used to facilitate the delivery of liposomes [24–28]. The application of heat is shown to enhance the extravasation of liposomes in a thermal dose dependent manner for up to six hours after heat treatment [29]. Additionally, this effect was also dependent on nanoparticle size where the larger systems exhibited the greatest increase in overall delivery with heat [30]. Precise control over heating, however, is necessary as vascular collapse and blood flow stasis is probable when temperatures rise above 43°C.
A major challenge with treating tumors with hyperthermia lies in the ability to effectively deliver the appropriate thermal dose in a site specific manner. Evolving technologies such as radiofrequency ablation as well as high-intensity focused ultrasound (HIFU) have proven useful in this regard [31], though these methods are not selective towards cancerous tissue and therefore rely on the physician to choose the regions which should receive thermal therapy. As the margins of tumor and normal tissue are often unknown, this adds greater risk of injury to healthy tissue. A recent method of selectively delivering heat to tumors takes advantage of the plasmonic properties of colloidal gold. Of special interest in this regard is the unique capacity of these colloids to scatter and absorb light. Under conditions of surface plasmon resonance (SPR), strong light absorption results in particle heating [32]. When located within a tumor mass, direct tissue heating can occur with laser light excitation by plasmonic photothermal therapy (PPTT) [33–35]. This heating process can be used as a means to selectively induce tumor hyperthermia with therapeutic intentions [36–38].
Recent studies have shown the utility of PPTT to improve the delivery of other nanomedicines [39–43] as well as radiotherapy [44]. In each of these studies, PPTT was applied to heat tumors between 42–45°C and a resulting increase in conjugate accumulation was observed. In a previous study, the tumor accumulation of heat shock targeted N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers was evaluated in combination with PPTT [40]. A peptide which has known affinity for an extracellular heat shock protein was incorporated in the polymer design to specifically target cancer cells treated with hyperthermia. PPTT for 10 minutes at 43°C caused a burst accumulation of the conjugates for up to 4 hours. After four hours, while the untargeted conjugates diffused back out of the tumor, the heat shock targeted conjugates were retained for an extended period of time (up to 12 hours) due to cell specific targeting [40]. These results provided evidence for the utility of this approach.
What remains unknown, however, is the tumor tissue distribution of HPMA copolymers after delivery enhancement with PPTT. This information is important because drug delivery is not evenly distributed due to tumor vascular heterogeneity, particularly for nanomedicines which are larger in size [45–46]. The objective of this study was to visualize the distribution of HPMA copolymers in prostate tumors after treatment with PPTT.
2. Materials and Methods
2.1 Synthesis and characterization of PEGylated GNRs
Poly(ethylene glycol) (PEG) coated gold nanorods (GNRs) were synthesized as described previously [40]. GNR size and shape were characterized by transmission electron microscopy (TEM) and the light absorption profile was measured by UV spectrometry. Zeta potential was calculated in deionized (DI) water by measuring its electrophoretic mobility using laser Doppler velocimetry (Zetasizer Nano ZS, Malvern Instruments Ltd, Worcestershire, UK).
2.2. Synthesis and characterization of HPMA copolymers
HPMA [47], aminopropylmethacrylamide-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (APMA-DOTA) [48], 5-[3-(methacryloylaminopropyl)thioureidyl] fluorescein (APMA-fluorescein) [49], and 3-[(N-methacryloylglycyl)glycyl]thiazolidine-2-thione (MA-GG-TT) [50] comonomers were synthesized as described previously. The precursor copolymer conjugates contained the reactive carboxyl groups (thiazolidine-2-thione) so that future studies with the same copolymer could incorporate targeting peptides into their design. In the present study, these groups were hydrolyzed to obtain untargeted conjugates. Copolymerization was performed by reversible addition-fragmentation chain-transfer (RAFT) polymerization using 2-cyano-2-propyl dodecyl trithiocarbonate as the chain transfer agent and VA-044 as the initiator in a DMF/MeOH (90:10) co-solvent at 50°C for 24 hrs. The unpurified product was then dissolved in DI water with gadolinium (Gd) (III) acetate hydrate (1.2 mol equivalent) and the pH was raised between 5.0–5.5. This solution was stirred overnight followed by addition of ethylenediaminetetraacetic acid (EDTA) to remove excess Gd (EDTA:GD, 1:1). The product was then filtered, dialyzed and lyophilized to obtain the final product. Mw, Mn, and Mw/Mn were estimated by size exclusion chromatography (SEC) using HPMA homopolymer fractions of known molecular weight. The Gd content was quantified by inductively coupled plasma mass spectrometry (ICP-MS) against a standard curve. Fluorescein labeled polymers were synthesized as described previously [40].
To calculate the conjugate’s longitudinal relaxivity, four different concentrations of copolymer (0.1 to 0.015 mM polymer) were prepared in DI water and placed in a Bruker BioSpec 7.1 T horizontal-bore MRI. T1 was measured by an inversion recovery fast spin-echo imaging sequence using inversion times of 50, 100, 300, 500, 800, 1000, 2000, 4000, 7000 and 8000 ms, echo time (TE) of 4.2 ms, and repetition time (TR) of 12000 ms. T1 for each vial was calculated using Bruker software and the relaxation rate (R1 = 1/T1) was plotted against Gd equivalent concentration. Relaxivity was measured as the slope of this plot.
2.3. Prostate tumor model
Animal experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of the University of Utah. Four-to-six week old female athymic (nu/nu) mice were anesthetized using 2% isoflurane and bilaterally inoculated with 107 DU145 prostate cancer cells in 200 μl phosphate buffered saline (PBS) on the flank of each animal. Animals were used in the study once the average tumor volume reached 50–100 mm3 (usually 10–21 days).
2.4. MR imaging
Prior to the experiment, those animals which were ultimately treated with PPTT received an intravenous dose of PEGylated GNRs (9.6 mg/kg) roughly 48 hrs before each experiment. This provided enough time for the GNRs to circulate and passively accumulate in the tumor tissue [51]. The animals in the laser only group did not receive GNRs. Each animal was then anesthetized with 2% isoflurane, placed within a Bruker BioSpec 7.1 T horizontal-bore MRI and an axial T1 flash pre-scan was taken. Each tumor was then swabbed with 50% propylene glycol to enhance laser penetration depth [52], and the Gd labeled HPMA copolymers were then intravenously administered (0.03 mmol Gd/kg) in saline. Immediately after injection, the right tumor was radiated for 10 minutes using an 808 nm fiber coupled laser diode (Oclaro Inc., San Jose, CA) with collimating lens (Thorlabs, Newton, NJ). Intratumoral temperature was monitored using a 33 gauge needle thermocouple (Omega, Stamford, CT), and the laser power (roughly 1.2 W/cm2) was directly controlled so that tumor temperature was maintained between 42°C and 43°C when treated with PPTT. Tumors on the left flank served as internal controls.
Immediately following laser treatment, the bed was placed back into the MRI and axial section T1 flash images of both tumors were taken. Two hours after laser treatment, a series of multislice T1 flash images were taken to provide anatomical information. Also, quantitative T1 MR images were obtained using an inversion recovery fast spin-echo pulse imaging sequence (slice thickness = 1 mm, number of slices = 8). This was done using the same acquisition parameters when the conjugate’s relaxivity was measured. In one animal, both T1 flash and T1 inversion recovery images were acquired before treatment (pre-scan), and then every 42 minutes for five hours after treatment. This was done to obtain time-dependent information on conjugate delivery.
2.5. Image analysis
The quantitative T1 relaxation maps were calculated from the inversion recovery data sets using QuickVol II, a plugin for Image J [53]. The images were then prepared in the following way. Using the T1 flash images, regions of interest (ROI) were drawn around both tumors and a mask was created for each slice. The mask was then used to isolate only the tumor data from the T1 relaxation maps. Due to some noise in the data, pixel outliers were removed using Image J. Each map was then overlayed onto its respective T1 flash image and a montage was created to display the whole tumor volume or the time dependent information as well as a 3D surface plot.
To compare the accumulation of polymers in both tumors, the average relaxation (R1 = 1/T1) was calculated in the skin around the tumor as well as the tumor’s center and periphery using Image J. This was done by first drawing ROIs on the T1 flash images (without the T1 map overlay) for both tumors. These ROIs were then used to calculate the average relaxation in each of these regions on the T1 relaxation map. The TER for each of these regions was then calculated by dividing R1 in the right tumor by R1 in the left tumor (TERPPTT = R1PPTT/R1control, or TERlaser only = R1laser only/R1control). This was done for each mouse, tumor and slice. A normalized histogram of R1 values for whole tumors (center plus periphery) was also obtained using Image J software. This data was then graphically represented using GraphPad Prism.
2.6. Histology
After MR imaging, the animals were euthanized by CO2 inhalation and the tumors were removed and fixed in neutral buffered formalin. Samples were then dehydrated, paraffin-embedded and cut into 4-micron thick sections. Immunohistochemical (IHC) analysis of Factor VIII expression, an endothelial cell marker, was then performed on some sections using a Factor VIII rabbit polyclonal antibody (Dako, Carpinteria, CA) and a biotinylated rabbit IgG secondary antibody. Positive signal was visualized using a streptavidin-HRP system, utilizing DAB (3,3′-diaminobenzidine) as the chromogen. All sections were then counterstained with hematoxylin. Washing with an iodine solution, followed by sodium thiosulfate, removed any precipitates. Finally, sections were dehydrated in alcohol, rinsed in xylene, coverslipped and imaged.
2.7. Fluorescence imaging
Prior to the experiment, animals received an intravenous dose of PEGylated GNRs (48 hrs before) and both tumors were swabbed with 50% propylene glycol (10 min before). Each animal (N = 4) was then intravenously administered 7.0 mg of FITC labeled HPMA copolymers and the right tumor was lased in the same way as the MRI experiment. Two hours after treatment with PPTT, each animal was then administered 5 mg of rhodamine labeled Concanavalin A (Vector Laboratories, Burlingame, CA) and euthanized 5 min later to visualize the vasculature. Both tumors were then collected, immediately placed in Tissue-Tek® OCT™ compound, frozen and cryo-sectioned into 12 μm thick sections (Leica CM3050, Wetzlar, Germany). Immediately before imaging, slides were dried and a cover slip was mounted using Cytoseal 60 diluted 1:5 in toluene. Large fluorescent imaging mosaics were acquired using a Nikon A1 confocal laser microscope system with a 10x objective.
2.8. Statistics
Statistical analyses were performed in GraphPad Prism. Comparisons between two groups (left vs. right tumors), were performed using a two-tailed, Welch-corrected unpaired t-test. P-values less than 0.05 were considered statistically significant. Data reported as mean ± SEM.
3. Results
3.1. GNR and HPMA copolymer synthesis and characterization
The GNRs were synthesized to be 58.6 ± 5.7 × 15.4 ± 0.8 nm in size which corresponds to an aspect ratio of 3.8 and a SPR peak at 800 nm (Fig. 1 A–B, Table 1). After PEGylation, the GNRs had a slightly negative zeta potential of −10 mV. These GNRs were found to be stable in a wide variety of buffers and solvents due to steric protection from aggregation.
Fig. 1. GNR characterization and HPMA copolymer schematic.

GNRs were synthesized to be 58.6 × 15.4 nm in size (A) with an SPR peak at 800 nm (B). Scale bar, 100 nm. Two HPMA copolymers were synthesized for this study (C). The first was copolymerized with HPMA (i), DOTA to chelate Gd to provide MRI contrast (ii), and a hydrolyzed reactive carboxyl group to enable targeting in future studies using the same copolymer (iv). The other was copolymerized with HPMA (i), APMA-FITC for fluorescent imaging (iii), and a hydrolyzed reactive carboxyl group for the same reasons (iv).
Table 1.
Physicochemical characteristics of GNRs and HPMA copolymers.
| Size (nm} | SPR (nm) | Zeta potential (mV) | |
|---|---|---|---|
| GISRs | 56.6 ± 5.7 × 15.4 ± 0.8 | 800 | −10 |
| HPMA (mol %) | APMA-DOTA-Gd (mol %) | APMA-FITC (mol %) | MA-GG-TT (mol%) | Gd content (mmol Gd/g polymer) | Apparent Mw (kDa) | Mw/Mn | Relaxivity (s−1 mM Gd−1) | |
|---|---|---|---|---|---|---|---|---|
| HPMA copolymer-Gd | 85 | 10 | 0 | 5 | 0.37 | 64.9 | 1.3 | 7.1 |
| HPMA copolymer-FITC | 93 | 0 | 2 | 5 | - | 62.4 | 1.4 | - |
The HPMA copolymers were synthesized by RAFT copolymerization to be roughly 65 kDa so that they were slightly above renal threshold to take advantage of the EPR effect (Fig. 1 C, Table 1). In order for the copolymers to be imaged by MRI, they contained APMA-DOTA comonomers which chelate Gd. For copolymers used for fluorescent imaging, APMA-FITC was used instead. An additional comonomer with a reactive carboxyl group, MA-GG-TT, was also incorporated so that future studies could incorporate receptor-mediated active targeting using the same copolymer if needed. In the present study, the TT group was hydrolyzed to obtain an untargeted conjugate. The copolymer Gd content was found to be similar to HPMA copolymer conjugates synthesized previously [48].
3.2. HPMA copolymer tumor delivery two hrs after treatment by MRI
In this experiment, half the animals were administered PEGylated GNRs (N = 3) and the other half saline (N = 3) 48 hrs before MR imaging. On the day of the experiment, each animal was administered HPMA copolymer-Gd conjugates prior to laser radiation of the right tumor for 10 minutes. The tumors in animals which were previously given GNRs exhibited rapid heating which was maintained near 43°C. Animals without GNRs (laser alone) were slightly heated which is consistent with previous results [39]. Two hours after laser treatment, quantitative MR imaging shows significantly enhanced copolymer delivery to tumors treated with PPTT (Fig. 2). While the left tumor (control) had some polymer accumulation, the right tumor (PPTT) displayed signs of greater copolymer accumulation. As expected, in both tumors the delivery was not evenly distributed likely due to vascular heterogeneity. However, the extent of distribution does appear greater in tumors treated with PPTT. In animals not previously given GNRs, differences between the laser radiated (right) and control (left) tumors were not apparent (Fig. 2).
Fig. 2. HPMA copolymer delivery two hours after treatment.

Laser treatment of the right tumor in animals previously administered GNRs (PPTT) facilitated significant enhancement of HPMA copolymer delivery in terms of both accumulation and overall tumor distribution (A, top row). Laser alone did not cause any increased delivery (A, bottom row). A 3D surface plot provides better visualization of this effect at a single slice (B).
When the relaxation rate (R1) was quantified for the skin around the tumors as well as the tumor’s center and periphery and expressed as a thermal enhancement ratio (TER, R1PPTT/R1control, or R1laser only/R1control), a clear trend was observed (Fig. 3A). Treatment with PPTT resulted in increased copolymer delivery to each of these regions. PPTT caused a 1.36-fold increase in delivery to the skin, though this difference was not statistically significant relative to the laser only group due to large variability in the data and some enhancement to the skin with laser alone (ns, p = 0.434). For the tumor’s center, PPTT significantly raised the copolymer concentration by 1.42-fold relative to those animals treated with laser only (*, p = 0.049). The periphery of the tumors, regions of the tumors not including their center, saw the greatest thermal enhancement of 1.54-fold and was the most significant (*, p = 0.016). When the right tumors were treated with laser alone, i.e. no GNRs present, the same effect was not observed (Fig. 3A). Treatment with laser alone did not cause any appreciable increase in copolymer delivery to the skin, tumor center or periphery.
Fig. 3. Image analysis of HPMA copolymer delivery.

Treatment of tumors with PPTT was capable of significantly enhancing the delivery of HPMA copolymers to the tumor’s center and periphery two hours after treatment (A, left). Treatment with laser alone, absence of GNRs, did not increase delivery. Representative ROIs for this analysis is also shown (A, right). A histogram of R1 values of both control and PPTT treated tumors is shown (B). This data shows the capability of PPTT to increase tumor mass distribution. *Indicates a statistically significant difference (p < 0.05) by t-test. Error bars represented as ±standard error of the mean.
Plotting a histogram of R1 values for control and PPTT treated tumors provides some additional information (Fig. 3B). In control tumors, the majority of its volume comprised of relatively low R1 and therefore polymer concentration values. This distribution was Gaussian (R2 = 0.83) and centered at roughly R1mean = 1.02 s−1 with a narrow standard deviation of σ = 0.28. For tumors treated with PPTT, the distribution was less Gaussian (R2 = 0.67), centered at R1mean = 2.31 s−1 and exhibited a much broader distribution of σ = 1.03. This indicates that the majority of the tumor volume received a greater and more variable distribution of copolymer delivery, though overall its entire volume received more copolymers. All regions in the PPTT histogram which do not overlap with the control histogram received some benefit of delivery due to therapy.
3.3. HPMA copolymer tumor delivery over time
To better understand the kinetics of delivery, one animal was imaged for five hours after treatment with PPTT (Fig. 4). Similar to previous findings [40], the majority of the dose was delivered to tumors within the first hour of treatment. Two-to-three hours after treatment accumulation increased with time. In the control tumor, copolymer delivery was observed as expected, though the increase in R1 was slight. In the right tumor treated with PPTT, large differences in R1 were observed mostly within the first hour.
Fig. 4. HPMA copolymer delivery over time.
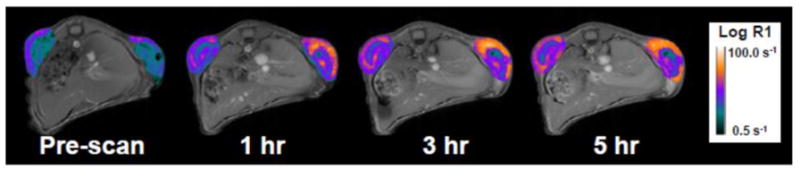
The majority of delivery occurs within the first hour of PPTT treatment.
3.4. Fluorescence imaging of HPMA copolymer delivery
When the tumor delivery two hours after treatment was visualized by fluorescent imaging, a similar trend was observed (Fig. 5). Treatment with PPTT facilitated more copolymer delivery overall. Delivery enhancement occurred mostly in the outer rim of the tumor, though increased delivery in the tumor’s center was also observed. By this method, overall enhancement of delivery was less pronounced than those results obtained by MRI. In both tumors, vascularization density appeared the same. Interestingly, HPMA copolymer delivery did not directly correlate with location of blood vessels.
Fig. 5. Fluorescent imaging of HPMA copolymer delivery.
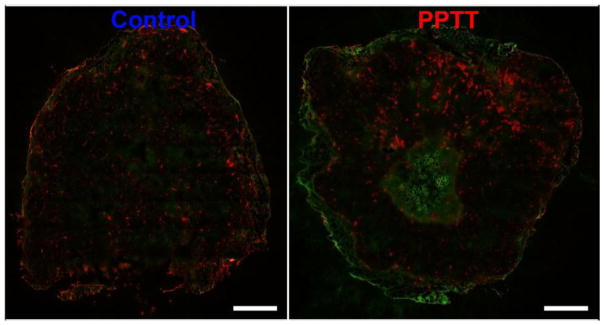
Two hours after treatment with PPTT, HPMA copolymer delivery (green) enhancement in the tumor’s periphery and center is observed relative to untreated controls. This effect is most pronounced in the tumor’s periphery. Blood vessel (red) density does not appear to be affected by treating with PPTT. Scale bar = 1 mm.
3.5. Histology of tumors after treatment with PPTT
After imaging, the right and left tumors were evaluated for damage by histology (Fig. 6). In both the control and PPTT treated tumors, many blood and lymphatic vessels were observed throughout and appeared to be intact (Fig. 6A–D). There was, however, one major difference between the groups. In all PPTT treated tumors, areas of tissue damage and cell death were observed (Fig. 6E–F). These regions of damage were typically less than 20% of the tumor’s total volume and were usually confined to the tumor’s center. In some cases, intact blood vessels were found in these areas indicating that vessel injury was not necessarily the reason for damage (Fig. 6F). Also, tumors treated with PPTT did not appear to have as many dividing cells further suggesting that some damage had occurred.
Fig. 6. Histology of control and PPTT treated tumors.

No differences between the tumor periphery of both control and PPTT treated tumors were observed (A–B, 10x objective, scale bar = 100 μm). IHC staining of blood vessels (BV) in the periphery did not provide evidence of damage in either group (C–D, 20x objective, scale bar = 50 μm). The center of tumors treated with PPTT had evidence of cell and tissue damage most likely due to excessive heating in this region (E, 10x objective, scale bar = 100 μm). A higher resolution view of this area shows the presence of capillary blood vessels which appear viable despite surrounding damage (F, 20x objective, scale bar = 50 μm).
4. Discussion
It was shown previously that PPTT can be used to effectively deliver greater numbers of nanocarriers to solid tumors [39–43]. But simply delivering more drugs to tumors may not necessarily improve overall delivery to cancerous cells. For example, excessive delivery of drug to only perivascular regions and not areas which are distant from viable vasculature may not improve overall treatment outcome. For this reason, delivery strategies which also increase tumor mass penetration are equally important [45].
There are many tumor tissue abnormalities that resist efficient transport of small and macromolecules to all cells. Most importantly, unregulated angiogenesis causes blood vessels to form with an abnormal and disorganized architecture [54–55]. The spatial distribution of blood vessels lacks order and continuity which ultimately generates a heterogenous distribution of tissue which is poorly perfused [56]. Upon entering the tumor microenvironment, efficient transport through the interstitium is further restricted due to several other abnormalities [57]. High extracellular matrix (ECM) densities in the tumor interstitium prevent large objects such as nanocarriers from diffusing freely [58]. Also, poor lymphatic drainage and related high interstitial fluid pressure (IFP) restricts fluid transport, particularly to the tumor’s center where pressure is the highest [59]. Finally, nonspecific binding to ECM or cellular components due to charge-charge or Van der Waals interactions restricts motion.
Using hyperthermia to improve the delivery of polymer-drug conjugates may offer distinct advantages towards maximizing nanocarrier delivery in the context of interstitial transport. To describe interstitial transport or flux, Ji, one must consider the contributions to convective, Jc, and diffusive, Jd, molecule transport [57, 60]:
| (1) |
where D is the diffusion coefficient of the nanocarrier, C and ∂C/∂x is the concentration and concentration gradient respectively, RF is the retardation factor, K is the tissue’s hydraulic conductivity, and ∂p/∂x is the pressure gradient in the tissue. During mild hyperthermia (T ≤43°C), it is known that tumor blood flow increases most likely due to increases in blood flow from the host vessels or increased cardiac output [18]. The result is increased microvascular pressure and therefore passive dilation of tumor blood vessels. Also during hyperthermia it has been found that IFP decreases [61]. The result of these two phenomenon is an increase in ∂p/∂x and therefore interstitial convective transport.
Regarding how such a combination might be beneficial from a diffusive standpoint, one must look at the polymer’s diffusion coefficient relative to other nanocarriers and the temperature of the environment. Using the Stokes-Einstein equation, D is calculated using the following relationship:
| (2) |
where k is Boltzmann’s constant, T is the absolute temperature, n is viscosity and R is the carrier’s hydrodynamic radius. Here, it is shown that diffusive transport is higher for smaller nanocarriers (lower R) and at elevated temperatures (higher T, lower n). Also, as shown previously [40], hyperthermia is able to increase the overall amount of copolymer delivery which therefore causes an increase in ∂C/∂x.
The above highlights a) the importance of nanocarrier size on interstitial diffusive transport, and b) the advantages that mild hyperthermia provides for improving both convective and diffusive transport. In this context, using heat to drive the distribution of polymer-drug conjugates which are typically less than 15 nm in hydrodynamic diameter may offer the best opportunity to increase tumor tissue delivery. This may also be true as flexible, linear polymers such as HPMA copolymers have been shown to have greater transport properties than branched or rigid systems [62–63]. Of course, a balance between favorable mass penetration and unfavorable renal clearance for these polymers is a necessary consideration in therapy design.
In this study, PPTT was used to selectively heat prostate tumors between 42–43°C to facilitate the delivery of HPMA copolymers. Using PPTT in this way decreases the chances of heating healthy tissue due to tumor specific delivery of GNRs by EPR and provides a high degree of control over heating. It is also possible that using tissue embedded antennas for energy generation, similar to brachytherapy, provides an advantage in terms of heat distribution. Also, PPTT may have other unknown benefits as it has clearly been shown to improve the delivery of both albumin [39] and HPMA copolymers [40], whereas previous studies using other methods to induce hyperthermia have not shown greater delivery of each of these [30, 64].
When PPTT was used to direct the delivery of Gd labeled HPMA copolymers to the right tumor of the animal and imaged by MRI, a clear difference between the tumors was observed (Fig. 2). The right tumor exhibited greater T1 contrast and therefore polymer concentration than the left, untreated tumor. In these images, it is observed that these treated tumors did not just receive higher amount of copolymer accumulation. Rather, more of the tumor volume overall received greater delivery suggesting greater tumor mass penetration of these copolymers due to PPTT. This is in stark contrast to tumors treated with laser but not previously administered GNRs, showing that PPTT is in fact responsible and not just laser radiation itself. Though it would be ideal to have an even distribution of high copolymer concentrations throughout the whole tumor volume, the images show that this is not the case. There still exist regions in the tumor that did receive less copolymer. These regions are likely to be the necrotic core of the tumor and are thus difficult to reach.
When these images were analyzed for thermal enhancement of delivery based on region (skin, tumor center and periphery), in each case thermal enhancement was observed (Fig. 3A). For the skin, this difference was not statistically significant from laser alone though enhanced delivery to the skin is expected during heating. Both the tumor’s center and periphery had a roughly 1.5-fold increase in delivery, where delivery to the center was barely significant and the periphery highly significant. This analysis further suggests that greater mass penetration to the tumor’s center was achieved. This interpretation should be taken with a degree of caution however. Using MRI, it is very difficult to delineate the viable from the necrotic regions of the tumor. The ROIs were drawn based on what appeared to be the tumor’s center and periphery, but this does not necessarily reflect the true location of the tumor’s necrotic core. Vascular heterogeneity often makes the location of the tumor’s core highly variable which explains why there is large variation in the data and limited significance in the tumor’s center.
A better representation of mass penetration is found is Fig. 3B. In this histogram, it is easy to observe that PPTT caused more of the tumor to have higher concentrations of copolymer than the control tumor. For example, let us consider the total percent area of the tumor (area under the curve) less than and greater than R1 = 1.34 s−1 for both tumors. In the control (untreated) tumors, 83.3% of its area had R1 values less than 1.34 s−1. In PPTT treated tumors, 46.5% of its area was less than 1.34 s−1 and 53.5% of its area had R1 values over this threshold. This data further suggests that higher concentrations of copolymers were able to more readily be transported through the tumor’s interstitium when treated with PPTT.
The fact that the majority of delivery occurred within the first hour of laser radiation is not surprising (Fig. 4). In the previous study where HPMA copolymer delivery was quantified, roughly 60–70% of the copolymers were delivered within the first 15 minutes after laser radiation [40]. This is likely due to two main reasons. First, clearance of the HPMA copolymers from the blood via the kidneys or reticuloendothelial system (RES) is likely due to the conjugate’s size and negative charge which makes the window of opportunity for delivery within the first few hours. Second, the differential increase in pressure, ∂p/∂x, which drives delivery during hyperthermia occurs over a short period of time (10–15 minutes). For this reason, future work aims to investigate longer periods of laser radiation to further improve delivery.
To corroborate the MRI results, FITC labeled HPMA copolymers were applied using the same experimental design to image distribution by fluorescence imaging. Because the location of copolymer delivery relative to vascularization is important, animals were administered rhodamine labeled lectin prior to euthanasia to stain the tumor’s vasculature. These results provide similar information in that PPTT increased delivery to both the tumor’s periphery and center. This observation was however less pronounced than the MRI results which may be due to the relative small z-direction cross-section; 0.012 mm vs. 1.0 mm for fluorescence and MR imaging respectively. It was anticipated that the location of blood vessels would directly correlate with the location of copolymer delivery. While this was indeed the case in the tumor’s outer rim where the blood vessels were of the greatest diameter (Fig. 6C–D), the inner capillaries did not permit much delivery. This is likely because of their small and constricted nature due to high cellular density and interstitial pressure in these regions.
Histological evaluation of the tumors provides some insight into the impact of mild PPTT on tumor viability. In both tumors, control and PPTT treated, the tumor’s periphery appeared undamaged with a high degree of vascularization (Fig. 6A–D). No signs of vascular damage was observed which is a concern when tumors are heated near the 43°C vascular damage threshold [65]. There was a difference though in the tumor’s center. In each of the PPTT treated tumors, areas of tissue damage were observed (Fig. 6E–F). This is likely to be because of the inability of the tumor’s center to dissipate heat effectively during hyperthermic treatment. Residual increases in temperature in these regions then cause direct cell death and tissue damage. What was interesting, however, was the presence of viable blood vessels found in these regions (Fig. 6F). These capillary blood vessels appeared to be intact, though further analysis of blood vessel viability in these regions is required to confirm this observation.
Effective transport of nanocarriers through the tumor’s interstitium remains a major challenge in nanomedicine. Large nanoparticles such as liposomes, micelles and inorganic nanoparticles are severely limited by this biological barrier, though strategies such as the one described in this study may help overcome this. An evolving and interesting concept to overcome the problem involves the design of multistage drug delivery platforms [66]. In these nanoparticle systems, large nanoparticles carry drugs to the site of the tumor followed by release of free drug in the interstitium due to external stimuli such as light and heat. In this way, such systems take full advantage of EPR and the high diffusivity of free drugs through the tumor’s interstitium.
5. Conclusions
This study shows by MR and fluorescence imaging that PPTT is capable of improving the tumor distribution of HPMA copolymers. During laser radiation of the tumor, heating may facilitate both convective and diffusive interstitial transport of these conjugates. PPTT was capable of not only providing greater amounts of copolymer delivery, but also more pervasive distribution throughout the whole tumor mass. This observation is important for more effective drug delivery to cancerous cells. Necrotic and unavailable regions of the tumor were still present even after treatment with PPTT, and future studies improving delivery to these regions remains a significant challenge.
Acknowledgments
The authors thank Osama Abdullah for his help with the MRI imaging, Jacob Hinkle for his help with the image analysis, as well as Sheryl Tripp and Dr. Mohamed Salama at ARUP Laboratories for their help with histology preparation and interpretation. This research was supported by a Department of Defense Prostate Cancer Predoctoral Training Award (PC094496) as well as the National Institutes of Health (EB-R01EB7171) and the Utah Science, Technology, and Research (USTAR) Initiative.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 2.Duncan R, Gaspar R. Nanomedicine (s) under the microscope. Mol Pharm. 2011;8:2101–2141. doi: 10.1021/mp200394t. [DOI] [PubMed] [Google Scholar]
- 3.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 4.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki M, Hori K, Abe I, Saito S, Sato H. A new approach to cancer chemotherapy: selective enhancement of tumor blood flow with angiotensin II. J Natl Cancer Inst. 1981;67:663–669. [PubMed] [Google Scholar]
- 7.Li C, Miyamoto Y, Kojima Y, Maeda H. Augmentation of tumour delivery of macromolecular drugs with reduced bone marrow delivery by elevating blood pressure. Br J Cancer. 1993;67:975–980. doi: 10.1038/bjc.1993.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noguchi A, Takahashi T, Yamaguchi T, Kitamura K, Tsurumi H, Takashina K, Maeda H. Enhanced tumor localization of monoclonal antibody by treatment with kininase II inhibitor and angiotensin II. Jpn J Cancer Res. 1992;83:240–243. doi: 10.1111/j.1349-7006.1992.tb00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100:2426–2430. doi: 10.1111/j.1349-7006.2009.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang J, Qin H, Nakamura H, Tsukigawa K, Shin T, Maeda H. Carbon monoxide, generated by heme oxygenase-1, mediates the enhanced permeability and retention effect in solid tumors. Cancer Sci. 2012;103:535–541. doi: 10.1111/j.1349-7006.2011.02178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aicher KP, Dupon JW, White DL, Aukerman SL, Moseley ME, Juster R, Rosenau W, Winkelhake JL, Brasch RC. Contrast-enhanced magnetic resonance imaging of tumor-bearing mice treated with human recombinant tumor necrosis factor α. Cancer Res. 1990;50:7376–7381. [PubMed] [Google Scholar]
- 12.Zhao L, Ching LM, Kestell P, Kelland LR, Baguley BC. Mechanisms of tumor vascular shutdown induced by 5, 6-dimethylxanthenone-4-acetic acid (DMXAA): Increased tumor vascular permeability. Int J Cancer. 2005;116:322–326. doi: 10.1002/ijc.21005. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 14.Chauhan VP, Stylianopoulos T, Martin JD, Popovi Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D, Jain RK. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383–388. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magzoub M, Jin S, Verkman A. Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. FASEB J. 2008;22:276–284. doi: 10.1096/fj.07-9150com. [DOI] [PubMed] [Google Scholar]
- 16.Vega VL, Charles W, De Maio A. A new feature of the stress response: increase in endocytosis mediated by Hsp70. Cell Stress Chaperones. 2009;15:517–527. doi: 10.1007/s12192-009-0165-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vega VL, De Maio A. Increase in phagocytosis after geldanamycin treatment or heat shock: role of heat shock proteins. J Immunol. 2005;175:5280–5287. doi: 10.4049/jimmunol.175.8.5280. [DOI] [PubMed] [Google Scholar]
- 18.Song CW. Effect of local hyperthermia on blood flow and microenvironment: a review. Cancer Res. 1984;44:4721s–4730s. [PubMed] [Google Scholar]
- 19.Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, Felix R, Riess H. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol. 2002;43:33–56. doi: 10.1016/s1040-8428(01)00179-2. [DOI] [PubMed] [Google Scholar]
- 20.Fajardo L, Schreiber A, Kelly N, Hahn G. Thermal sensitivity of endothelial cells. Radiat Res. 1985;103:276–285. [PubMed] [Google Scholar]
- 21.Chen B, Zhou M, Xu L. Study of vascular endothelial cell morphology during hyperthermia. J Therm Biol. 2005;30:111–117. [Google Scholar]
- 22.Lefor AT, Makohon S, Ackerman NB. The effects of hyperthermia on vascular permeability in experimental liver metastasis. J Surg Oncol. 1985;28:297–300. doi: 10.1002/jso.2930280412. [DOI] [PubMed] [Google Scholar]
- 23.Fujiwara K, Watanabe T. Effects of hyperthermia, radiotherapy and thermoradiotherapy on tumor microvascular permeability. Pathol Int. 2008;40:79–84. doi: 10.1111/j.1440-1827.1990.tb01546.x. [DOI] [PubMed] [Google Scholar]
- 24.Kong G, Dewhirst MW. Hyperthermia and liposomes. Int J Hyperthermia. 1999;15:345–370. doi: 10.1080/026567399285558. [DOI] [PubMed] [Google Scholar]
- 25.Huang SK, Stauffer PR, Hong K, Guo JWH, Phillips TL, Huang A, Papahadjopoulos D. Liposomes and hyperthermia in mice: increased tumor uptake and therapeutic efficacy of doxorubicin in sterically stabilized liposomes. Cancer Res. 1994;54:2186–2191. [PubMed] [Google Scholar]
- 26.Weinstein J, Magin R, Yatvin M, Zaharko D. Liposomes and local hyperthermia: selective delivery of methotrexate to heated tumors. Science. 1979;204:188–191. doi: 10.1126/science.432641. [DOI] [PubMed] [Google Scholar]
- 27.Gaber MH, Wu NZ, Hong K, Huang SK, Dewhirst MW, Papahadjopoulos D. Thermosensitive liposomes: extravasation and release of contents in tumor microvascular networks. Int J Radiat Oncol Biol Phys. 1996;36:1177–1187. doi: 10.1016/s0360-3016(96)00389-6. [DOI] [PubMed] [Google Scholar]
- 28.Matteucci ML, Anyarambhatla G, Rosner G, Azuma C, Fisher PE, Dewhirst MW, Needham D, Thrall DE. Hyperthermia increases accumulation of technetium-99m-labeled liposomes in feline sarcomas. Clin Cancer Res. 2000;6:3748–3755. [PubMed] [Google Scholar]
- 29.Kong G, Braun RD, Dewhirst MW. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res. 2001;61:3027–3032. [PubMed] [Google Scholar]
- 30.Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: effect of particle size. Cancer Res. 2000;60:4440–4445. [PubMed] [Google Scholar]
- 31.Ma G, Jiang G. Review of tumor hyperthermia technique in biomedical engineering frontier. 2010; 3rd International Conference on Biomedical Engineering and Informatics (BMEI); IEEE. 2010. pp. 1357–1359. [Google Scholar]
- 32.Link S, El-Sayed MA. Shape and size dependence of radiative, non-radiative and photothermal properties of gold nanocrystals. Int Rev Phys Chem. 2000;19:409–453. [Google Scholar]
- 33.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci USA. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Stern JM, Stanfield J, Kabbani W, Hsieh JT, Cadeddu JA. Selective prostate cancer thermal ablation with laser activated gold nanoshells. J Urol. 2008;179:748–753. doi: 10.1016/j.juro.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Jain PK, El-Sayed IH, El-Sayed MA. Gold nanoparticles: interesting optical properties and recent applications in cancer diagnostics and therapy. Nanomed. 2007;2:681–693. doi: 10.2217/17435889.2.5.681. [DOI] [PubMed] [Google Scholar]
- 37.Jain PK, El-Sayed IH, El-Sayed MA. Au nanoparticles target cancer. Nano Today. 2007;2:18–29. [Google Scholar]
- 38.Huang X, Jain PK, El-Sayed IH, El-Sayed MA. Plasmonic photothermal therapy (PPTT) using gold nanoparticles. Lasers Med Sci. 2008;23:217–228. doi: 10.1007/s10103-007-0470-x. [DOI] [PubMed] [Google Scholar]
- 39.Gormley AJ, Greish K, Ray A, Robinson R, Gustafson JA, Ghandehari H. Gold nanorod mediated plasmonic photothermal therapy: A tool to enhance macromolecular delivery. Int J Pharm. 2011;415:315–318. doi: 10.1016/j.ijpharm.2011.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gormley AJ, Larson N, Sadekar S, Robinson R, Ray A, Ghandehari H. Guided delivery of polymer therapeutics using plasmonic photothermal therapy. Nano Today. 2012;7:158–167. doi: 10.1016/j.nantod.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc Natl Acad Sci USA. 2010;107:981–986. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park JH, Maltzahn Gv, Ong LL, Centrone A, Hatton TA, Ruoslahti E, Bhatia SN, Sailor MJ. Cooperative nanoparticles for tumor detection and photothermally triggered drug delivery. Adv Mater. 2010;22:880–885. doi: 10.1002/adma.200902895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Von Maltzahn G, Park JH, Lin KY, Singh N, Schwöppe C, Mesters R, Berdel WE, Ruoslahti E, Sailor MJ, Bhatia SN. Nanoparticles that communicate in vivo to amplify tumour targeting. Nat Mater. 2011;10:545–552. doi: 10.1038/nmat3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diagaradjane P, Shetty A, Wang JC, Elliott AM, Schwartz J, Shentu S, Park HC, Deorukhkar A, Stafford RJ, Cho SH. Modulation of in vivo tumor radiation response via gold nanoshell-mediated vascular-focused hyperthermia: characterizing an integrated antihypoxic and localized vascular disrupting targeting strategy. Nano Lett. 2008;8:1492–1500. doi: 10.1021/nl080496z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Mirovascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]
- 47.Strohalm J, Kopecek J. Poly N-(2-hydroxypropyl) methacrylamide. 4. Heterogeneous polymerization, Angew. Makromol Chem. 1978;70:109–118. [Google Scholar]
- 48.Zarabi B, Borgman MP, Zhuo J, Gullapalli R, Ghandehari H. Noninvasive monitoring of HPMA copolymer-RGDfK conjugates by magnetic resonance imaging. Pharm Res. 2009;26:1121–1129. doi: 10.1007/s11095-009-9830-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Omelyanenko V, Kopecková P, Gentry C, Kopecek J. Targetable HPMA copolymer-adriamycin conjugates. Recognition, internalization, and subcellular fate. J Control Release. 1998;53:25–37. doi: 10.1016/s0168-3659(97)00235-6. [DOI] [PubMed] [Google Scholar]
- 50.Šubr V, Ulbrich K. Synthesis and properties of new N-(2-hydroxypropyl) methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React Funct Polym. 2006;66:1525–1538. [Google Scholar]
- 51.Gormley AJ, Malugin A, Ray A, Robinson R, Ghandehari H. Biological evaluation of RGDfK-gold nanorod conjugates for prostate cancer treatment. J Drug Target. 2011;19:915–924. doi: 10.3109/1061186X.2011.623701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang RK, Tuchin VV. Enhance light penetration in tissue for high resolution optical imaging techniques by the use of biocompatible chemical agents. J X Ray Sci Tech. 2002;10:167–176. [PubMed] [Google Scholar]
- 53.Schmidt KF, Ziu M, Ole Schmidt N, Vaghasia P, Cargioli TG, Doshi S, Albert MS, Black PML, Carroll RS, Sun Y. Volume reconstruction techniques improve the correlation between histological and in vivo tumor volume measurements in mouse models of human gliomas. J Neurooncol. 2004;68:207–215. doi: 10.1023/b:neon.0000033364.43142.bf. [DOI] [PubMed] [Google Scholar]
- 54.Konerdingi M, Fait E, Gaumann A, Dimitropouloui C, Malkusch W. Scanning electron microscopy of corrosion casts in the study of tumor. In: Maragoudakis ME, editor. Angiogenesis: models, modulators, and clinical applications. Plenum Press; New York, USA: 1998. pp. 429–447. [Google Scholar]
- 55.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–30. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 56.Baish JW, Jain RK. Fractals and cancer. Cancer Res. 2000;60:3683–3688. [PubMed] [Google Scholar]
- 57.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 58.Ramanujan S, Pluen A, McKee TD, Brown EB, Boucher Y, Jain RK. Diffusion and convection in collagen gels: implications for transport in the tumor interstitium. Biophys J. 2002;83:1650–1660. doi: 10.1016/S0006-3495(02)73933-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 1989;37:77–104. doi: 10.1016/0026-2862(89)90074-5. [DOI] [PubMed] [Google Scholar]
- 60.Swabb EA, Wei J, Gullino PM. Diffusion and convection in normal and neoplastic tissues. Cancer Res. 1974;34:2814–2822. [PubMed] [Google Scholar]
- 61.Leunig M, Goetz AE, Dellian M, Zetterer G, Gamarra F, Jain RK, Messmer K. Interstitial fluid pressure in solid tumors following hyperthermia: possible correlation with therapeutic response. Cancer Res. 1992;52:487–490. [PubMed] [Google Scholar]
- 62.Deen W, Bohrer M, Epstein N. Effects of molecular size and configuration on diffusion in microporous membranes. AIChE J. 1981;27:952–959. [Google Scholar]
- 63.Sadekar S, Ray A, Janat-Amsbury M, Peterson C, Ghandehari H. Comparative biodistribution of PAMAM dendrimers and HPMA copolymers in ovarian-tumor-bearing mice. Biomacromolecules. 2011;12:88–96. doi: 10.1021/bm101046d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lammers T, Peschke P, Kühnlein R, Subr V, Ulbrich K, Debus J, Huber P, Hennink W, Storm G. Effect of radiotherapy and hyperthermia on the tumor accumulation of HPMA copolymer-based drug delivery systems. J Control Release. 2007;117:333–341. doi: 10.1016/j.jconrel.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 65.Song CW, Kang MS, Rhee JG, Levitt SH. Effect of hyperthermia on vascular function in normal and neoplastic tissues. Ann NY Acad Sci. 1980;335:35–47. doi: 10.1111/j.1749-6632.1980.tb50735.x. [DOI] [PubMed] [Google Scholar]
- 66.Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popovi Z, Jain RK, Bawendi MG, Fukumura D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci USA. 2011;108:2426–2431. doi: 10.1073/pnas.1018382108. [DOI] [PMC free article] [PubMed] [Google Scholar]