

Abstract
Background
Aspirin or dual antiplatelet therapy (DAPT) with aspirin and clopidogrel is standard therapy for patients at increased risk for cardiovascular events. However, the genetic determinants of variable response to aspirin (alone and in combination with clopidogrel) are not known.
Methods and Results
We measured ex-vivo platelet aggregation before and after DAPT in individuals (n=565) from the Pharmacogenomics of Antiplatelet Intervention (PAPI) Study and conducted a genome-wide association study (GWAS) of drug response. Significant findings were extended by examining genotype and cardiovascular outcomes in two independent aspirin-treated cohorts: 227 percutaneous coronary intervention (PCI) patients, and 1,000 patients of the International VErapamil SR/trandolapril Study (INVEST) GENEtic Substudy (INVEST-GENES). GWAS revealed a strong association between single nucleotide polymorphisms on chromosome 1q23 and post-DAPT platelet aggregation. Further genotyping revealed rs12041331 in the platelet endothelial aggregation receptor-1 (PEAR1) gene to be most strongly associated with DAPT response (P=7.66×10−9). In Caucasian and African American patients undergoing PCI, A-allele carriers of rs12041331 were more likely to experience a cardiovascular event or death compared to GG homozygotes (hazard ratio = 2.62, 95%CI 0.96-7.10, P=0.059 and hazard ratio = 3.97, 95%CI 1.10-14.31, P=0.035 respectively). In aspirin-treated INVEST-GENES patients, rs12041331 A-allele carriers had significantly increased risk of myocardial infarction compared to GG homozygotes (OR=2.03, 95%CI 1.01-4.09, P=0.048).
Conclusions
Common genetic variation in PEAR1 may be a determinant of platelet response and cardiovascular events in patients on aspirin, alone and in combination with clopidogrel.
Clinical Trial Registration Information
clinicaltrials.gov; Identifiers: NCT00799396 and NCT00370045
Keywords: pharmacogenomics, platelets, percutaneous coronary intervention, PEAR1, CYP2C19
Aspirin or dual antiplatelet therapy (DAPT) with aspirin and clopidogrel is standard care for patients at increased risk for cardiovascular events. For patients with acute coronary syndrome undergoing percutaneous coronary intervention (PCI), DAPT is effective in reducing recurrent cardiovascular events1, 2. However, inter-individual variation in platelet response to aspirin, alone or in combination with clopidogrel, has been well documented3, 4, and patients who demonstrate higher on-treatment platelet reactivity on DAPT have increased risk of experiencing an ischemic event5-9. While the causes of inter-individual variability in response to DAPT are multifactorial10, the determinants of most of this variation remain unknown.
Recently, our group and others identified the loss-of-function CYP2C19*2 variant (rs4244285) as a major determinant of clopidogrel response as measured by adenosine diphosphate- (ADP) stimulated platelet aggregation11-16. Although this variant accounts for approximately 12% of the variation in inhibition of ADP-stimulated platelet aggregation by clopidogrel, and is associated with an approximately 1.5 to 2-fold risk of recurrent cardiovascular events in PCI patients, most of the variation potentially attributable to genetics remains unknown. For aspirin response, alone or in combination with clopidogrel, many genes have been examined10, 17-19; however, few if any have been reproducibly shown to be associated with response. In an attempt to identify the genetic determinants of platelet aggregation in response to aspirin in the setting of DAPT, we measured collagen-stimulated ex-vivo platelet aggregation at baseline, after clopidogrel administration, and after clopidogrel and aspirin administration in 565 healthy subjects of the Pharmacogenomics of Anti-Platelet Intervention (PAPI) Study, and conducted a genome-wide association study (GWAS). Significant findings in this group were replicated and extended by examining the relationship between genotype and cardiovascular outcomes in two independent populations: 227 patients recruited from a cardiac catheterization laboratory at Sinai Hospital in Baltimore, Maryland, and 1,000 patients of the International VErapamil SR/trandolapril Study (INVEST) GENEtic Substudy (INVEST-GENES).
Methods
Study Populations
The Amish PAPI Study (NCT0079936) recruited 668 healthy Caucasian individuals aged 20 years or older and has been described in detail previously11. This report includes 565 PAPI Study subjects who completed the intervention and in whom a complete set of validated collagen-stimulated platelet aggregation tests were available. Further information on this cohort can be found in the Supplemental Material. Briefly, platelet-rich plasma (PRP) was isolated from blood samples of participants and baseline platelet function was assessed by optical aggregometry using a PAP8E Aggregometer (Bio/Data Corporation, Horsham, Pennsylvania) after stimulation with collagen (5 μg/ml)11. Participants were then given a 300 mg loading dose of clopidogrel followed by 75 mg/d for 6 days. One hour following the last dose of clopidogrel, platelet aggregation measurements were repeated. To measure aspirin response in the presence of clopidogrel, platelet aggregation measurement was performed later the same day, 1 hour after a single dose of aspirin (324 mg).
The Sinai Hospital of Baltimore Study (NCT00370045) enrolled 227 patients older than 18 years and undergoing nonemergent PCI. Characteristics of these patients have been described previously11. Of the enrolled patients, 140 (61.7%) were Caucasian, 83 (36.6%) were African American, and 4 (1.8%) were of other race/ethnicity. Immediately before the PCI, patients received either a 600 mg (n = 112) or a 300 mg (n = 25) loading dose of clopidogrel; 90 patients were already receiving maintenance therapy with a 75 mg daily dose at the time of PCI and received no loading dose. Patients also received 81 to 325 mg of aspirin daily for at least 1 week prior to PCI and 325 mg on the day of the procedure. Platelet aggregation was assessed in PRP after stimulation with 2 μg/ml collagen using a Chronolog Lumi-Aggregometer (Model 490-4D; Chronolog, Havertown, Pennsylvania), as described previously8. Further information on this cohort can be found in the Supplemental Material.
Patients were contacted at the end of 1 and 12 months post-PCI to determine the occurrence of post-discharge cardiovascular ischemic events. A physician, blinded to the study results of the patient, adjudicated all end points through review of source documents obtained from medical records. Post-discharge ischemic events were defined as myocardial infarction (MI) (the occurrence of ischemic symptoms and troponin I value greater than the upper-limits of normal), ischemic stroke, stent thrombosis (definite stent thrombosis according to the Academic Research Consortium criteria), unplanned target vessel revascularization (revascularization of vessel treated at the time of study enrollment), hospitalization for coronary ischemia with revascularization (hospitalization for chest pain with evidence of ischemia on electrocardiogram and no evidence of myocardial infarction as measured by troponin I values), and death secondary to any cardiovascular cause.
The INVEST-GENES study has been previously described20. Briefly, DNA from 5,979 INVEST patients with hypertension and stable coronary artery disease residing in the mainland United States and Puerto Rico was collected. Using these samples, a nested case-control group consisting of 361 patients who experienced death, nonfatal myocardial infarction, or nonfatal stroke and 639 age-, sex-, and race-frequency-matched controls were evaluated. Components of the primary outcome were adjudicated by an independent adjudication committee21. Among these patients, 570 (56.8%) participants were Caucasian, 154 (15.3%) were African American, and 276 (27.5%) were Hispanic. Of these individuals, 499 were taking aspirin (dose unknown) and 501 were not. Further information on this cohort can be found in the Supplemental Material.
Study protocols were approved by the respective institutional review boards. Written informed consent was obtained from each participant; participants were compensated for their participation.
Genotype Analysis
Genotyping in the PAPI Study was performed with the Affymetrix GeneChip Human Mapping 500K or 1 M (version 6.0) arrays according to the manufacturer’s instructions (Affymetrix Inc., Santa Clara, California). Genotype calls were performed using BRLMM (500K array) or Birdseed version 2 (1 M array). Single nucleotide polymorphisms (SNPs) with minor allele frequencies greater than 1% present on both arrays were included in our analyses. Mean genotype call rate of these 400,230 SNPs was 98.7%. Follow-up genotyping of platelet endothelial aggregation receptor 1 (PEAR1) SNP rs12041331 in the replication/extension cohorts was performed using a TaqMan SNP genotyping assay (Applied Biosystems, Foster City, California). Genotype concordance rate of this polymorphism in a subset of duplicate samples was 100%.
Statistical Analysis
All Cohorts
Summary statistics and frequencies for the Amish PAPI Study, Sinai Hospital of Baltimore cohort, and INVEST-GENES were generated using SAS version 9.1 (SAS Institute Inc., Cary, North Carolina) or SPSS version 11.5 (Chicago, Illinois). Since the allele frequency of rs12041331 differed substantially among African Americans and Caucasians, we performed all analyses stratified by race. All tests evaluating association with collagen-stimulated platelet aggregation were conducted assuming an additive genetic model. Measures of Hardy-Weinberg equilibrium were calculated using a χ2 test. For the GWAS, we considered a P-value < 5 × 10−8 as evidence for genome-wide significance. For all other analyses, P-values less than 0.05 were considered statistically significant.
Amish PAPI Study
Correlates of aspirin response (e.g. age, sex, body mass index [BMI], lipid levels, and blood pressure) were evaluated using a regression-based approach as implemented in SOLAR version 4.07 (Southwest Foundation for Biomedical Research, San Antonio, Texas). Relatedness among study participants was accounted for by including a polygenic component as a random effect. Triglyceride levels were logarithm-transformed for analysis and back-transformed for presentation. Distribution analyses were generated in SAS. All statistical tests were 2-sided.
Genome-wide association analyses between SNPs and collagen-stimulated platelet aggregation at baseline, after clopidogrel, and after both clopidogrel and aspirin treatment were performed under a variance component model that assesses the effect of genotype as an additive effect on the quantitative trait, while simultaneously estimating the effects of age, age2, sex, baseline platelet aggregation (for post-clopidogrel and post-clopidogrel and aspirin analyses), and the aforementioned polygenic component. The polygenic component was modeled using the relationship matrix derived from the complete Amish pedigree structure available through the Anabaptist Genealogy Database22, 23. Follow-up analyses of SNPs of interest were performed using an additive model. Heritability of baseline platelet aggregation and aspirin response corresponds to the proportion of the trait variance accounted for by the polygenic component. The genomic control λ coefficient was 1.06; thus, the P-values reported were unadjusted. To determine if the PEAR1 SNP rs12041331 accounted for the chromosome 1q23 GWAS signal, we estimated the independent effects of both rs2148135 (the most strongly associated SNP with aspirin response from the genome-wide association analysis) and rs12041331 on platelet aggregation by including both in the model simultaneously.
Power calculations in the PAPI Study population (n=565) indicated 80% power at ά = 5.0 × 10−8 to detect SNPs with minor allele frequencies ranging from 0.2 to 0.4 with effect sizes from 0.43 to 0.35 SD, respectively, accounting for 6% of the phenotypic variation. Pairwise linkage disequilibrium (LD) statistics (D’ and r2) were calculated using Haploview version 4.2 (http://www.broadinstitute.org/haploview/).
Sinai Hospital of Baltimore Study
We estimated the effect of PEAR1 SNP rs12041331 genotype on post-aspirin collagen-stimulated platelet aggregation under an additive genetic model. The genotype effect was estimated using analysis of variance with adjustment for age, sex, race, study, and treatment group (clopidogrel dose and use of eptifibatide).
Survival curves were generated to evaluate 1-year cardiovascular event-free survival between patients with (n = 71) and without (n = 156) the PEAR1 rs12041331 A-allele. Relative hazard associated with having the A-allele on subsequent ischemic events was calculated using the proportional hazards model and was adjusted for age and sex. Survival analyses based on rs12041331 were performed both with and without collagen-stimulated platelet aggregation included as a mediator variable
INternational VErapamil SR/trandolapril STudy GENEtic Substudy
Characteristics of INVEST-GENES participants were compared by rs12041331 genotype using analysis of variance for quantitative traits or χ2 test for dichotomous traits. Association analyses of rs12041331 genotype on the occurrence of cardiovascular outcomes were calculated using logistic regression. Analyses stratified by aspirin use were also conducted. Interaction terms between rs12041331 genotype and aspirin use for each analysis were calculated. All tests were adjusted for age, sex, BMI, history of myocardial infarction, heart failure, diabetes, and smoking.
Results
Amish PAPI Study
Characteristics of the PAPI participants are shown in Table 1. Clopidogrel therapy alone resulted in mildly reduced collagen-induced platelet aggregation compared to baseline (% change in maximal aggregation ± SD = 17 ± 23%; P < 0.001); however, with the addition of aspirin, there was a marked effect (% change in maximal aggregation ± SD = 73 ± 16%; P < 0.001) (Supplementary Figure 1). Poorer DAPT response was associated with increasing age (12.1% of the variance; 95% confidence interval [CI], 11.9%-12.3%; P < 0.001) and female sex (2.5% of the variance; 95% CI 2.47%-2.58%; P < 0.001) (Supplementary Table 1). The estimated heritability of collagen-stimulated platelet aggregation at baseline, in response to clopidogrel, and in response to both clopidogrel and aspirin was 0.12 (SE, 0.10; 95% CI, 0.00-0.22; P = 0.81), 0.39 (SE, 0.12; 95% CI, 0.16-0.61; P < 0.001), and 0.65 (SE, 0.10; 95% CI, 0.46-0.84; P < 0.001) respectively.
Table 1.
Characteristics of Amish PAPI Study and Sinai Hospital of Baltimore Study Participants
| Amish PAPI Study* | Sinai Hospital of Baltimore PCI Patients |
INVEST Study Patients | ||||
|---|---|---|---|---|---|---|
| Characteristic (units) | Men | Women | Men | Women | Men | Women |
| Number (n) | 285 | 293 | 136 | 91 | 510 | 494 |
| Age ± SD (yr) | 43.9 ± 12.6 | 46.3 ± 13.9 | 62.5 ± 11.4 | 67.0 ± 11.0 | 66.7±9.8 | 68.4±10.1 |
| BMI ± SD (kg/m2) | 26.0 ± 3.6 | 28.0 ± 5.3 | 30.1 ± 6.3 | 30.9 ± 7.1 | 28.9±4.9 | 29.3±6.3 |
| African American (%) | 0.0 | 0.0 | 29.4 | 47.3 | 10.6 | 19.0 |
| Systolic blood pressure ± SD (mm Hg) | 115.8 ± 11.5 | 116.3 ± 13.7 | 138.0 ± 19.9 | 144.6 ± 19.9 | 147.6±18.0 | 152.0±17.9 |
| Diastolic blood pressure ± SD (mm Hg) | 70.4 ± 7.4 | 69.2 ± 7.6 | 73.6 ± 13.8 | 70.3 ± 14.2 | 84.8±11.3 | 86.1±10.9 |
| Hypertension (%)† | 4.2 | 5.8 | 75.0 | 79.1 | 100% | 100% |
| Total cholesterol ± SD (mg/dl) | 207.0 ± 43.8 | 212.5 ± 50.5 | NA | NA | NA | NA |
| LDL-cholesterol ± SD (mg/dl) | 138.2 ± 40.5 | 135.7 ± 46.7 | NA | NA | NA | NA |
| HDL-cholesterol ± SD (mg/dl) | 55.2 ± 14.7 | 61.7 ± 15.1 | NA | NA | NA | NA |
| Triglycerides ± SD (mg/dl)‡ | 67.8 ± 38.2 | 75.7 ± 43.9 | NA | NA | NA | NA |
| Hypercholesterolemia (%)§ | 23.5 | 24.3 | 81.6 | 79.1 | 62.6 | 52.0 |
| Taking aspirin (%) | 0.0 | 0.0 | 100 | 100 | 58.2 | 41.1 |
| Self-reported diabetes (%) | 0.7 | 0.7 | 28.7 | 48.4 | 24.5 | 28.1 |
| Hematocrit ± SD (%) | 41.5 ± 2.4 | 37.6 ± 2.2 | 41.8 ± 5.0 | 37.9 ± 4.5 | NA | NA |
| White blood cell count ± SD (n × 1,000) | 6.1 ± 1.4 | 6.1 ± 1.5 | 6.8 ± 1.3 | 7.5 ± 1.5 | NA | NA |
| Platelet count ± SD (n × 100,000) | 237.5 ± 43.6 | 246.9 ± 51.7 | 223.6 ± 68.5 | 252.2 ± 71.4 | NA | NA |
| Current smoker (%)∥ | 20.5 | 0.0 | 29.4 | 19.8 | 13.3 | 7.1 |
Abbreviations: BMI, body mass index; HDL, high-density lipoprotein; LDL, low-density lipoprotein; NA, not applicable; PAPI, Pharmacogenomics of Antiplatelet Intervention; SD, standard deviation.
SI conversion factors: To convert HDL-cholesterol, LDL-cholesterol, and total cholesterol values to mmol/L, multiply by 0.0259; triglycerides to mmol/L, multiply by 0.0113.
For PAPI Study, all participants were withdrawn from prescription and nonprescription medications, vitamins, and supplements 7 days prior to and for the duration of the study. Participants taking aspirin, antihypertensive, lipid-lowering, and diabetes medications accounted for less than 4% of participants.
Defined as systolic blood pressure greater than 140 mm Hg or diastolic blood pressure greater than 90 mm Hg or taking prescription medication for previously diagnosed hypertension.
Logarithm-transformed for analysis and back-transformed for presentation.
Defined as LDL-cholesterol greater than 160 mg/dl or taking prescription medication for previously diagnosed hypercholesterolemia.
Self-reported history of smoking cigarette, pipe, or cigar.
A GWAS of collagen-induced platelet aggregation revealed a cluster of 5 SNPs (rs2148135, rs11264825, rs6696137, rs7534239 and rs1176535) in high LD and in Hardy-Weinberg equilibrium (P > 0.05) spanning approximately 1 Mb on chromosome 1q23 showing strong association with DAPT response, with P values <10−7 (Figure 1, Supplementary Table 2). These SNPs were not associated with measures of platelet aggregation at baseline, and only nominal evidence of association was observed after clopidogrel treatment alone (P-value range = 2.62 × 10−2 – 7.58 × 10−4). No other regions throughout the genome revealed evidence of association with collagen-induced platelet aggregation after DAPT treatment that met or exceeded criteria for genome-wide significance (see Supplementary Table 2 for all SNPs with P-value < 10−5).
Figure 1.
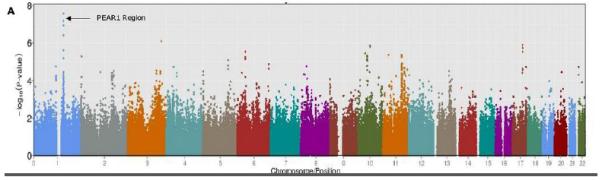
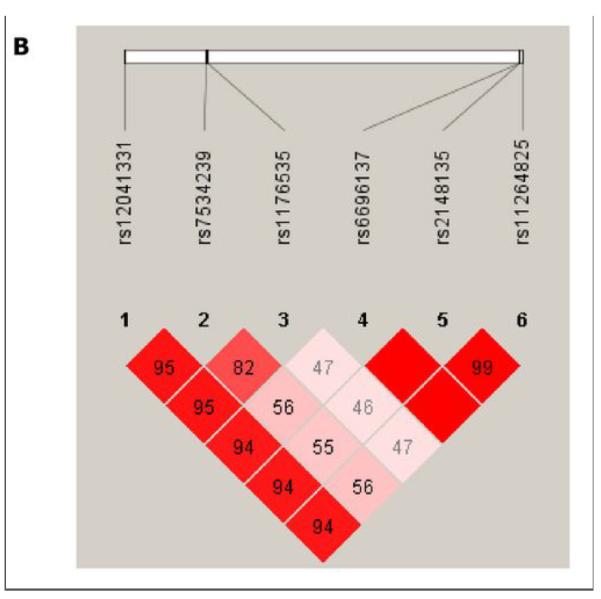
Genome-Wide Association Study of Collagen-Stimulated Platelet Aggregation in Response to Aspirin and Clopidogrel Treatment in the Pharmacogenomics of Antiplatelet Intervention (PAPI) Study. A. Association (plotted as –log10 P value) of individual single-nucleotide polymorphisms distributed across the 22 autosomes. B. Linkage disequilibrium (LD) between PEAR1 SNP rs12041331 and the cluster of chromosome 1q23 SNPs associated with aspirin response (P < 1.0 × 10−7). The color of each square indicates the D’ value between two variants and the extent of LD lessens as the shading gets lighter. The r2 values between variants are shown in each square; no value denotes an r2 = 1.0. This figure was generated using Haploview 4.2 using standard color schemes.
Rs2148135 lies in a gene-rich region on chromosome 1q23. Based on prior publications19, 24-26 as well as reported biological and genetic information regarding genes in the chromosome 1q23 region involved in platelet function, further SNP genotyping was performed in the PEAR1 gene (rs12041331), which encodes a type 1 membrane receptor important in platelet aggregation. Analyses revealed that rs12041331 (minor allele frequency = 0.10, Hardy Weinberg P > 0.05) in intron 1 of PEAR1 was the most highly associated SNP with collagen-induced platelet aggregation after DAPT administration (P = 7.66 × 10−9). This SNP was not associated with baseline platelet aggregation measures (P = 0.89) and was only nominally associated with platelet aggregation after clopidogrel alone (P = 0.008) (Figure 2). Despite its distance from rs2148135 (0.91 MB), rs12041331 was in tight LD with the cluster of 1q23 SNPs identified in the GWAS (D’ = 1.0 and r2 = 0.94 with rs2148135), possibly due to extended LD in this young founder population (Figure 1). After including rs12041331 genotype as a covariate, the association of the most strongly associated 1q23 SNP rs2148135 and DAPT response was markedly attenuated (P = 4.85 × 10−8 without adjustment to P = 2.11 × 10−4 with adjustment).
Figure 2.
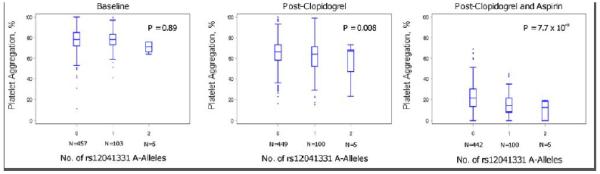
Association of PEAR1 variant rs12041331 with collagen-stimulated platelet aggregation before and after aspirin and clopidogrel administration in participants of the Pharmacogenomics of Antiplatelet Intervention (PAPI) Study. The horizontal line within each box indicates the median; the top and bottom borders of each box indicate the inter-quartile range (IQR). The whiskers extending from each box indicate plus/minus 1.5 IQRs and the points beyond the whiskers indicate outliers beyond 1.5 IQRs.
In PAPI participants with 0, 1, and 2 copies of the rs12041331 minor allele (A), collagen-stimulated platelet aggregation was reduced to 25.0%, 14.6%, and 7.0% of baseline in response to DAPT, respectively. Rs12041331, present in its heterozygous and homozygous state in 18.3% and 0.9% of the PAPI study population respectively, accounted for 5.1% of the total variation in DAPT response (P < 0.001). Rs12041331 was not associated with other cardiovascular or metabolic traits including blood pressure, carotid intima media thickness, or fasting serum lipid levels (Supplementary Table 3).
Sinai Hospital of Baltimore Study
Findings in the PAPI study were extended by examining the relationship between rs12041331 genotype and platelet reactivity as well as cardiovascular outcomes in a group of patients from Sinai Hospital of Baltimore who underwent PCI. Characteristics of these patients are shown in Table 1. Given the difference in rs12041331 minor allele between race/ethnicity, stratified analyses were performed. All patients were taking aspirin prior to and at the time of PCI. Similar to the PAPI study, increasing copies of the rs12041331 A-allele in aspirin-treated Caucasian subjects tended to result in lower collagen-induced platelet aggregation (58.5 ± 3.5, 32.0 ± 4.3, and 28.4 ± 3.4 for GG, GA, and AA genotypes, respectively, P = 0.05). In aspirin-treated African American patients, the frequency of the A allele of rs12041331 was higher than in Caucasians (0.30 versus 0.12, respectively) and there was no evidence for association of rs12041331 with platelet aggregation (32.4 ± 3.9, 34.4 ± 4.6, and 24.5 ± 6.6 for GG, GA, and AA genotypes, respectively, P = 0.96).
In the Sinai Hospital cohort, A-allele carriers of rs12041331 tended to experience a cardiovascular event more often compared to GG homozygotes in both Caucasian (23.1% vs. 9.5%, hazard ratio [HR] = 2.62, 95% CI 0.96-7.10, P = 0.059) and African American (26.7% vs. 7.5%, HR = 3.97, 95% CI 1.10-14.31, P = 0.035) patients after 1 year of follow up. In analyses in which the effects of collagen-stimulated platelet aggregation was included as a covariate, attenuation of the association between rs12041331 and cardiovascular events was observed in both Caucasian (HR = 1.66, 95% CI 0.41-6.76, P = 0.48) and African American (HR = 2.70, 95% CI 0.70-10.46, P = 0.15) patients. In addition, the effect of rs12041331 was independent of and additive to the increased risk of cardiovascular events observed in patients carrying the loss of function CYP2C19*2 allele, a well-documented determinant of post-clopidogrel ADP-stimulated platelet aggregation (Figure 3).
Figure 3.
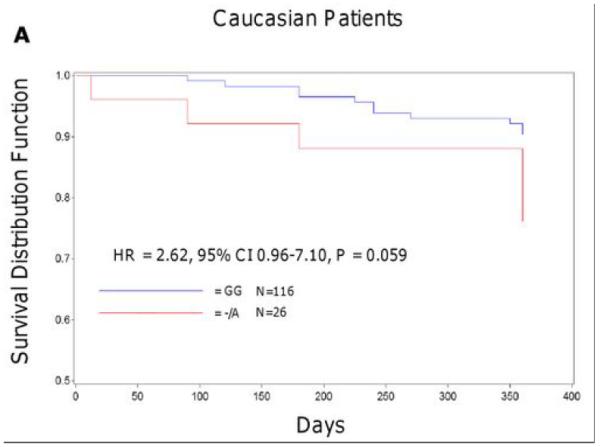
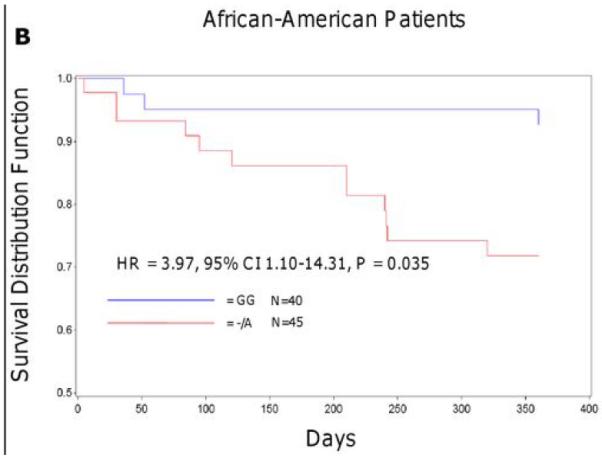
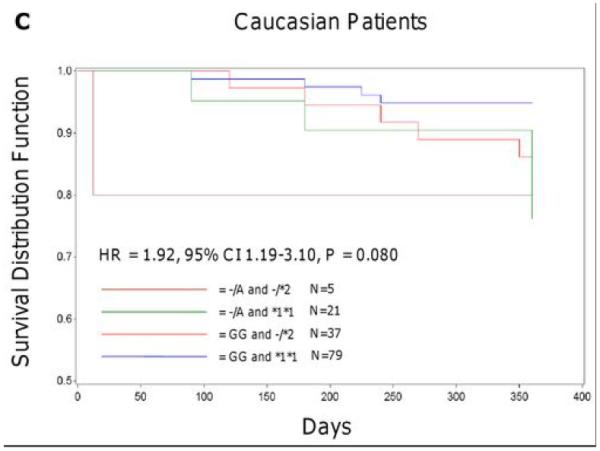
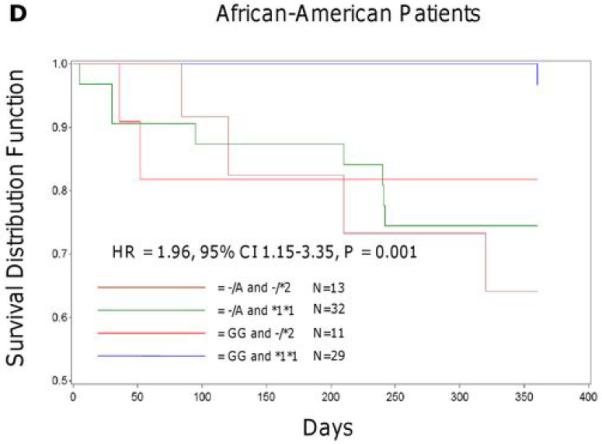
Event-free survival over 1 year of follow-up in Sinai Hospital of Baltimore patients treated with aspirin and clopidogrel following percutaneous coronary intervention, stratified by PEAR1 rs12041331 genotype (A, B) and by PEAR1 rs12041331 and CYP2C19*2 genotype (C, D). Post-discharge ischemic events were defined as myocardial infarction (the occurrence of ischemic symptoms and troponin I value greater than the upper-limits of normal), ischemic stroke, stent thrombosis (definite stent thrombosis according to the Academic Research Consortium criteria), unplanned target vessel revascularization (revascularization of vessel treated at the time of study enrollment), hospitalization for coronary ischemia with revascularization (hospitalization for chest pain with evidence of ischemia on electrocardiogram and no evidence of myocardial infarction as measured by troponin I values), and death secondary to any cardiovascular cause. All analyses were adjusted for age and sex.
INVEST-GENES Cohort
Characteristics of the INVEST-GENES participants are shown in Table 1 and Supplementary Table 4. In the entire cohort, rs12041331 was not associated with the primary outcome, which consisted of death, nonfatal MI, or nonfatal stroke (odds ratio [OR] = 0.95, 95% CI 0.71-1.28, P = 0.73). Similarly, when analyses were conducted stratified by race, no evidence of association between rs12041331 and the primary outcome was observed in Caucasian (OR = 1.19, 95% CI 0.76-1.86, P = 0.45), African American (OR = 1.00, 95% CI 0.46-2.15, P = 0.99), or Hispanic patients (OR = 0.83, 95% CI 0.47-1.46, P = 0.52). However, in aspirin-treated Caucasian subjects of this study, rs12041331 A-allele carriers were significantly more likely to experience a fatal or non-fatal MI compared to GG homozygotes (OR = 2.03, 95% CI 1.01-4.09, P = 0.048). This was not observed in those not taking aspirin (Figure 4). In addition, Caucasian rs12041331 A-allele carriers tended to experience death, nonfatal MI, or nonfatal stroke more often than GG homozygotes when on aspirin therapy (OR = 1.62, 95% CI 0.91-2.90, P = 0.10) compared to those who were not on aspirin (OR = 0.54, 95% CI 0.22-1.31, P = 0.16); although this interaction was not statistically significant (P = 0.056). No evidence of association between rs12041331 genotype and cardiovascular events was observed in either the African American or Hispanic populations, irrespective of aspirin treatment (Supplementary Figure 2). Other cardiovascular and metabolic characteristics were similar when compared by rs12041331 genotype, with the exception of heart failure, where Caucasian A-allele carriers had a higher prevalence of heart failure compared to GG homozygotes (19% vs. 10%, respectively, P = 0.01).
Figure 4.
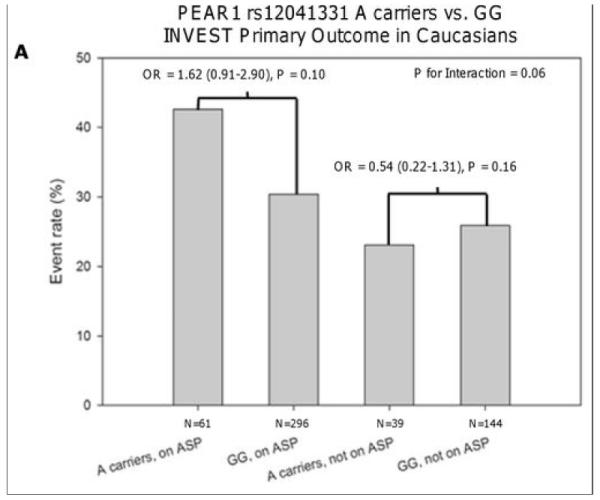
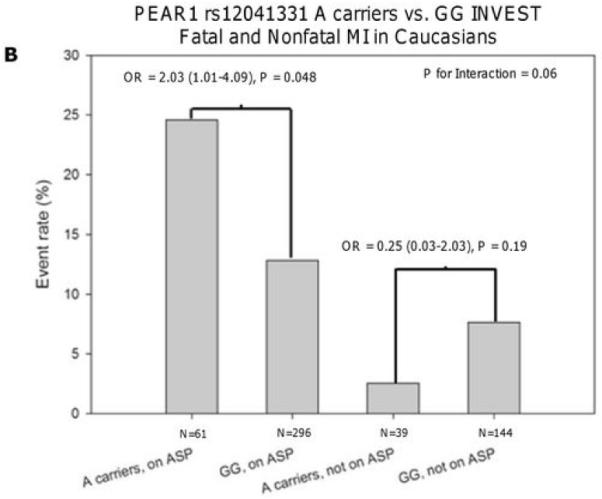
Association between PEAR1 SNP rs12041331 A-allele carrier status and the primary outcome (A) or fatal and non-fatal myocardial infarction (B) in aspirin-treated and non-treated Caucasian patients of INVEST-GENES. All analyses were adjusted for age, sex, history of heart failure, and diabetes.
Discussion
In the PAPI Study, we found collagen-stimulated platelet aggregation in response to aspirin in the presence of clopidogrel, the most commonly prescribed DAPT regimen, to be normally distributed with no distinct cutoff in which to define resistance.
Our GWAS and subsequent genotyping identified a common intronic SNP in PEAR1 (rs12041331) accounting for 5.1% of the phenotypic variation of platelet response to aspirin in the presence of clopidogrel. These findings were similar in patients of the Sinai Hospital of Baltimore Study strengthening our confidence in the initial results and also demonstrating generalizability to outbred populations with significant atherosclerotic disease.
Given that rs12041331 was not associated with baseline measures of platelet aggregation and that post-aspirin measures were associated with genotype even after adjusting for baseline measures, this polymorphism seems to exert a true pharmacogenetic effect. In addition, rs12041331 was only marginally associated with post-clopidogrel collagen-induced platelet aggregation, suggesting that the strong association in the presence of both clopidogrel and aspirin reflects largely an effect on aspirin response. While a synergic effect of both anti-platelet agents is possible, another study reported that following 2 weeks of aspirin treatment (81 mg/d), rs12041331 was significantly associated with platelet aggregation in response to several agonists, suggesting that indeed this variant may be relevant in patients treated with aspirin alone26. A GWAS meta-analysis of baseline platelet aggregation identified rs12041331 as the most strongly associated SNP with both ADP- and epinephrine-induced platelet aggregation suggesting a role of this variant in baseline platelet aggregation via pathways other than those mediated by collagen 25. We measured the effect of rs12041331 on ADP-stimulated platelet aggregation pre- and post-clopidogrel treatment in both PAPI Study participants and PCI patients from Sinai Hospital and found no evidence of association in either population using this agonist (data not shown). This observation suggests that the effect of rs12041331 on post-aspirin platelet aggregation is mediated through collagen receptor pathways and not ADP-dependent pathways.
It is important to note that rs12041331 may not be the causal variant and may, in fact, be in LD with the true and, as of yet, unidentified risk variant. Indeed, the genomic region identified by GWAS contained several genes other than PEAR1. However, in addition to conditional analyses performed in this investigation, which provide evidence that rs12041331 accounted for most of the initial GWAS signal, preliminary RNA sequencing (N = 2 samples) and Western blot data in our laboratory suggest that of the genes in this region (i.e. FCRL1, FCRL2, FCRL3, FCRL4, FCRL5, ETVL3, ARHGEF11, C1orf92, and PEAR1), only PEAR1 is expressed in platelets (data not shown). It is important to note, however, that rs2148135 did show nominal evidence of association with collagen-stimulated platelet aggregation after adjusting for rs12041331 genotype (P = 2.11 × 10−4). Therefore, we cannot rule out the possibility that other variants in any one or more of the genes at this locus may contribute to differences in aspirin response. Furthermore, exome sequencing of 60 subjects with and without the risk haplotype did not reveal any variants that were more highly associated with post-aspirin platelet aggregation than rs2148135 or rs12041331. These data are consistent with the work of Faraday and colleagues in which sequencing of the entire PEAR1 gene in 104 subjects at the extremes of platelet aggregation distribution revealed no variant more strongly associated with platelet aggregation than rs1204133126. In addition, these authors also observed rs12041331-allele specific differences in PEAR1 protein expression by Western blot, ELISA, and luciferase reporter assay, potentially suggesting a mechanism to explain the association between this SNP and platelet aggregation/cardiovascular outcomes26.
The PEAR1 SNP rs12041331 is a common variant in several ethnically diverse populations. In European, Hispanic, and African populations, approximately 21%, 38%, and 67% of individuals have at least one copy of the risk A-allele, respectively, which may have potentially important clinical implications. We observed that both Caucasian and African American PCI patients on DAPT who were rs12041331 A-allele carriers had decreased event-free survival. Furthermore, our data suggest that differences in genotype-specific event-free survival are influenced by platelet reactivity as analyses that included collagen-stimulated platelet aggregation as a mediator variable showed no significant difference in event rates between rs12041331 GG homozygotes and A-allele carriers in both Caucasian and African American patients. However, while Caucasian PCI patients who carried the A-allele of PEAR1 rs12041331 tended to have lower levels of platelet aggregation compared to GG homozygotes, we did not observe this association in African American PCI patients from Sinai Hospital. This is in contrast to prior investigations by Faraday et al. and Johnson et al., which suggest that rs12041331 explains a greater amount of variability in platelet aggregation in African Americans compared to Caucasians25, 26. This apparent discrepancy in findings may be attributed to differences in study design (for example, our study measured aspirin response on the background of clopidogrel therapy), patient population, and/or inadequate power in our sample. Another limitation of the PCI cohort is the differing regiments of antiplatelet agents received in the acute setting (1-3 days). However, stratified analyses of acute management did not detect significant effects on the long-term outcomes investigated. Furthermore, adverse bleeding events could not be evaluated, again, due to small sample size.
In the INVEST-GENES study, we also observed a significant influence of PEAR1 SNP rs12041331 on cardiovascular outcomes in the presence of aspirin treatment. Caucasian rs12041331 A-allele carriers who were taking aspirin were more likely to experience a fatal or non-fatal MI compared to GG homozygotes. This was not observed in the absence of aspirin administration, again suggesting a true pharmacogenomic effect. Interestingly, rs12041331 A-allele carriers on aspirin appeared to have poorer outcomes than A-allele carriers not on aspirin, suggesting that this variant may identify a subgroup of patients in whom aspirin may be contraindicated. Additional studies will be required to examine this finding further. No association was observed between rs12041331 and cardiovascular events in aspirin-treated African-American or Hispanic subjects. However, power to detect an association was limited by the small number of cases (n < 20 for each subgroup).
The rs12041331 A-allele was associated with lower post-aspirin collagen-stimulated platelet aggregation and increased cardiovascular events. As we replicated platelet aggregation measures in the PAPI and Sinai cohorts and cardiovascular events in Sinai and INVEST cohorts, these findings are unlikely to be statistical artifacts. The biological basis for this seemingly paradoxical finding is unknown. We do acknowledge, however, that in some of our analyses the sample size(s) of comparison groups are small and further replication in independent cohorts is warranted. PEAR1 is a type 1 membrane protein expressed in platelets and endothelial cells. While its function is not fully known, phosphorylation of PEAR1 appears to facilitate αIIbβ3-mediated platelet-platelet contacts24. In addition, Kauskot and colleagues demonstrated the functional role of PEAR1 in platelet activation/aggregation by showing that phosphorylation of this receptor results in PI3K and AKT activation, amplification of αIIbβ3 signaling, extensive platelet degranulation, and irreversible aggregation reactions27. It is possible that decreased expression of PEAR1 on platelets (and perhaps also endothelial cells), mediated by rs12041331, results in decreased ex vivo-measured platelet aggregation but unstable thrombi in vivo, leading to increased cardiovascular events. Alternatively, while there is a growing body of evidence that demonstrate that rs12041331 strongly influences platelet aggregation, it is possible that differences in cardiovascular outcomes could be the result of rs12041331-mediated effects on the vascular endothelium where PEAR1 is expressed 6-fold higher than in platelets24. Finally, it is also possible that two different SNPs in partial LD with rs12041331 but in minimal LD with each other may be independently mediating these seemingly paradoxical effects. Detailed biochemical and signaling characterization of PEAR1 will be necessary to understand the mechanisms by which this variant exerts its effects.
In summary, we identified a common variant in PEAR1 (rs12041331) that was reproducibly influenced collagen-induced platelet aggregation in response to aspirin in the presence of clopidogrel. This finding was replicated in PCI patients on DAPT, and extended to include cardiovascular outcomes in this population as well in an independent population with stable coronary artery disease treated with aspirin alone. The findings appear to be independent from and additive to the increased risk of cardiovascular events in DAPT-treated PCI patients carrying the loss of function CYP2C19*2 variant. Further studies will be necessary to define the precise role of PEAR1 genotyping, perhaps in combination with CYP2C19 genotyping in prescribing the most effective individualized antiplatelet therapy to reduce recurrent cardiovascular events in high-risk patients.
Supplementary Material
Acknowledgments
We gratefully acknowledge our Amish liaisons and field workers and the extraordinary cooperation and support of the Amish community, without which these studies would not have been possible.
Funding Sources: This study was supported by National Institutes of Health grants NIH U01 GM074518, U01 HL105198, U01 HL084756, U01 GM074492, R01074730, and K23 GM102678, the Mid-Atlantic Nutrition and Obesity Center (P30 DK072488), the University of Maryland General Clinical Research Center (M01 RR 16500), the Baltimore Veterans Administration Geriatric Research and Education Clinical Center, BASF Pharmaceuticals, Abbott Laboratories, the University of Florida Opportunity Fund, and Sinai Hospital of Baltimore.
Footnotes
Conflict of Interest Disclosures: Dr. Gurbel receives grant support from Astra Zeneca, Daiichi-Sankyo, Bayer Healthcare, Eli Lilly, Portola Pharmaceuticals, Haemonetics, Pozen, and Sanofi-Aventis; and Honoraria/Consulting income from Accumetrics, Astra Zeneca, Bayer Healthcare, Boehringer Ingelheim, Daiichi-Sankyo, Eli Lilly, Merck, Pozen, Portola/Novartis, and Sanofi-Aventis; stock ownership in Merck, Pfizer, and Medtronic. Dr. Shuldiner receives grant support from NIH to study the pharmacogenomics of anti-platelet therapy. He is also a consultant for Bristol Myers-Squibb/Sanofi Aventis. Dr. Lewis has pending grant support from the NIH to study the pharmacogenomics of anti-platelet therapy.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.King SB, 3rd, Smith SC, Jr., Hirshfeld JW, Jr., Jacobs AK, Morrison DA, Williams DO, et al. 2007 focused update of the acc/aha/scai 2005 guideline update for percutaneous coronary intervention: A report of the american college of cardiology/american heart association task force on practice guidelines: 2007 writing group to review new evidence and update the acc/aha/scai 2005 guideline update for percutaneous coronary intervention, writing on behalf of the 2005 writing committee. Circulation. 2008;117:261–295. doi: 10.1161/CIRCULATIONAHA.107.188208. [DOI] [PubMed] [Google Scholar]
- 2.Kushner FG, Hand M, Smith SC, Jr., King SB, 3rd, Anderson JL, Antman EM, et al. 2009 focused updates: Acc/aha guidelines for the management of patients with st-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and acc/aha/scai guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update): A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Catheter Cardiovasc Interv. 2009;74:E25–68. doi: 10.1002/ccd.22351. [DOI] [PubMed] [Google Scholar]
- 3.King SB, 3rd, Smith SC, Jr., Hirshfeld JW, Jr., Jacobs AK, Morrison DA, Williams DO, et al. 2007 focused update of the acc/aha/scai 2005 guideline update for percutaneous coronary intervention: A report of the american college of cardiology/american heart association task force on practice guidelines. J Am Coll Cardiol. 2008;51:172–209. doi: 10.1016/j.jacc.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Feher G, Feher A, Pusch G, Koltai K, Tibold A, Gasztonyi B, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol. 2010;2:171–186. doi: 10.4330/wjc.v2.i7.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tantry US, Mahla E, Gurbel PA. Aspirin resistance. Prog Cardiovasc Dis. 2009;52:141–152. doi: 10.1016/j.pcad.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Bonello L, Tantry US, Marcucci R, Blindt R, Angiolillo DJ, Becker R, et al. Consensus and future directions on the definition of high on-treatment platelet reactivity to adenosine diphosphate. J Am Coll Cardiol. 2010;56:919–933. doi: 10.1016/j.jacc.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 7.Michelson AD, Cattaneo M, Eikelboom JW, Gurbel P, Kottke-Marchant K, Kunicki TJ, et al. Aspirin resistance: Position paper of the working group on aspirin resistance. J Thromb Haemost. 2005;3:1309–1311. doi: 10.1111/j.1538-7836.2005.01351.x. [DOI] [PubMed] [Google Scholar]
- 8.Gurbel PA, Bliden KP, Hiatt BL, O’Connor CM. Clopidogrel for coronary stenting: Response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation. 2003;107:2908–2913. doi: 10.1161/01.CIR.0000072771.11429.83. [DOI] [PubMed] [Google Scholar]
- 9.Cattaneo M. Aspirin and clopidogrel: Efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004;24:1980–1987. doi: 10.1161/01.ATV.0000145980.39477.a9. [DOI] [PubMed] [Google Scholar]
- 10.Zuern CS, Schwab M, Gawaz M, Geisler T. Platelet pharmacogenomics. J Thromb Haemost. 2010;8:1147–1158. doi: 10.1111/j.1538-7836.2010.03791.x. [DOI] [PubMed] [Google Scholar]
- 11.Shuldiner AR, O’Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB, et al. Association of cytochrome p450 2c19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302:849–857. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- 13.Hulot JS, Bura A, Villard E, Azizi M, Remones V, Goyenvalle C, et al. Cytochrome p450 2c19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108:2244–2247. doi: 10.1182/blood-2006-04-013052. [DOI] [PubMed] [Google Scholar]
- 14.Frere C, Cuisset T, Morange PE, Quilici J, Camoin-Jau L, Saut N, et al. Effect of cytochrome p450 polymorphisms on platelet reactivity after treatment with clopidogrel in acute coronary syndrome. Am J Cardiol. 2008;101:1088–1093. doi: 10.1016/j.amjcard.2007.11.065. [DOI] [PubMed] [Google Scholar]
- 15.Mega JL, Simon T, Collet JP, Anderson JL, Antman EM, Bliden K, et al. Reduced-function cyp2c19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for pci: A meta-analysis. JAMA. 2010;304:1821–1830. doi: 10.1001/jama.2010.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulot JS, Collet JP, Silvain J, Pena A, Bellemain-Appaix A, Barthelemy O, et al. Cardiovascular risk in clopidogrel-treated patients according to cytochrome p450 2c19*2 loss-of-function allele or proton pump inhibitor coadministration: A systematic meta-analysis. J Am Coll Cardiol. 2010;56:134–143. doi: 10.1016/j.jacc.2009.12.071. [DOI] [PubMed] [Google Scholar]
- 17.Feher G, Feher A, Pusch G, Lupkovics G, Szapary L, Papp E. The genetics of antiplatelet drug resistance. Clin Genet. 2009;75:1–18. doi: 10.1111/j.1399-0004.2008.01105.x. [DOI] [PubMed] [Google Scholar]
- 18.Agundez JA, Martinez C, Perez-Sala D, Carballo M, Torres MJ, Garcia-Martin E. Pharmacogenomics in aspirin intolerance. Curr Drug Metab. 2009;10:998–1008. doi: 10.2174/138920009790711814. [DOI] [PubMed] [Google Scholar]
- 19.Herrera-Galeano JE, Becker DM, Wilson AF, Yanek LR, Bray P, Vaidya D, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol. 2008;28:1484–1490. doi: 10.1161/ATVBAHA.108.168971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beitelshees AL, Gong Y, Wang D, Schork NJ, Cooper-Dehoff RM, Langaee TY, et al. Kcnmb1 genotype influences response to verapamil sr and adverse outcomes in the international verapamil sr/trandolapril study (invest) Pharmacogenet Genomics. 2007;17:719–729. doi: 10.1097/FPC.0b013e32810f2e3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pepine CJ, Handberg EM, Cooper-DeHoff RM, Marks RG, Kowey P, Messerli FH, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The international verapamil-trandolapril study (invest): A randomized controlled trial. JAMA. 2003;290:2805–2816. doi: 10.1001/jama.290.21.2805. [DOI] [PubMed] [Google Scholar]
- 22.Agarwala R, Biesecker LG, Hopkins KA, Francomano CA, Schaffer AA. Software for constructing and verifying pedigrees within large genealogies and an application to the old order amish of lancaster county. Genome Res. 1998;8:211–221. doi: 10.1101/gr.8.3.211. [DOI] [PubMed] [Google Scholar]
- 23.Agarwala R, Biesecker LG, Tomlin JF, Schaffer AA. Towards a complete north american anabaptist genealogy: A systematic approach to merging partially overlapping genealogy resources. Am J Med Genet. 1999;86:156–161. doi: 10.1002/(sici)1096-8628(19990910)86:2<156::aid-ajmg13>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Nanda N, Bao M, Lin H, Clauser K, Komuves L, Quertermous T, et al. Platelet endothelial aggregation receptor 1 (pear1), a novel epidermal growth factor repeat-containing transmembrane receptor, participates in platelet contact-induced activation. J Biol Chem. 2005;280:24680–24689. doi: 10.1074/jbc.M413411200. [DOI] [PubMed] [Google Scholar]
- 25.Johnson AD, Yanek LR, Chen MH, Faraday N, Larson MG, Tofler G, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42:608–613. doi: 10.1038/ng.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faraday N, Yanek LR, Yang XP, Mathias R, Herrera-Galeano JE, Suktitipat B, et al. Identification of a specific intronic pear1 gene variant associated with greater platelet aggregability and protein expression. Blood. 2011;118:3367–3375. doi: 10.1182/blood-2010-11-320788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kauskot A, Di Michele M, Loyen S, Freson K, Verhamme P, Hoylaerts MF. A novel mechanism of sustained platelet alphaiibbeta3 activation via pear1. Blood. 2012;119:4056–4065. doi: 10.1182/blood-2011-11-392787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.