

Abstract
Background
Ethical concerns about randomly assigning patients to suboptimal or placebo arms and the paucity of willing participants for randomization into control and experimental groups have renewed focus on the use of historical controls in clinical trials. Although databases of historical controls have been advocated, no published reports have described the technical and informatics issues involved in their creation.
Objective
To create a historical controls database by leveraging internal clinical trial data at Pfizer, focusing on patients who received only placebo in randomized controlled trials.
Methods
We transformed disparate clinical data sources by indexing, developing, and integrating clinical data within internal databases and archives. We focused primarily on trials mapped into a consistent standard and trials in the pain therapeutic area as a pilot.
Results
Of the more than 20 000 internal Pfizer clinical trials, 2404 completed placebo controlled studies with a parallel design were identified. Due to challenges with informed consent and data standards used in older clinical trials, studies completed before 2000 were excluded, yielding 1134 studies from which placebo subjects and associated clinical data were extracted.
Conclusions
It is technically feasible to pool portions of placebo populations through a stratification and segmentation approach for a historical placebo group database. A sufficiently large placebo controls database would enable previous distribution calculations on representative populations to supplement, not eliminate, the placebo arm of future clinical trials. Creation of an industry-wide placebo controls database, utilizing a universal standard, beyond the borders of Pfizer would add significant efficiencies to the clinical trial and drug development process.
Keywords: Historical Controls Database, ePlacebo, ePlacebo Database
Background and introduction
The limited availability of patient populations eligible for and willing to participate in clinical trials, coupled with increased attrition rates of novel therapies during drug development, has motivated pharmaceutical companies and regulatory agencies to re-examine how investigational therapies are evaluated and brought to the clinic. An illustrative example of these challenges is the ongoing search for novel treatment options for patients with amyotrophic lateral sclerosis. Despite more than 30 clinical trials including almost 10 000 patients, many of whom received either suboptimal comparators or placebo, only one modestly effective treatment, riluzole, for the treatment of amyotrophic lateral sclerosis has been identified.1 In this context, and other scenarios like it, an ethical dilemma is raised for subsequent studies; whether to expose patients again to either suboptimal comparator or placebo. The ethical dilemmas posed by active control trials, placebo controlled trials (either when a reasonable treatment is available2 3 or even when no treatment is available),3 4 and clinical equipoise have been heavily debated.4–8 This ongoing debate suggests both idealized ethical and scientific rigor for randomized controlled trials (RCT) are difficult to satisfy due to complexities of clinical trials.9 However, the consensus from medical and research ethics literature indicates a focus to minimize harm in the clinical trial process, either by minimizing exposure to scientifically questionable comparator treatments or minimizing exposure to placebo.9 The lack of efficiency, coupled with ethical concerns about randomly assigning thousands of patients to suboptimal or placebo arms, has renewed focus on the use of historical controls to optimize (not eliminate) concurrent placebo and active control arms, which are essential components of RCT.
Clinical trials with historical control designs that supplement, but do not do away with, concurrent or placebo groups have been successfully utilized in important ways. A meta-analysis of antieplictic drugs (AED) demonstrated that patients randomly assigned to (low-dose) suboptimal treatments in all previous trials had similar outcomes thus allowing for the creation of a pool of historical controls for AED trials.10 A methodology utilizing these historical controls was recently approved by the US Food and Drug Administration (FDA) to compare novel AED in future trials.10 As a result, the first historical control design trial in epilepsy, which evaluated lamotrigine XR for conversion to monotherapy, was completed and published in 2012.11 Historical controls have also been used in vaccine safety trials. For example, historical controls matched for age at vaccination, season, sex and geographical area were used to monitor new onset of chronic illnesses within 6 months of vaccination.12 Those studies, and others in cancer, surgical interventions, devices, and biomarker validation,13–16 demonstrate that historical controls have a place in the hierarchy of evidence-based investigation and that a sufficiently large and flexible database of historical controls would have a population representative enough to calculate a previous distribution to augment the placebo arm of future clinical trials.
Historical information is available in a variety of forms (paper, electronic, images), from different sources, and has been widely utilized in retrospective and meta-analyses.17–19 Innovative trial designs using historical controls have the potential to be used effectively in clinical trials if the historical control data are available in sufficient quantity and quality. The practical and statistical concerns for prospective use, design, and analysis of clinical trials such as summarizing historical control information and power and sample size calculation in the design and analysis of a new trial with historical control data have recently been investigated and discussed elsewhere.20–23
The creation of a historical placebo database presents an opportunity to preserve historical institutional information and knowledge in a way that can be accessed, shared, and re-used for the benefit of current and future research. There are many potential benefits in creating a database of patients from the comparator arms of past clinical trials including, but not limited to, reduction in the number of new patients recruited into a RCT, ensuring sufficient/accurate numbers are represented in the placebo arm, and reduction of time and expense associated with executing clinical trials. A large enough ‘ePlacebo’ database has the capacity to produce requisite numbers of patients to reach higher levels of statistical power for historical controls and to match a host of parameters (eg, diagnostic criteria, inclusion/exclusion criteria, stage or severity of disease, concomitant treatments, methods of assessing outcomes, etc) to reduce heterogeneity for matched controls.24 Furthermore, a historical controls database would help with challenges facing orphan or rare diseases that have limited access to patients for random assignment into both experimental and control groups. Approval of drugs for rare diseases are, on average, based on a smaller number of clinical studies, with lower numbers of study participants, and at times with surrogate clinical endpoints. Therefore, careful post-market monitoring of safety and efficacy is needed and re-assessment of efficacy in RCT may be warranted in some cases. A large historical controls database, with data collected over a greater period of time, would provide an important resource for orphan drug discovery.
The creation of such an ‘ePlacebo’ database, however, presents several informatics challenges, such as decisions to include or exclude data sources and indexing data in a value-added and accessible format. The major informatics challenge in creating a harmonized database lies in combining data from disparate clinical trials, particularly those from legacy sources in which different data collection standards were used. Different trials collect different data points, so a historical placebo database would either need an expansive schema or a minimum dataset that would be used as a subset of elements collected for clinical trials. Here we describe our institutional experience at Pfizer during the first phase of creating a ‘ePlacebo’ or historical placebo control database.
Methods
We set out to assess whether existing clinical trial data from disparate sources within Pfizer can be integrated in an efficient manner to create a database of patients who have received only placebo during clinical trials. All data were de-identified and informed consent documents were reviewed to ensure data could be re-used for research purposes. To identify placebo assets, data flow analyses were performed on patient data systems, drug distribution systems, and patient-level datasets. A road map (figure 1) was established for the ‘ePlacebo’ database, which included a search of repositories for appropriate clinical trials in therapeutic and disease areas, collection and separation of comparator arms from trials, standardized representation of patients and data points, and loading and integration of data to create an ‘ePlacebo’ database.
Figure 1.

Road map and data access methods for ePlacebo database. Methods, systems, and algorithms employed to extract data along with decision algorithms and filters applied at each step are depicted. CCTR, corporate clinical trial registry; CDARS, clinical data analysis and reporting system; Demog, demography; OC, Oracle Clinical; PDS, Pfizer data standard; TA, therapeutic area.
For the initial determination of placebo assets, we searched for active data in several Pfizer systems including clinical reporting and portfolio systems, patient data systems, and distribution systems. Due to the multiple technical challenges involved, we did not analyze data in archived sources. During this search, it was discovered that patient level data would be required to determine which study arm each patient was randomly assigned into, thereby enabling a summary of patients at the study, disease area, and therapeutic area levels. The initial determination of placebo assets at Pfizer was determined by searching protocol descriptions and drug types that included the term ‘placebo’ and similar terms in study authoritative sources (in this case, the corporate clinical trial registry; CCTR). Once these studies were identified, we searched patient databases, clinical trials repositories, and clinical data reporting systems to determine if placebo arm patient level data existed for these trials. Trial data within clinical data analysis and reporting systems (CDARS) are stored in different legacy standards and are stored at a submission level, making integration for a single query highly complex, and in some cases unfeasible. There were challenges associated with accessing and integrating these data, particularly when source data were backed up in legacy data standards.
We interrogated a number of systems that contained data of interest, starting with an internal clinical trials information repository, which is organized by drug project/product. This repository stores multiple datasets in one location that are mapped to a single data standard within the repository. For a majority of the trials in this assessment, patient level data originated and were extracted from Oracle Clinical (OC).25 OC contains raw data captured in response to the case report form questions during the trial. However, this database does not contain value-added data, such as associating treatment data with laboratory and/or adverse event (AE) data, first and last active dates of therapy for a subject, imputed AE dates, flagged baseline laboratory values and other derived data. Therefore, CDARS contains patient-level data in individual SAS26 datasets in a UNIX environment with minimal metadata was also used to enhance the ‘ePlacebo’ database.
Multiple datasets are stored for a protocol supporting various deliverables in the lifecycle of a clinical trial. The diversity in data standards make integration across multiple programs challenging. Careful consideration was given to selecting the target data standard (in this case the Pfizer data standard; PDS). If source data are directly from the clinical database or an archived version of the clinical database the de-normalization, or joining of data tables step, can be very complex or almost impossible to perform. Due to the challenges of integrating data from old clinical data standards and data accessibility for older trials, newer studies were preferred over older studies due to a higher likelihood of compliance with current PDS. Although mapping the data was an option, due to resources, time, and upfront institutional commitment and investment required, we performed all analyses on data in the existing data standard. The selected PDS meets the requirement of being able to reverse-engineer historical data and to de-normalize and transpose the data depending on source structures. In addition to challenges of data standards, review of legal informed consent documents before 2000 did not allow for, or explicitly state, secondary use of data from these trials. Furthermore, studies before 2000 were operationally executed differently and thus had different data structures. Retrieving older studies would require additional resources such as space, un-archiving, and mapping data that are prohibitive in the absence of a likelihood of success in joining data structures and tables with newer studies. Therefore, based on these specific challenges of data standard, operational differences in clinical trial execution, and constraints of legal informed consent before 2000, all studies completed before 2000 were eliminated from the scope of this analysis.
Aggregation of data for specific queries required SAS routines to crawl across hundreds of directory structures to acquire and integrate data. Once placebo data assets were identified and integrated into the ‘ePlacebo’ database, we sought to implement and provide an example use case for this asset. We looked for therapeutic areas where there was a high number of patients with a relatively few number of studies. The best candidates for analysis were: neuroscience, cardiovascular and metabolic and endocrine diseases, inflammation, and pain (figure 2). The ‘pain’ disease area was chosen to assess the feasibility of clustering the data because they contained a manageable amount of studies for the pilot, and contained quality controlled data previously utilized for other analyses.
Figure 2.

Basic characteristics of subjects in ePlacebo database. (A) Histogram of subject counts in ePlacebo database from randomized controlled trials completed after 2000 for five therapeutic areas. (B–D) Counts of placebo subjects in pain by disease area (B) and snapshot of demographics for pain placebo patients by age (C), and race (D) with given gender breakdowns. Chronic, severe chronic pain; CvMeD, cardiovascular metabolic; Fibro, fibromyalgia; GenUr, genitourinary; Infl, inflammation; I. Pain, inflammatory pain; Neuro, neuroscience; Neurop, neuropathic pain; OA, osteoarthritis; OAP, osteoarthritis pain.
Results
We integrated, via SAS, placebo patient data across studies, compounds, and drug programs to generate single datasets per clinical domain. The specific data types collected in the ‘ePlacebo’ database are listed in table 1. Instead of aiming to create a single harmonized database of clinical data for all placebo subjects we took a stratification and segmentation approach in which clinical placebo clusters were developed. A cluster is defined as a pool of patients for a disease area that is defined by a single dataset for each of the major clinical data domains (demography, safety, etc). Through this approach, patients can be pooled into matched statistically significant groups, with enough power to address clinical and scientific questions.
Table 1.
PDS clinical domains
| Domain | Description |
|---|---|
| ADVERSE (AE, SAE) | Adverse events |
| CONDRUG | Concomitant/previous medications |
| CONTRT | Concomitant/previous treatment |
| DEMOG (CHILD, ALLERGY) | Demography (childbearing potential, allergy) |
| ECG | Electrocardiogram |
| EFFICACY | Efficacy of treatment—there are separate efficacy modules for each therapy area. Existing efficacy modules exist for allergy and respiratory (A&R), cardiovascular meds (CVMED, CVMED HYPO), gastrointestinal (GI), genitourinary (GU), infectious diseases (ID), inflammation, neuroscience, ophthalmology, pain |
| FINAL | Subject summary |
| LABS | Laboratory |
| MEDHIST | Medical history |
| PHYEXAM | Physical examination |
| PK CONCENTRATION | Pharmacokinetic concentration-time |
| PK PARAMETERS | Pharmacokinetic parameters and satistics |
| PRIMARY DIAGNOSIS | Primary diagnosis |
| SECONDARY DIAGNOSIS | Secondary diagnosis |
| TREATMENT (RANDOM) | Treatment/randomization |
| VITAL SIGNS | Vital sgns |
AE, adverse event; PDS, Pfizer data standard; PK, pharmacokinetic; SAE, serious adverse event.
As detailed in table 2, we first identified all studies that contain placebo data (4075 studies) and then identified all placebo studies that were completed and that had a parallel design (2404 studies). Due to the challenges of integrating data from old clinical data standards (see Methods for details) and constraints of legal informed consent before 2000, studies completed before 2000 were excluded, yielding 1134 studies available for analysis. In our data finding step it was uncovered that several studies were created in CCTR but were not present in OC. These studies were either planned but never initiated, terminated early, or did not meet criteria of a clinical trial. As hierarchically OC is the system of record for patient data at Pfizer, table 2 only includes studies that were run in OC (692 studies). These studies were then grouped by therapeutic area and disease area (figure 2A, B). Moreover, as assessed by subjective measures, therapeutic areas have similarities in inclusion/exclusion criteria, and trials within a therapeutic area/disease area are more likely to collect similar data, thus proving easier to align across studies. However, more explicit and quantitative steps to match inclusion/exclusion criteria for populations can be taken in the future for a specific subset of the database to study defined populations. Next, as a means to pilot the database and attempt to identify a ‘pure/idealized’ placebo population we asked whether we could apply filters for crossover design and duration of the study. We identified 326 studies in which there were no concomitant drugs (eg, aspirin) or a crossover design (table 2). Of these, 203 studies were identified to have a duration greater than at least 2 weeks (table 2).
Table 2.
Algorithm and identification of clinical trials for ePlacebo database
| Category | No of clinical trials |
|---|---|
| Total studies in population (combined Pfizer, legacy Wyeth, and legacy Pharmacia) | 24 581 |
| Total studies that have a status of ‘completed’ and where placebo was administered | 4075 |
| No of above studies with a parallel design (placebo+completed+parallel) | 2406 |
| No of above studies completed since 1999 (placebo+completed+parallel+last 11 years) | 1134 |
| No of above studies in (OC) (placebo+completed+parallel+last 11 years) | 692 |
| No of above studies with placebo arm AND without concomitant drug (eg, aspirin) and crossover design (placebo+completed+parallel+last 11 years+only OC studies+no drug arm) | 326 |
| No of above studies with duration greater than 2 weeks (placebo+completed+parallel+last 11 years+only OC studies+no drug arm+ >2 weeks) | 203 |
OC, Oracle Clinical.
We next sought a mechanism to integrate these data sources and focused on creating two datasets; an observations dataset and a roll-up dataset. The observation dataset is composed of three different sources and contains raw or subject level data, including number of visits, therapeutic areas, laboratory values, study ID, drug name, and other relevant clinical domain observations. The roll-up dataset, also composed of multiple sources, is an aggregated dataset that consists of number of subjects screened, randomly assigned, discontinued, completed, and placebo subjects. The integration of these datasets was done in an external staging area. We found these identified studies included two data standards, a legacy standard and the current PDS. For this phase of the ‘ePlacebo’ database, only PDS studies (ie, completed in the past 11 years) were considered for the consolidated database and all studies with legacy standards were excluded. Therefore, the first phase of the ‘ePlacebo’ database contains studies that are placebo controlled, with a parallel design, completed after the year 2000, and are in one data standard, the PDS.
Applying subjective inclusion and exclusion filters and using SAS, TOAD for Oracle, our own external data evaluation environment, and Tibco Spotfire27 28 we interrogated the ‘ePlacebo’ database to build a profile of historical placebo controls for the pain therapeutic area (figure 2B–D). Demography, treatment, and AE datasets were generated for the pain therapeutic area using placebo patients from only included studies. In addition, laboratory data were aggregated for placebo patients in Pfizer clinical trials. Among patients in the pain cluster, 432 434 laboratory values were collected across multiple laboratory test types. The most commonly collected values are indicated in table 3. Utilizing this resource it was possible to isolate individual patient laboratory values across a therapeutic area and rapidly plot distributions of the data (figure 3). Therefore, the ‘ePlacebo’ database is capable of quickly answering scientific and medical questions that are relevant to the design and analysis of current and future clinical trials.
Table 3.
Common laboratory data collected in pain cluster
| No of observations | Laboratory test name |
|---|---|
| 9741 | CREATININE |
| 9654 | HEMOGLOBIN |
| 9651 | HEMATOCRIT |
| 9649 | PLATELETS |
| 9434 | ALANINE AMINOTRANSFERASE (ALT) |
| 9430 | BILIRUBIN (TOTAL) |
| 9425 | ALKALINE PHOSPHATASE |
| 9380 | WHITE BLOOD CELLS |
| 8234 | RED BLOOD CELLS |
| 7856 | PROTEIN (TOTAL) |
| 7856 | POTASSIUM |
| 7855 | SODIUM |
| 7786 | BLOOD UREA NITROGEN |
| 7782 | CALCIUM |
| 7740 | ALBUMIN |
| 7547 | CHLORIDE |
| 7431 | URIC ACID |
| 7370 | NEUTROPHILS (ABSOLUTE) |
| 7227 | CREATINE KINASE |
| 7078 | EOSINOPHILS (ABSOLUTE) |
| 7078 | MONOCYTES (ABSOLUTE) |
| 7078 | BASOPHILS (ABSOLUTE) |
| 7062 | LYMPHOCYTES (ABSOLUTE) |
| 6709 | ASPARTATE AMINOTRANSFERASE (AST) |
| 5434 | LYMPHOCYTES (%) |
| 5434 | MONOCYTES (%) |
| 5434 | EOSINOPHILS (%) |
| 5434 | NEUTROPHILS (%) |
| 5434 | BASOPHILS (%) |
| 5299 | GLUCOSE |
Figure 3.
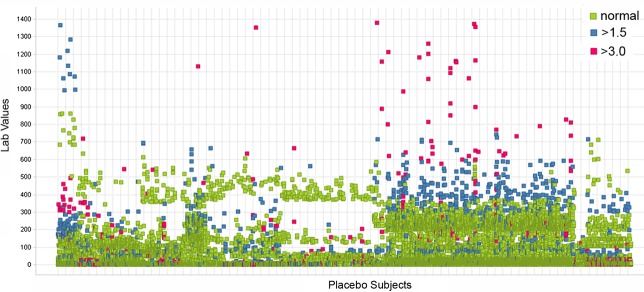
Scatter plot of laboratory values from ePlacebo database. Scatter plot depicts normal (green), greater than 1.5 times upper normal limit (blue), and greater than 3 times upper normal limit (red) values. Subjects are plotted on x-axis and normalized laboratory values are plotted on y-axis. Note multiple data points with same value can appear as one data point.
From our experience creating the ‘ePlacebo’ database, we estimate a total of approximately 300 h and the effort of one full-time equivalent data analyst with moderate to advanced programming proficiency and one full time equivalent (FTE) informatician to design, implement, and test a similar database. Once created and tested, maintenance does not require significant additional resources. We were able to execute this proof of concept with a moderate level of effort because we restricted our scope to clinical trials in which the data were already mapped to a single standard. We expect an undertaking to aggregate data across the industry would require significantly more effort to map the disparate standards into a single, industry-wide, data standard. Currently, efforts are underway at Pfizer to map our clinical trial data to a CDISC29 format and thus Pfizer would contribute or share these data in CDISC to a forum with satisfactory legal and security provisions.
Discussion
We have reported our experience transforming historical clinical trial data from disparate sources to create a database of patients who received placebo during Pfizer clinical trials, an ‘ePlacebo’ database. The intent of creating an ‘ePlacebo’ database is not to replace or eliminate concurrent or placebo controls, but rather to augment these control groups and drive efficiencies in the clinical trial process. Going forward, clinical trial data from all future Pfizer studies will be released to and collected in a single clinical trials information repository that will allow seamless preservation, access, and integration of new data. The creation of a larger industry-wide ‘ePlacebo’ database would require an agreed-upon standard, such as CDISC,29 and a legal and consent framework that would allow any given number of objective, subjective, or consensus criteria to be applied to obtain idealized controls from placebo studies thus improving clinical trial efficiencies. The ‘ePlacebo’ database has many potential uses as a research utility including, but not limited to, studying the natural course of diseases, developing safety models and detecting safety signals, designing more efficient trials, and reducing the time and costs associated with clinical trials.
During the creation of this ‘ePlacebo’ database, we identified several nuances between and within systems when aggregating and profiling clinical trials data from different sources. It was our experience that systems such as OC function well for analytics within a study and as a transactional system; however, challenges were encountered when pooling individual placebo subjects from OC because it is not an ideal repository to perform analyses across multiple studies. To perform multiple study analyses, data from each study need to be pulled into a staging area before such analyses can be performed. Ultimately databases that contain datasets in a SAS format in a UNIX environment were utilized to pool placebo patients for the ‘ePlacebo’ database. When bringing together the actual clinical data, CDARS was preferred over source clinical database systems (eg, OC). Challenges were also encountered when comparing observations that were collected at different time durations and frequencies, for example protocols require collecting information such as laboratory values at different intervals; for example, collection of values at 30-day and 90-day intervals within the same clinical development program. Moreover, a gap analysis was performed to determine what similarities or differences (expected or unexpected) were present in the data gathered in our ePlacebo database. This analysis revealed that full demographic information was captured in over 99% of subjects and routine laboratory results (alanine aminotransferase, hemoglobin) were collected in over 90% of subjects in the pain cluster. However, it should be noted that differences in data captured across therapeutic/disease areas were also uncovered, as to be expected, due to differences in clinical trial design. Therefore, this database is limited by the data points and domains captured, and available, across therapeutic/disease areas. Thus, in cross study analyses, a formal mechanism is required to equate these observations to ensure that the interpretations of the data are correct.
Although clinical trials can identify prominent safety signals, incorporation of ‘ePlacebo’ data, by calculating a previous distribution effect, would allow fewer patients to take placebo, more patients to take test drugs, and help develop better safety and efficacy models that are more informative to designers of future clinical trials. The FDA already has clinical databases of approved products for safety monitoring such as the AE reporting system that have been utilized in historical control studies and in data-driven predictions of drug interactions.30 There are limitations to self-reported or voluntary reporting databases and these have been well documented.31 However, a large industry- agency ‘ePlacebo’ database can help give context to safety observations by providing documented observations in a controlled clinical setting, limiting uncertainties associated with underreporting, biases reporting trends, and the unknowns of the total number of exposed subjects.
While historical controls are a valuable resource for research, it is important that future studies incorporating historical controls be consistent with the studies that make up the original dataset for factors such as study design, evaluation criteria, and analysis plan. There are several caveats that stipulate why historical control data should be approached and utilized with caution.32 We acknowledge there are factors that an ‘ePlacebo’ database may not be able to overcome (eg, clinical reasons for heterogeneity, extent of protocol implementation, etc) and therefore do not advocate replacement of the placebo or concurrent control arm in clinical trials, rather point to the benefits of inclusion of historical controls in a thoughtful, prospective, and data leveraged manner, via a ‘ePlacebo’ database, which can maximize clinical trial efficiencies. Therefore, evaluation of potential sources and reasons for heterogeneity among RCT, to be included in planned analyses, should be a requisite step for future applications of the ‘ePlacebo’ database. The next phase for the ‘ePlacebo’ database is to produce integrated (cross-protocol) datasets for other clinical data domains (eg, efficacy), run simulations for statistical handling of historical placebo data, and test whether results of past clinical analysis can be reproduced using historical control data. This would provide the opportunity for a dialog with regulators for guidance about the broader viability of this research tool.
A review of the FDA's current policies and practices on driving biomedical innovation indicates a major focus by the agency is on harnessing the potential of data mining and information sharing, while protecting patient privacy, to improve products for patients.33 34 We believe an industry-wide ‘ePlacebo’ database would help further the agency's innovation agenda. Ideally hosted by a neutral third-party (eg, the Critical Path Institute),35 this database should require questions in advance of granting access, to prevent misuse, and approximately 10% turnover of the data per year by continuous addition of new data. The challenges going forward for a large public–private collaborative historical placebo control database will be access to, and analysis of, data for compounds that have failed in late development, a legal framework that protects patient privacy, and a legal framework that promotes inter-company cooperation. During creation of the ‘ePlacebo’ database, review of informed consents from older clinical trials excluded their inclusion in this analysis because informed consent documents did not explicitly stipulate data re-use for research other than that related to the original study. A legal framework that will protect patient privacy, allow appropriate downstream use of data, and third party data sharing provisions in informed consent of clinical trials is essential to the successful creation of a more expansive historical controls database.
Conclusion
The challenges of creating a harmonized, single standard, database of clinical data for all placebo subjects can be mitigated through a stratification and segmentation approach in which comparable clinical placebo clusters are developed. This ‘ePlacebo’ database will allow access to and encourage data re-use and enable researchers to ask and answer research questions in an intuitive manner. Our internal database provides a starting point, but ultimately a shared repository of placebo data, utilizing a universal standard such as CDISC, managed by a third party, and with input from regulatory agencies, would add value to the clinical trial and drug development process.
Acknowledgments
The authors would like to thank Debra Bremer, David Isom, Cheryl Feiner, CTIR, and Pfizer Worldwide Research and Development for support and guidance. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Footnotes
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Miller RG, Moore DH, Forshew DA, et al. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis. Neurology 2011;77:973–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polman CH, Reingold SC, Barkhoff F, et al. Ethics of placebo-controlled clinical trials in multiple sclerosis: a reassessment. Neurology 2008;70:1134–40 [DOI] [PubMed] [Google Scholar]
- 3.Fleischhacker WW, Czobor P, Hummer M, et al. Placebo or active control trials of antipsychotic drugs? Arch Gen Psychiatry 2003;60:458–64 [DOI] [PubMed] [Google Scholar]
- 4.Splawinski J, Kuzniar J. Clinical trials: active control vs placebo—what is ethical? Sci Eng Ethics 2004;10:73–9 [DOI] [PubMed] [Google Scholar]
- 5.Gupta S, Fugh-Berman AJ, Scialli A. Ethics and eplerenone considerations. J Med Ethics 2013;39:110–4 [DOI] [PubMed] [Google Scholar]
- 6.Rothman KJ, Michels KB. The continuing unethical use of placebo controls. N Engl J Med 1994;331:394–8 [DOI] [PubMed] [Google Scholar]
- 7.Ellenberg SS, Temple R. Placebo-controlled trials and active-control trials in the evaluation of new treatments. II. Practical issues and specific cases. Ann Intern Med 2000;133:464–70 [DOI] [PubMed] [Google Scholar]
- 8.Miller FG, Joffe S. Equipoise and the dilemma of randomized clinical trials. N Engl J Med 2011;364:476–80 [DOI] [PubMed] [Google Scholar]
- 9.Emanuel EJ, Miller FG. The ethics of placebo-controlled trials—a middle ground. N Engl J Med 2001;345:915–19 [DOI] [PubMed] [Google Scholar]
- 10.French JA, Wang S, Warnock B, et al. Historical control monotherapy design in the treatment of epilepsy. Epilepsia 2010;51:1936–43 [DOI] [PubMed] [Google Scholar]
- 11.French JA, Temkin NR, Shneker BF. Lamotrigine XR conversion to monotherapy: first study using a historical control group. Neurotherapuetics 2012;9:176–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berlin JA, Colditz GA. The role of meta-analysis in the regulatory process for foods, drugs, and devices. JAMA 1999;281:830–4 [DOI] [PubMed] [Google Scholar]
- 13.Klein NP, Hansen J, Lewis E, et al. Post-marketing safety evaluation of a tetanus toxoid, reduced diphtheria toxoid and 3-component acellular pertussis vaccine administered to a cohort of adolescents in a United States health maintenance organization. Pediatr Infect Dis J 2010;29:613–17 [DOI] [PubMed] [Google Scholar]
- 14.Wagner JA, Wright EC, Enni MM. Utility of adiponectin as a biomarker predictive of glycemic efficacy is demonstrated by collaborative pooling of data from clinical trials conducted by multiple sponsors. Clin Pharmacol Ther 2009;86:619–25 [DOI] [PubMed] [Google Scholar]
- 15.Tan SB, Machin D, Tai BC, et al. A Bayesian re-assessment of two phase II trials of gemcitabine in metastatic nasopharyngeal cancer. Br J Cancer 2002;86:843–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaba RC, Ansari SA, Roy SS, et al. Embolization of intracranial aneurysms with hydrogel-coated coils versus inert platinum coils: effects on packing density, coil length and quantity, procedure performance, cost, length of hospital stay, and durability of therapy. Stroke 2006;37:1443–50 [DOI] [PubMed] [Google Scholar]
- 17.Watts NB, Brown JP, Cline G. Risedronate on 2 consecutive days a month reduced vertebral fracture risk at 1 year compared with historical placebo. J Clin Densitom 2010;13:56–62 [DOI] [PubMed] [Google Scholar]
- 18.Temple R. Meta-analysis and epidemiologic studies in drug development and postmarketing surveillance. JAMA 1999;281:841–4 [DOI] [PubMed] [Google Scholar]
- 19.Hammad TA, Pinheiro SP, Neyarapally GA. Secondary use of randomized controlled trials to evaluate drug safety: a review of methodological considerations. Clin Trials 2011;8:559–70 [DOI] [PubMed] [Google Scholar]
- 20.Baker SG, Linderman KS. Rethinking historical controls. Biostatistics 2002;2:383–96 [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Cao J, Ahn C. Calculating sample size in trials using historical controls. Clin Trials 2010;7:343–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong X, Tan M, Bovett J. A sequential procedure for monitoring clinical trials against historical controls. Stat Med 2007;26:1497–511 [DOI] [PubMed] [Google Scholar]
- 23.Neuenschwander B, Capkun-Niggli G, Branson M, et al. Summarizing historical information on controls in clinical trials. Clin Trials 2010;7:5–18 [DOI] [PubMed] [Google Scholar]
- 24. 2006 U.S. Food and Drug Administration. FDA/CDER Guidance for the use of Bayesian statistics in medical device clinical trials – Draft guidance for industry and FDA staff. [Google Scholar]
- 25.Oracle® Clinical http://www.oracle.com/us/industries/life-sciences/046720.html (accessed Apr 2012). [Google Scholar]
- 26.SAS® http://www.sas.com/ (accessed Apr 2012). [Google Scholar]
- 27.TOAD® http://www.quest.com/toad-for-oracle/ (accessed Apr 2012). [Google Scholar]
- 28.Tibco® Spotfire® http://spotfire.tibco.com/products/spotfire-professional/exploratory-data-analysis.aspx (accessed Apr 2012). [Google Scholar]
- 29.Kuchinke W, Aerts J, Semler SC, et al. CDISC standard-based electronic archiving of clinical trials. Methods Inf Med 2009;48:408–13 [DOI] [PubMed] [Google Scholar]
- 30.Tatonetti NP, Ye PP, Daneshjou R, et al. Data-driven prediction of drug effects and interactions. Sci Transl Med 2012;4:125–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bate A, Evans SJ. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf 2009;18:427–36 [DOI] [PubMed] [Google Scholar]
- 32.Juilous SA, Mulle MA. Confounding and the Simpson's paradox. BMJ 1994;309:1480–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.February. 2010. U.S. Food and Drug Administration. FDA/CDER Guidance for Industry: adaptive Design Clinical Trials for Drugs and Biologics.
- 34.October. 2011. U.S. Food and Drug Administration. FDA Driving Biomedical Innovation: Initiatives to Improve Products for Patients.
- 35.Critical Path Institute http://c-path.org (accessed Jul 2012).