

Abstract
In demyelinating diseases, such as multiple sclerosis, remyelination offers the potential to recover function of viable denuded axons by restoring saltatory conduction and/or protecting from further damage. Mice with genetic reduction of fibroblast growth factor 2 (Fgf2) or Fgf receptor 1 (Fgfr1) exhibit dramatically improved remyelination following experimental demyelination with cuprizone. The current studies are the first to test neurobehavioral outcomes with these gene deletions that improved remyelination. The cuprizone protocols used did not produce overt abnormalities but did reduce bilateral sensorimotor coordination (complex wheel task) and increase sociability (two chamber apparatus with novel mouse). A significant effect of genotype was observed on the complex wheel task but not in the sociability apparatus. Specifically, complex wheel velocities for Fgf2 nulls improved significantly after removal of cuprizone from the diet. This improvement in Fgf2 null mice occurred following either acute (6 wk) or chronic (12 wk) demyelination. Plp/CreERT:Fgfr1fl/fl mice administered tamoxifen at 10 wks of cuprizone treatment to induce Fgfr1 knockdown also showed improved recovery of running velocities on the complex wheels. Therefore, constitutive deletion of Fgf2 or Fgfr1 knockdown in oligodendrocyte lineage cells is sufficient to overcome impairment of sensorimotor coordination after cuprizone demyelination.
Keywords: remyelination, corpus callosum, wheel activity, sociability, growth factor, multiple sclerosis, cuprizone
INTRODUCTION
Remyelination, which may restore nerve conduction and protect axons, is significantly greater in early stage multiple sclerosis (MS) lesions than in chronic disease [5, 6]. Failed differentiation of oligodendrocyte lineage cells (OLCs) may contribute to poor remyelination in chronic MS lesions and prolonged neurological deficits [9]. Multiple molecular signaling pathways inhibit differentiation of oligodendrocyte progenitor (OP) cells and limit remyelination in experimental models [1]. Modifying inhibitory signals in lesion areas could potentially enhance functional recovery in MS patients by improving the remyelination capacity of immature OLCs that persist in MS lesions [4, 9].
Our earlier studies showed increased expression of FGF2 and FGFR1 in demyelinated lesions [2, 3, 12, 15]. OP differentiation is increased during remyelination in Fgf2 null mice [14]. FGF2 inhibits OP differentiation through activation of FGFR1 [20]. Genetic reduction of Fgf2 and Fgfr1signaling increased oligodendrogenesis and remyelination and also reduced axon damage in the corpus callosum after chronic cuprizone demyelination [2, 18, 21].
The current studies are the first to evaluate recovery of function in Fgf2 null mice as well as in mice with Fgfr1 knockdown in OLCs after cuprizone demyelination. A complex wheel task differentiates cuprizone treated mice due to impaired bilateral sensorimotor coordination that reflects corpus callosum dysfunction [7, 16]. This complex wheel assessment detects deficits during acute and chronic cuprizone demyelination and with latent axon loss in the corpus callosum [7, 10, 11]. In addition, social interaction is evaluated as a distinct behavioral domain that is altered with cuprizone demyelination [7].
MATERIALS AND METHODS
Mice and cuprizone demyelination
All procedures were approved by the USUHS IACUC. Fgf2 and Plp/CreERT:Fgfr1fl/fl mice were bred, fed cuprizone and treated with tamoxifen as previously described [2, 21].
Wheel running
Cohorts of up to 12 mice were tested on the wheels simultaneously as previously detailed [7].
Sociability
A sociability apparatus (Ugo Basile, Italy) along with AnyMaze software (Stoelting Co., Wood Dale, IL) was used to quantify sociability [13]. During a 15-min habituation period, mice explore between two chambers (top and bottom) - each has one side empty and the other contains a wire cage mouse carrier. An unfamiliar mouse is placed within the carrier in the top chamber during a subsequent 15-min social period.
Myelination status
Immunohistochemistry for myelin oligodendrocyte glycoprotein (MOG) was performed and quantified as described previously [2].
Data Analysis
Prism (GraphPad Software, La Jolla, CA) was used to perform ANOVA followed by Tukey's multiple comparison test for statistical analyses. Cohorts were run simultaneously for gene deletion and control comparisons by investigators blinded to condition. Behavioral cohorts included 3-6 mice per condition.
RESULTS
Complex wheel assessment of bilateral sensorimotor coordination in Fgf2 null mice
Cohorts of Fgf2 null and control mice were assessed on the complex wheel task (Figures 1 and 2). Fgf2 wild type mice show reduced maximum velocities during acute cuprizone treatment that do not improve significantly after return to normal chow (Figure 2A), consistent with our results in C57BL/6 mice [7]. In contrast, Fgf2 null mice improve significantly during the recovery period on normal chow (Figure 2B). With chronic cuprizone demyelination, both Fgf2 wild type and null mice show impairment during acute and chronic cuprizone treatment periods (Figure 2C-D). Notably, only Fgf2 null mice exhibit significant improvement following this chronic demyelination (Figure 2D).
Figure 1. Experimental design.

A: The running wheel was configured either as a “training wheel” with all rungs in place (left) or as a “complex” wheel with a non-uniform rung distribution (right) to increase the task difficulty. B: Fgf2 mice behavioral assessment timelines showing weeks with training (T) or complex (C) wheels and sociability (S) testing. C: Fgfr1 mice behavioral assessments shown weekly along body weight record. Cuprizone ingestion significantly reduced body weight in Plp/CreERT:Fgfr1fl/fl mice (p < 0.001 “cup” vs “no cup”) and Fgfr1fl/fl mice (p < 0.001 compared to Plp/CreERT:Fgfr1fl/fl “no cup”). Tamoxifen (tam) administration did not adversely affect body weight.
Figure 2. Fgf2 null mice show significant improvement of running velocity during the recovery period following acute or chronic cuprizone demyelination.

A-B: Control mice (A; Fgf2+/+) and Fgf2 null mice (B; Fgf2−/−) show reduced maximum running velocities on the complex wheel after acute cuprizone demyelination (red, *p < 0.05 vs pre-cup). Fgf2 null mice (B), but not controls (A), show significant improvement of running velocity during the recovery period on normal chow (green, #p < 0.05 vs acute cup). C-D: Controls (C) and Fgf2 null (D) mice show reduced running velocities on the complex wheel after both acute and chronic cuprizone demyelination (red, *p < 0.05 vs. pre-cup). Fgf2 null mice (D) show significant improvement during the recovery period (green, #p < 0.05 vs both acute cup and chronic cup) that is not observed in controls (C).
An additional cohort of Fgf2 null mice was fed 0.3% cuprizone attempting to further differentiate the demyelination deficits from the recovery phase (data not shown). As with 0.2% cuprizone, this 0.3% dose did not produce overt changes in behavior or appearance. Similar to the data presented for 0.2% cuprizone (Figure 2), significant improvement was again observed with return to normal chow after chronic demyelination with 0.3% cuprizone in Fgf2 null mice (p < 0.05 for recovery vs. chronic). A similar extent of remyelination was also observed. After chronic demyelination and a 6-week recovery period on normal chow, 87.45% ± 1.18 of the area of the corpus callosum immunostained for MOG in Fgf2 null mice treated with 0.3% cuprizone as compared to 87.84% ± 4.72 with 0.2% cuprizone (p = 0.9308).
Sociability assessment in Fgf2 null mice
Using a sociability apparatus, Fgf2 mice were tested after 12 weeks of chronic cuprizone followed by a 6-week recovery period (Figure 3). During the habituation period, without a second mouse present, both Fgf2 null and control mice spent equivalent time exploring each quadrant (Figure 3A). During the social phase, the mice spent more time exploring the quadrant with the novel mouse (Figure 3B; top “mouse” vs bottom “carrier”; p < 0.05). However, there was no difference between genotypes (Figure 3B).
Figure 3. Absence of Fgf2 does not alter sociability.

Comparison of sociability between Fgf2 genotypes after chronic cuprizone demyelination (12 weeks) and recovery (6 weeks). A: During the habituation period, mice explore between top and bottom chambers (each with one quadrant empty and the other containing a wire cage mouse carrier). Mice spend more time near the carrier device as compared to an empty quadrant (p < 0.05 for top and bottom comparisons within each genotype). B: During a subsequent social period, test mice spend significantly more time near the second mouse (p < 0.05 for mouse vs carrier). Fgf2 null (Fgf2−/−) and control genotypes (Fgf2+/+ and Fgf2+/−) are not significantly different between quadrants.
Complex wheel assessment of bilateral sensorimotor coordination in Plp/CreERT:Fgfr1fl/fl mice
Plp/CreERT:Fgfr1fl/fl mice administered tamoxifen at 10 weeks of cuprizone show enhanced remyelination after chronic cuprizone treatment [21]. Therefore, Plp/CreERT:Fgfr1fl/fl mice were tested on the complex wheel task to evaluate behavioral effects (Figure 4). To avoid confounding variables, tamoxifen was administered during cuprizone week 10 before placement of the training wheels for week 11 and complex wheels during week 12. Plp/CreERT:Fgfr1fl/fl mice fed normal chow throughout served as age matched non-demyelination controls and showed consistent maximum wheel velocities across time points with the exception of a reduction following tamoxifen administration (Figure 4A). In Plp/CreERT:Fgfr1fl/fl mice fed cuprizone (Figure 4B), a dose of 0.3% was used to ensure a deficit on the complex wheels while the training wheel period for stabilization of running behavior was shortened to accommodate tamoxifen administration. Plp/CreERT:Fgfr1fl/fl mice show significantly reduced running velocities during the acute cuprizone treatment (Figure 4B). After tamoxifen administration, running velocities improved significantly even though the mice were still ingesting cuprizone and velocities remained elevated after a return to normal chow (Figure 4B). Fgfr1fl/fl mice without Plp/CreERT were used as a cuprizone-treated comparison so that tamoxifen could be administered without deletion of Fgfr1 (Figure 4C). These mice had significantly reduced running velocities during acute cuprizone treatment that continued during the chronic cuprizone period with progressive recovery after return to normal chow (Figure 4C).
Figure 4. Conditional deletion of Fgfr1 improves running velocity in the chronic cuprizone demyelination model.

All mice were administered tamoxifen during week 10 of chronic cuprizone ingestion or the age-matched equivalent. A: Plp/CreERT:Fgfr1fl/fl mice administered tamoxifen but not fed cuprizone (i.e. non-demyelinated controls with Fgfr1 knockdown) show consistent maximum running velocities across the time course except during the testing period following tamoxifen administration (*p < 0.05 vs. pre-cup). B: Plp/CreERT:Fgfr1fl/fl mice given tamoxifen have reduced velocities during the acute cuprizone period (*p < 0.05 vs. pre-cup). During chronic cuprizone demyelination maximum velocities are still reduced (*p < 0.05 vs. precup) but are significantly increased relative to the acute period (#p < 0.05 vs. acute) and remain elevated during the subsequent recovery period on normal chow (#p < 0.05 vs. acute). C: Fgfr1fl/fl mice without the Plp/CreERT transgene were run in parallel and administered tamoxifen as a control without Fgfr1 gene deletion, i.e. Fgfr1 wild type. These Fgfr1 control mice show reduced velocities during both the acute and chronic cuprizone periods (*p < 0.05 vs. pre-cup) followed by a progressive increase of values which reaches significance during the recovery period (#p < 0.05 vs. acute).
Sociability assessment in Plp/CreERT:Fgfr1fl/fl mice
Sociability was tested in Plp/CreERT:Fgfr1fl/fl mice throughout the cuprizone time course to compare the response longitudinally in Plp/CreERT:Fgfr1fl/fl mice before and after tamoxifen induction of Fgfr1 knockdown (Figure 5). Plp/CreERT:Fgfr1fl/fl mice fed normal chow and administered tamoxifen exhibited consistent time spent with the second mouse. Acute cuprizone treatment of Plp/CreERT:Fgfr1fl/fl mice and Fgfr1fl/fl mice significantly increased time spent with the second mouse. These cohorts of cuprizone treated mice were similar before tamoxifen when both genotypes are essentially Fgfr1 wild type. These cohorts of cuprizone treated mice also behaved similarly after tamoxifen to induce Fgfr1 knockdown in the Plp/CreERT:Fgfr1fl/fl mice which indicates Fgfr1 genotype did not alter sociability after cuprizone demyelination.
Figure 5. Fgfr1 genotype does not alter sociability.
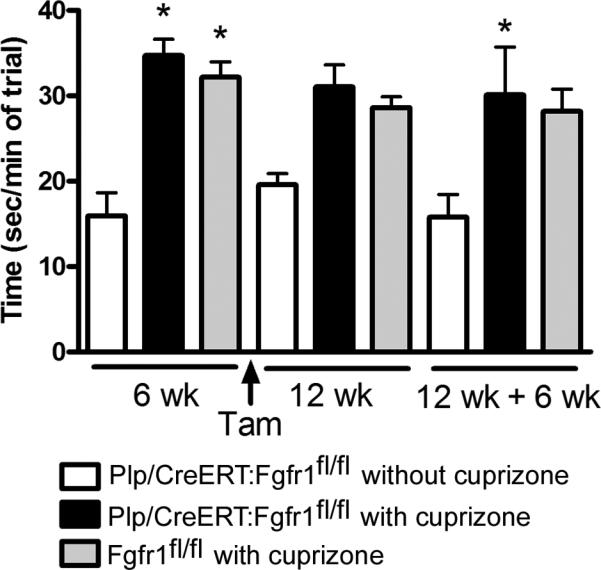
Sociability testing before and after tamoxifen administration to induce conditional Fgfr1 gene deletion. Values show the time spent exploring the quadrant with a second mouse present. The Plp/CreERT:Fgfr1fl/fl mice that were not fed cuprizone (white bars) are similar across testing periods. Plp/CreERT:Fgfr1fl/fl mice (black bars) and Fgfr1fl/fl mice (gray bars) both show a significant increase in sociability with acute cuprizone treatment (*p < 0.05, with vs. without cuprizone) prior to tamoxifen, which is therefore due to cuprizone and not due to Fgfr1 knockdown. Correspondingly, values before and after tamoxifen are similar between Plp/CreERT:Fgfr1fl/fl (black bars) and Fgfr1fl/fl (gray bars).
Corpus callosum remyelination after chronic demyelination with 0.3% cuprizone in Plp/CreERT:Fgfr1fl/fl mice
Our previous studies in Plp/CreERT:Fgfr1fl/fl mice used 0.2% cuprizone [21]. Remyelination following the current 0.3% dose of cuprizone was evaluated after the completion of behavioral assessments (Figure 6). Plp/CreERT:Fgfr1fl/fl mice administered tamoxifen showed significantly greater MOG immunolabeling after chronic demyelination and recovery (Figure 6D-F). Additional littermates (Fgfr1fl/fl and Fgfr1fl/wt mice) were fed 0.3% cuprizone in parallel with the wheel cohorts to confirm effective demyelination after 6 weeks (42.82% ± 2.61, p < 0.05) and 12 weeks (50.79% ± 1.39, p < 0.05) of 0.3% cuprizone as compared to Plp/CreERT:Fgfr1fl/fl mice fed normal chow (90.53% ± 2.16)(Figure 6B-C).
Figure 6. Conditional induced deletion of Fgfr1 increases remyelination after chronic demyelination with 0.3% cuprizone.

Immunohistochemistry for myelin oligodendrocyte glycoprotein (MOG) in coronal sections through the corpus callosum. A: Normal myelination in a Plp/CreERT:Fgfr1fl/fl mouse that did not receive cuprizone. B-C: Demyelination of the corpus callosum after an acute (B, 6 week) or chronic (C, 12 week) period of 0.3% cuprizone. D: Limited remyelination is observed with a 6-week recovery period following chronic demyelination in Fgfr1fl/fl mice without Plp/CreERT to enable Fgfr1 deletion. E: Robust remyelination is observed in Plp/CreERT:Fgfr1fl/fl mice with tamoxifen induced Fgfr1 deletion. Scale bar for A-E, shown in A = 250 μm. F: Quantification of MOG immunostaining to estimate corpus callosum myelination. Normal levels of MOG immunolabeling are found in Plp/CreERT:Fgfr1fl/fl mice administered tamoxifen and fed normal chow (white bar). In Fgfr1fl/fl mice without Plp/CreERT (gray bar) significant demyelination persists (*p < 0.05 vs no cuprizone) after ingestion of 0.3% cuprizone for 12 weeks with only partial remyelination during the 6-week recovery period on normal chow. Significant remyelination is observed in Plp/CreERT:Fgfr1fl/fl mice which undergo Fgfr1 deletion after tamoxifen administration (black bar; 12 wks cuprizone followed by 6-week recovery;*p < 0.05 vs. Fgfr1fl/fl).
DISCUSSION
The current studies show that genetic reduction of either Fgf2 or Fgfr is sufficient to improve sensorimotor coordination after cuprizone demyelination. Furthermore, Plp/CreERT conditional deletion of Fgfr1 targets this functional recovery to OLCs after tamoxifen administration at 10 weeks of chronic cuprizone demyelination.
Our findings in Fgf2 null and Fgfr1 transgenic mice demonstrate behavioral deficits on the complex wheel task with cuprizone demyelination, confirming the usefulness of this assay as identified in C57BL/6 mice [7, 10, 11]. Importantly, Fgf2 and Fgfr1 genotype affected running behavior; cuprizone deficits were reduced in mice with genetically reduced Fgf2 and Fgfr1 signaling. Cuprizone also alters social behavior [7]. The current studies use a sociability apparatus which is more objective than the video analysis of social interaction used in C57BL/6 mice [7] yet similarly shows an increase of social behavior in cuprizone treated mice. However, sociability did not show an effect of Fgf2 or Fgfr1 genotype effect. This sociability assessment may be less sensitive than the complex wheel assessment in our experiments for multiple reasons, including sampling time of minutes versus days, not assessed in a home environment, or less dependent on corpus callosum myelination.
Cuprizone is a systemically administered neurotoxicant so that specifically connecting a complex behavior to a given neuroanatomical change is challenging. Cuprizone treatment produces widespread demyelination of the corpus callosum with a reproducible rostrocaudal pattern [19]. Cuprizone can demyelinate the hippocampus and cerebral cortex [7, 8, 17] which was observed in C57BL/6 mice with complex wheel deficits indicating potential involvement of these sites, in contrast to cerebellum [7] or spinal cord (data not shown). Importantly, Fgf2 null mice and mice with Fgfr1 knockdown in OLCs show significantly improved remyelination in the corpus callosum based on immunohistochemical detection of myelin as well as electron microscopic analysis of myelin thickness and the percentage of myelinated axons [18, 21]. Furthermore, this increased remyelination in the corpus callosum corresponded with reduced axon damage which may also be an important factor contributing to improved behavioral performance in the current study.
CONCLUSIONS
These studies of FGF2 and FGFR1 roles in experimental models provide evidence that removing inhibitory signals can enhance the repair capacity of endogenous cells sufficiently to enable recovery of neurological functions impaired by demyelinating disease.
HIGHLIGHTS.
Cuprizone demyelination reduces sensorimotor coordination and increases social interaction.
Fgf2 null mice have improved running speed on complex wheels after acute or chronic demyelination.
Fgfr1 knockdown in the oligodendrocyte lineage during chronic demyelination improves running.
Fgf2 deletion and Fgfr1 knockdown do not alter social interaction.
Fgf2 deletion and Fgfr1 knockdown promote remyelination and improve functional recovery.
ACKNOWLEDGEMENTS
This work was supported by funding from the National Multiple Sclerosis Society (RG3515; RG4224), the National Institutes for Health (NS39293), and the Center for Neuroscience and Regenerative Medicine (DoD).
Abbreviations
- Cup
cuprizone
- FGF2
fibroblast growth factor 2 ligand
- Fgf2
gene encoding murine fibroblast growth factor 2
- FGFR1
fibroblast growth factor receptor 1
- Fgfr1
gene encoding murine fibroblast growth factor receptor 1
- fl
floxed gene
- MOG
myelin oligodendrocyte protein
- MS
multiple sclerosis
- OLCs
oligodendrocyte lineage cells
- Plp/CreERT
transgene encoding the murine proteolipid protein promoter driving Cre recombinase fused to mutated estrogen receptor
- Tam
tamoxifen
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- 1.Armstrong RC. Growth factor regulation of remyelination: behind the growing interest in endogenous cell repair of the CNS. Future Neurol. 2007;2:689–697. doi: 10.2217/14796708.2.6.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong RC, Le TQ, Flint NC, Vana AC, Zhou YX. Endogenous cell repair of chronic demyelination. J Neuropathol Exp Neurol. 2006;65:245–256. doi: 10.1097/01.jnen.0000205142.08716.7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22:8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- 5.Franklin RJ, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nature reviews. Neurology. 2012;8:624–634. doi: 10.1038/nrneurol.2012.200. [DOI] [PubMed] [Google Scholar]
- 6.Goldschmidt T, Antel J, Konig FB, Bruck W, Kuhlmann T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology. 2009;72:1914–1921. doi: 10.1212/WNL.0b013e3181a8260a. [DOI] [PubMed] [Google Scholar]
- 7.Hibbits N, Pannu R, John Wu T, Armstrong RC. Cuprizone demyelination of the corpus callosum in mice correlates with altered social interaction and impaired bilateral sensorimotor coordination. ASN Neuro. 2009;1 doi: 10.1042/AN20090032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koutsoudaki PN, Skripuletz T, Gudi V, Moharregh-Khiabani D, Hildebrandt H, Trebst C, Stangel M. Demyelination of the hippocampus is prominent in the cuprizone model. Neurosci Lett. 2009;451:83–88. doi: 10.1016/j.neulet.2008.11.058. [DOI] [PubMed] [Google Scholar]
- 9.Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- 10.Liebetanz D, Merkler D. Effects of commissural de- and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Experimental neurology. 2006;202:217–224. doi: 10.1016/j.expneurol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 11.Manrique-Hoyos N, Jurgens T, Gronborg M, Kreutzfeldt M, Schedensack M, Kuhlmann T, Schrick C, Bruck W, Urlaub H, Simons M, Merkler D. Late motor decline after accomplished remyelination: impact for progressive multiple sclerosis. Ann Neurol. 2012;71:227–244. doi: 10.1002/ana.22681. [DOI] [PubMed] [Google Scholar]
- 12.Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62:241–256. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes, brain, and behavior. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 14.Murtie JC, Zhou YX, Le TQ, Vana AC, Armstrong RC. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis. 2005;19:171–182. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Redwine JM, Armstrong RC. In vivo proliferation of oligodendrocyte progenitors expressing PDGFalphaR during early remyelination. J Neurobiol. 1998;37:413–428. doi: 10.1002/(sici)1097-4695(19981115)37:3<413::aid-neu7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 16.Schalomon PM, Wahlsten D. Wheel running behavior is impaired by both surgical section and genetic absence of the mouse corpus callosum. Brain Res Bull. 2002;57:27–33. doi: 10.1016/s0361-9230(01)00633-5. [DOI] [PubMed] [Google Scholar]
- 17.Skripuletz T, Lindner M, Kotsiari A, Garde N, Fokuhl J, Linsmeier F, Trebst C, Stangel M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. The American journal of pathology. 2008;172:1053–1061. doi: 10.2353/ajpath.2008.070850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobin JE, Xie M, Le TQ, Song SK, Armstrong RC. Reduced axonopathy and enhanced remyelination after chronic demyelination in fibroblast growth factor 2 (fgf2)-null mice: differential detection with diffusion tensor imaging. J Neuropathol Exp Neurol. 2011;70:157–165. doi: 10.1097/NEN.0b013e31820937e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie M, Tobin JE, Budde MD, Chen CI, Trinkaus K, Cross AH, McDaniel DP, Song SK, Armstrong RC. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol. 2010;69:704–716. doi: 10.1097/NEN.0b013e3181e3de90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou YX, Flint NC, Murtie JC, Le TQ, Armstrong RC. Retroviral lineage analysis of fibroblast growth factor receptor signaling in FGF2 inhibition of oligodendrocyte progenitor differentiation. Glia. 2006;54:578–590. doi: 10.1002/glia.20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou YX, Pannu R, Le TQ, Armstrong RC. Fibroblast growth factor 1 (FGFR1) modulation regulates repair capacity of oligodendrocyte progenitor cells following chronic demyelination. Neurobiol Dis. 2012;45:196–205. doi: 10.1016/j.nbd.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]