

SUMMARY
Background
Primary sclerosing cholangitis (PSC) is characterised by progressive inflammatory and fibrotic destruction of the biliary ducts. There are no effective medical therapies and presently high dose ursodeoxycholic acid is no longer recommended due to significant adverse events in a recent clinical trial. Cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction is associated with PSC in both children and adults. Since CFTR dysfunction leads to altered fatty acid metabolism, specifically reduced docosahexaenoic acid (DHA), we hypothesised that DHA supplementation might be an effective therapy for patients with PSC.
Aim
To determine the safety and efficacy of oral DHA supplementation for the treatment of PSC.
Methods
We conducted a 12 month open-label pilot study to evaluate safety of oral DHA and its effects on serum alkaline phosphatase as a primary outcome measure in 23 patients with PSC. DHA was administered orally at 800 mg twice per day. Secondary outcomes included changes in other liver function tests and fibrosis biomarkers.
Results
A 1.7-fold increase in serum DHA levels was observed with supplementation. The mean alkaline phosphatase level (±S.E.) at baseline was 357.8 ± 37.1 IU compared to 297.1 ± 23.7 IU (P < 0.05) after 12 months of treatment. There were no changes in other liver function tests and fibrosis biomarkers. No adverse events were reported.
Conclusions
Oral DHA supplementation is associated with an increase in serum DHA levels and a significant decline in alkaline phosphatase levels in patients with PSC. These data support the need for a rigorous trial of DHA therapy in PSC.
INTRODUCTION
Primary sclerosing cholangitis (PSC) is characterised by a slowly, progressive inflammatory and fibro-obliterative destruction of the intra- and extrahepatic biliary tree. Patients often present with chronic cholestasis, jaundice, hepatomegaly and pruritus. There are no established, effective therapies with many of the treatments directed at ameliorating the complications of the disease such as excessive pruritus and biliary duct strictures. Of greater concern is the demonstration that high-dose ursodeoxycholic acid is not only ineffective, but recently shown in a large NIH trial to lead to increased complications and thus is no longer a therapeutic option.1
The aetiology of PSC is not known, although, patients are more likely to be men, middle-aged and have concomitant inflammatory bowel disease, most notably ulcerative colitis.2–4 Furthermore, many patients with PSC also have increased autoantibody markers. However, immunosuppressive therapies have not been effective and the significance of increased auto antibodies remains to be elucidated.2 In 6–20% of patients, progressive cirrhosis develops leading to end-stage liver disease necessitating liver transplantation. The median time from diagnosis to death or liver transplantation is 9–18 years.2–4
Our laboratory has shown that there is an increased prevalence of cystic fibrosis transmembrane conductance regulator (CFTR) abnormalities in adults with PSC as demonstrated by genotype and phenotype analyses. Analysis of the CFTR gene in PSC patients compared with disease controls demonstrated a significantly increased number of single allelic mutations in the PSC group (37% vs. 9% of disease controls).5 In addition, studies in children with PSC demonstrated that 11 of 19 subjects with PSC had abnormal CFTR function (>2 standard deviations above median levels in individuals with inflammatory bowel disease alone).6
The significance of determining an increase in CFTR dysfunction in patients with PSC is several-fold. First, it has been established that CFTR expression in the hepatobiliary system is restricted to the apical membrane of the intrahepatic and extrahepatic bile duct epithelial cells. CFTR dysfunction causes decreased chloride secretion into the bile canaliculi with subsequent decrease in osmotic extrusion of water into the lumen. Impaired function of this cyclic-AMP dependent chloride channel results in hyperconcentration and acidification of bile leading to obstruction of intrahepatic bile ductules, secondary inflammation and eventual focal biliary cirrhosis.7, 8 Secondly, CFTR dysfunction is associated with innate immune defects and leads to an excessive host inflammatory response.9–11 Our group has demonstrated this increased inflammatory response is not only true in patients homozygous for CFTR, but also for healthy obligate heterozygotes.11 Thus, a single CFTR allele is sufficient to lower the threshold for inflammation. Third, CFTR dysfunction is linked to fatty acid alterations, specifically a decrease in docosahexaenoic acid (DHA) and an increase in arachidonic acid (AA).12, 13 We have shown that correction of this fatty acid abnormality reverses the organ-specific pathology seen in cftr−/−knockout mice including the development of bile duct injury.9, 14, 15 The mechanism of DHA action is mediated at least in part, through induction of peroxisome proliferator-activated receptor alpha (PPARα).16 However, DHA independent of CFTR dysfunction inhibits inflammation through several pathways including decreasing AA levels and downstream eicosanoid production as well as the direct biogenesis of resolvins/do-cosatrienes to terminate the inflammatory response.17
We, therefore, hypothesised that CFTR dysfunction may contribute to the pathogenesis of PSC and that correction of CFTR-related fatty acid abnormalities, and its associated defects in immune defences and inflammatory responses, with oral DHA supplementation might be an effective therapy.
The objective of this study was to conduct a pilot, open-labelled, clinical trial to determine whether or not DHA is safe and a potentially efficacious therapy for PSC. Due to the fact that PSC is a slowly progressive disease and definitive outcome measures, such as time to transplantation or death, could take many years to show a statistical difference, short-term outcome measures likely to show a difference during the treatment period of 1 year were examined.
PATIENTS AND METHODS
Study type and study locations
This pilot study was an open-label trial of oral DHA supplementation for the treatment of PSC conducted over the course of 12 months at Beth Israel Deaconess Medical Center (BIDMC) Boston, MA and The Mayo Clinic, Rochester, MN. The Institutional Review Boards at both study sites approved the study and written informed consent was obtained from subjects prior to enrolment. This study was registered on the clinicaltrials.gov website (NCT00325013).
Subject enrolment
Inclusion criteria
Subjects were eligible for inclusion if they met the following criteria: (i) had a diagnosis of PSC as defined by chronic cholestatic liver disease of at least 6 months’ duration; a serum alkaline phosphatase (AP) level at least 1.5 times the upper limit of normal; cholangiographic findings of intrahepatic or extrahepatic biliary-duct obstruction, beading, or narrowing consistent with PSC; and a liver biopsy in the previous 12 months with compatible findings, and (ii) no evidence of secondary cholangitis or other liver disease (primary biliary cirrhosis, alcoholic liver disease, autoimmune hepatitis and chronic viral hepatitis).
Exclusion criteria
Subjects were excluded from enrolment if they possessed any of the following criteria: (i) age less than 18 years or more than 80 years, (ii) history of previous bile duct surgery, previous choledocholithiasis, recurrent ascending cholangitis, previous history of variceal haemorrhage, or cholangiocarcinoma, (iii) PSC stage III (fibrosis) or IV (cirrhosis) based on the criteria of Ludwig et al.,18 (iv) treatment with corticosteroids, ciclosporin or methotrexate within the preceding 3 months, or (v) anticipated need for liver transplantation within 1 year.
Intervention
Primary intervention
Recruited subjects received oral algal-DHA, 800 mg (four capsules) twice per day for 52 weeks. DHA was provided as the commercially available triglyceride (Crypthecodiniumcohnii, DHASCO) from Martek Bioscience Corporation (Columbia, MD, USA).
Secondary intervention
Subjects were instructed not to take fish oil supplements while participating in the study. In addition, subjects were instructed to maintain a diet high in omega-3 and low in omega-6 fatty acids. These dietary instructions were provided in detail by the General Clinical Research Center at BIDMC by the bionutritionists and included the recommended use of olive oil (52% omega-9) instead of other vegetable oils and minimal consumption of red meats and certain nuts.
Patients already receiving 5-aminosalicylate preparations, azathioprine, or ursodeoxycholic acid at the time of enrolment were asked to remain on these preparations for the duration of the trial. Since ursodeoxycholic acid can affect the liver function tests depending on the dose, all subjects on this medication were asked to continue this medication during the study at their current dose. Note that this study was conducted prior to the National Institutes of Health study showing that high dose ursodeoxycholic acid can have severe adverse effects.1
Outcome measures
Primary outcome measure
The primary outcome measure was change in serum AP levels. Serum AP levels were obtained on all subjects at baseline and every 8 weeks for the first 6 months (visit at month 2, 4 and 6), then every 12 weeks until the study completion (month 9 and 12). Serum AP levels were measured in the hospital clinical chemistry laboratories. Outcomes in AP levels were defined as ‘highly positive’, ‘positive’, ‘no change’, ‘negative’ or ‘highly negative’. ‘Highly positive’ was defined as a reduction in the 12 month AP level by greater than 50% compared to the baseline level, a ‘positive’ change was a reduction by greater than 25% but less than 50%, ‘no change’ was a reduction of less than 25% or an increase by less than 25%, a ‘negative’ change was an increase by greater than 25% but less than 50%, and a ‘highly negative’ change was an increase in the serum AP level at 12 months by greater than 50% compared to baseline.
Secondary outcome measures
Secondary outcome measures included: (i) changes in other liver function tests, (ii) changes in serum fatty acid profiles and (iii) changes in measures of liver fibrosis markers. In addition to AP, alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyltranspeptidase (GGT), total bilirubin and albumin levels were obtained in all enrolled subjects at baseline, and 2, 4, 6, 9, and 12 months. Pro-thrombin time (PT) was measured at all timepoints for BIDMC enrolled patients and at baseline and 12 months for patients enrolled at Mayo Clinic.
For patients enrolled at BIDMC, serum and fatty acid profiles [DHA, AA, linoleic acid (LA), and eicosapentaenoic acid (EPA)] were analysed at all time points by gas chromatography/mass spectroscopy. Fatty acids were isolated and methylated using a modified Folch method as previously described.12 Gas chromatography-mass spectroscopy analysis of fatty acids was performed on a Hewlett-Packard Series II 5890 chromatograph coupled to an HP-5971 mass spectrometer equipped with a supelcowax SP-10 capillary column. Peak identification was based upon comparison of both retention time and mass spectra of unknown peak to that of known standards within the GC-MS database library. Fatty acid methyl ester (FAME) mass was determined by comparing areas of unknown FAMEs to that of a fixed concentration of 17:0 internal standard. Individual fatty acids are expressed as per cent of the total fatty acid mass (mol%).
Finally, serum liver fibrosis markers were analysed including hyaluronic acid (HA), amino-terminal propeptide-of-type-III-collagen (PIIINP) and tissue-inhibitor of matrix-metalloproteinase-1 (TIMP-I). An overall fibrosis score was determined by an algorithm of these three fibrosis markers called the Enhanced Liver Fibrosis (ELF) test. Samples were analysed for these markers and an ELF score was determined at baseline, at 4 months and at 9 months for patients enrolled at BIDMC. Samples were analysed by iQur Limited, Southhampton, UK using proprietary assays by Siemens Healthcare Diagnostics Inc. (Tarrytown, NY, USA).
Statistics
Sample size and power
If it is assumed that at most 5% of subjects would have experienced a highly positive reduction in AP levels (>50% reduction) without treatment, then 28 patients would be needed to detect a highly positive reduction in 20% of subjects with 80% power and a 5% level of significance.
Data analysis
Since our main focus is the change in outcomes, the paired t-test was used to compare initial and final measurements of all the variables listed including the primary outcome variable of serum AP levels. We interpreted results as significant if DHA therapy is associated with a highly positive reduction in AP levels in >20% of patients.
To evaluate differences in baseline characteristics and secondary outcome measures by primary outcome group, a sub-analysis was performed. For this sub-analysis, the two ‘positive’ and the two ‘negative’ response groups were collapsed for a final categorisation of change in AP levels in response to oral DHA supplementation into three groups: ‘positive, n = 8; ‘no change’, n = 13, and ‘negative’, n = 2. With two subjects in the ‘negative’ group, one of whom developed cholangiocarcinoma, comparisons were made between the ‘positive’ and ‘no change’ groups only, although data are still shown for the ‘negative’ group.
All analyses were performed using STATA statistical software, version 11 (StataCorp) and GRAPHPAD Prism version 5.00 for Windows, GraphPad Software, San Diego, CA, USA, http://www.graphpad.com.
RESULTS
Patient characteristics
Fourteen subjects were screened at each site for a total of 28 subjects. After screening, one subject at BIDMC and two subjects from Mayo were found not to meet all inclusion criteria for a final enrolment of 13 subjects at BIDMC and 12 subjects at the Mayo Clinic. After enrolment, two subjects from Mayo were not compliant with the intervention. Thus, the study enrolment for final analysis included 13 subjects from BIDMC and 10 subjects from Mayo for a total of 23 subjects.
The patient characteristics at the time of enrolment are shown in Table 1. The average age was 47.9 years, 52% of the subjects were men, and the average body mass index was 27.4. Ninety six per cent of the subjects had inflammatory bowel disease, and 32% were on Ursodiol prior to study enrolment. The average Mayo PSC risk score was 1.1 placing the cohort in an ‘intermediate’ risk group.
Table 1.
Patient characteristics
| Characteristic | |
|---|---|
| n | 23 |
| Age, years, mean (±s.d.) | 47.9 (±15.7) |
| BMI, mean (±s.d.) | 27.4 (±6.4) |
| Gender, male, % (n) | 52% (12/23) |
| IBD, % (n) | 96% (22/23) |
| Ursodiol, % (n) | 32% (7/22)* |
| Mayo PSC Risk Score† | 1.1 (±1.0) |
s.d., standard deviation; BMI, Body Mass Index; PSC, primary sclerosing cholangitis.
One subject with unknown Ursodiol status prior to study enrollment.
The Mayo PSC Risk Score estimates risk of death up to 4 years of assessment. A score of ≤0 is ‘low’ risk, >0 but <2 is ‘intermediate’ risk, ≥2 is ‘high’ risk.
Change in serum alkaline phosphatase levels
Over the course of oral DHA treatment the study cohort demonstrated a decline in serum AP levels (Figure 1). At baseline, the mean AP level was 357.8 ± 37.1 IU/L (Table 2). The mean serum AP levels at 9 months (275.6 ± 36.3 IU/L) and at 12 months (297.1 ± 37.9 IU/L) were significantly lower compared to the mean baseline value. By subject, five (22%) demonstrated a highly positive change in serum AP levels, three (13%) showed a ‘positive’ change, 13 (56%) had ‘no change’ in their AP levels, and two (9%) had a ‘highly negative’ change (Figure 2). One of the two with a ‘highly negative’ response was diagnosed with cholangiocarcinoma while enrolled in the study.
Figure 1.

Baseline and subsequent serum alkaline phosphatase (AP) levels over the course of treatment. Significant reductions in serum AP levels are observed at 9 and 12 months.
Table 2.
Changes in mean values of other, secondary outcome, liver function tests over the course of treatment
| LFTs, units (normal range) | Baseline | 2 months | 4 months Mean (±S.E.) | 6 months | 9 months | 12 months |
|---|---|---|---|---|---|---|
| ALT, IU/L (0–40) | 122.7 (±31.9) | 127.5 (±29.5) | 118.2 (±27.9) | 127.8 (±30.5) | 113.3 (±26.7) | 128.5 (±27.3) |
| AST, IU/L (0–40) | 79.4 (±14.6) | 88.3 (±16.8) | 80.1 (±12.7) | 83.1 (±14.6) | 78.4 (±15.2) | 93.1 (±17.2) |
| GGT, IU/L (5–61) | 376.7 (±64.5) | 411.5 (±73.8) | 431.4 (±93.0) | 388.1 (±65.4) | 325.9 (±73.4) | 405.0 (±75.0) |
| Bilirubin, mg/dL (0–1.5) | 1.0 (±0.2) | 1.1 (±0.2) | 1.1 (±0.2) | 1.2 (±0.2) | 1.4 (±0.3) | 1.7 (±0.4)* |
| Baseline ≤ 1.5, n = 18 | 0.6 (±0.1) | 0.7 (±0.1) | 0.8 (±0.1) | 0.8 (±0.2) | 0.8 (±0.2) | 1.1 (±0.2)* |
| Baseline >1.5, n = 5 | 2.5 (±0.5) | 2.3 (±0.4) | 2.4 (±0.5) | 2.5 (±0.5) | 3.4 (±0.8) | 3.8 (±1.3) |
| Albumin, g/dL (3.5–5.2) | 4.2 (±0.1) | 4.3 (±0.1) | 4.3 (±0.1) | 4.2 (±0.1) | 4.3 (±0.1) | 4.3 (±0.1) |
| PT, seconds†(10.4–13.4) | 11.6 (±1.1) | 13.2 (±1.1) | 11.9 (±0.2) | 13.0 (±0.9) | 12.0 (±0.3) | 10.9 (±0.7) |
S.E., standard error of the mean; LFTs, liver function tests; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma glutamyltransferase; PT, prothrombin time.
P < 0.05; paired t-test, 12 month value compared to baseline value.
BIDMC subjects contributed to all time points; Mayo subjects only for baseline and 12 months.
Figure 2.
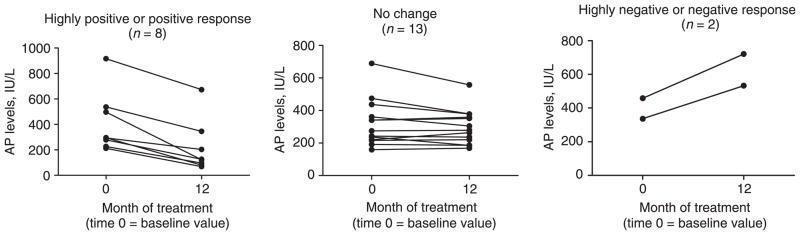
Individual alkaline phosphatase (AP) levels at baseline and 12 months by response to DHA treatment. Response is defined by the change in the AP level at 12 months compared to the baseline value. ‘Highly positive’ = a reduction by greater than 50%; ‘Positive’ = a reduction by greater than 25% but less than 50%; ‘No Change’ = a reduction of less than 25% or an increase by less than 25%; ‘Negative’ = an increase by greater than 25% but less than 50%, and ‘Highly Negative’ = an increase by greater than 50%.
Changes in other liver function tests
There were no significant changes over time in ALT, AST, GGT or albumin levels (Table 2). The 12 month bilirubin level of 1.7 ± 0.4 mg/dL was significantly increased from baseline (1.0 ± 0.2 mg/dL). The 12 month PT of 10.9 ± 0.7 s was similar to a baseline value of 11.6 ± 1.1 s. The difference in the PT values from baseline to 12 months is not clinically significant with both means within the normal range; whereas for the entire cohort, the values in bilirubin levels over time surpassed the upper limit of normal (1.5 mg/dL). However, when stratified by baseline bilirubin levels, the mean change over time in patients with a starting bilirubin ≤ 1.5 mg/dL remained less than this threshold.
Changes in liver fibrosis markers
Analysis of the BIDMC subjects up to 9 months demonstrated no significant change in the individual fibrosis markers or the overall fibrosis score, ELF, over time (Table 3).
Table 3.
Fibrosis markers*
| Markers, ng/mL | Baseline (n = 12) | 4 months (n = 12) Mean (S.E.) | 9 months (n = 13) |
|---|---|---|---|
| HA | 154.7 (±40.3) | 134.1 (±37.9) | 147.9 (±35.1) |
| PIIINP | 13.7 (±1.9) | 12.6 (±1.8) | 15.4 (±2.2) |
| TIMP-1 | 811.5 (±94.2) | 703.2 (±68.3) | 747.5 (±84.9) |
| ELF Score (normal range: 0–6.6) | 10.1 (±.48) | 10.1 (±.32) | 10.2 (±.40) |
S.E., standard error of the mean; HA, hyaluronic acid; PIIINP, amino-terminal propeptide-of-type-III-collagen; TIMP-1, tissue-inhibitor of matrix-metaloproteinas-1; ELF, enhanced liver fibrosis.
These data are from the Beth Israel Deaconess Medical Center subjects only.
Changes in fatty acid profiles
Serum total DHA levels significantly increased from baseline values while on oral DHA supplementation (Figure 3a). This increase was evident by month 2 and was significantly higher compared to baseline for the total duration of the study. Similarly, AA levels were significantly decreased from baseline values (Figure 3b) and this change was also evident by 2 months and lasted for the total duration of the study. There were no changes in EPA or LA levels with oral DHA supplementation at any time point compared to baseline values (Figures 3c and 3d).
Figure 3.

Baseline and subsequent serum levels, expressed as mol%, of select fatty acids over the course of treatment for BIDMC subjects only. At the 2 month follow-up visit, DHA is significantly higher, while, AA is lower compared to baseline. No changes are observed at 12 months compared to baseline for EPA and LA.
Comparison of secondary outcomes measures by primary outcome group
Finally, we sought to determine differences in baseline characteristics and secondary outcome measures by primary outcome group (Table 4). There was no difference in age, and gender at enrolment between response groups. We found that at baseline there was a trend for the ‘positive response’ group to have an overall lower severity of illness as demonstrated by a lower Mayo PSC risk score and a lower fibrosis score, with the latter score in the ‘mild to moderate’ range vs. ‘moderate to severe’ in the ‘no change’ group. However, these trends in baseline severity measures were not statistically significant. Similarly, there were no differences in the baseline DHA levels. However, the ‘no change’ group did have lower AA levels at baseline. With intervention, the ‘positive’ response group trended to higher DHA levels and lower AA levels, although this did not achieve statistical significance. The ‘positive’ response group had a greater reduction in other liver function tests, specifically, ALT, AST and GGT (P = 0.01, 0.005 and 0.03, respectively). There was a trend for a greater increase in liver synthetic measures such as albumin and PT in the ‘positive’ response group, although this was not statistically significant. No adverse events attributable to DHA were observed.
Table 4.
Change in select fatty acids and liver function tests by level of response to intervention
| Positive response | No change | Negative response | P (positive response vs. no change) | |
|---|---|---|---|---|
| n | 8 | 13 | 2 | |
| Baseline patient characteristics | ||||
| Age, years, mean (±S.E.) | 48.8 (±4.8) | 46.1 (±4.7) | 56.5 (±14.5) | 0.7 |
| Gender, male (%) | 63 | 54 | 0 | |
| Mayo PSC risk score (±S.E.) | 0.8 (±0.4) | 1.2 (±0.2) | 1.8 (±1.0) | 0.4 |
| Ursodiol, % | 25 | 33 | 50 | |
| Change in fatty acids, LFTs, and fibrosis ELF score | ||||
| Fatty acids* mol% (±S.E.) | ||||
| DHA | ||||
| Baseline | 1.7 (±0.5) | 1.8 (±0.3) | 1.2† | 0.9 |
| 12 months | 3.9 (±0.7) | 3.3 (±0.3) | 3.2† | 0.4 |
| % change, mean (±S.E.) | 280 (±136.4) | 88 (±48.5) | 161† | 0.2 |
| AA | ||||
| Baseline | 7.6 (±0.6) | 5.7 (±0.6) | 6.4† | 0.04 |
| 12 months | 5.6 (±0.6) | 4.7 (±0.5) | 3.1† | 0.3 |
| % change, mean (±S.E.) | −26 (±6.1) | −7 (±13.7) | −52† | 0.2 |
| LA | ||||
| Baseline | 28.2 (±3.2) | 23.7 (±0.9) | 24.8† | 0.1 |
| 12 months | 29.5 (±1.6) | 24.1 (±2.4) | 26.3† | 0.1 |
| % change, mean (±S.E.) | 5 (±12.9) | 5 (±9.0) | 6† | 1.0 |
| LFTs, (normal range), mean (±S.E.) | ||||
| AP (35–130 IU/L) | ||||
| Baseline | 406.9 (±84.0) | 321.5 (±40.4) | 397.0 (±61.0) | 0.3 |
| 12 months | 214.5 (±72.8) | 297.2 (±30.0) | 627.0 (±94.0) | 0.2 |
| % change | −53 (±6.9) | −5 (±3.5) | 58 (±0.6) | <0.00001 |
| ALT (0–40 IU/L) | ||||
| Baseline | 93.8 (±37.9) | 144.0 (±51.5) | 100.0 (±59.0) | 0.5 |
| 12 months | 69.5 (±30.8) | 139.8 (±29.4) | 294.5 (±228.5) | 0.1 |
| % change | −26 (±8.4) | 24 (±12.5) | 145 (±84.0) | 0.01 |
| AST (0–40 IU/L) | ||||
| Baseline | 72.9 (±30.4) | 84.9 (±18.9) | 69.5 (±1.5) | 0.7 |
| 12 months | 63.1 (±30.0) | 92.5 (±14.9) | 217 (±117.0) | 0.3 |
| % change | −18 (±7.6) | 19 (±7.8) | 209 (±161.7) | 0.005 |
| GGT (5–61 IU/L) | ||||
| Baseline | 313.6 (±113.0) | 386.6 (±84.9) | 569.5 (±274.5) | 0.6 |
| 12 months | 193.0 (±72.3) | 504.2 (±102.9) | 708 (±354.0) | 0.04 |
| % change | −28 (±16.0) | 31 (±18.3) | 23 (±2.9) | 0.03 |
| Bilirubin (0–1.5 mg/dL) | ||||
| Baseline | 1.0 (±0.4) | 1.1 (±0.2) | 0.8 (±0.2) | 0.9 |
| 12 months | 1.6 (±0.9) | 1.7 (±0.4) | 2.2 (±0.1) | 0.5 |
| % change | 21 (±15.0) | 114 (±49.2) | 188 (±78.3) | 0.2 |
| Albumin (3.5–5.2 g/dL) | ||||
| Baseline | 4.3 (±0.1) | 4.2 (±0.1) | 3.9 (±0.6) | 0.5 |
| 12 months | 4.4 (±0.08) | 4.2 (±0.1) | 4.0 (±0.5) | 0.07 |
| % change | 3 (±2.7) | −8 (±7.9) | 3 (±3.0) | 0.3 |
| PT (35–130 s) | ||||
| Baseline | 10.9 (±0.6) | 12.3 (±1.8) | 9.9 (±1.4) | 0.6 |
| 12 months | 10.8 (±0.6) | 11.1 (±1.4) | 10.2 (±1.1) | 0.9 |
| % change | 2 (±1.7) | −14 (±13.7) | 4 (±3.5) | 0.4 |
| Fibrosis ELF score (normal 0–6.6), mean (±S.E.) | ||||
| Baseline | 9.1 (±0.9) | 10.5 (±0.5) | 12.3† | 0.2 |
| 12 months | 9.4 (±0.8) | 10.6 (±0.3) | 12.0† | 0.2 |
| % change | 7 (±3.0) | 3 (±5.7) | −2† | 0.6 |
S.E., standard error of the mean; DHA, docosahexaenoic acid; AA, arachidonic acid; LFTs, liver function tests; AP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma glutamyltransferase; PT, prothrombin time.
Positive response indicates a reduction in AP of >25%. No change indicates a reduction in AP of <25% or an increase in AP of <25%. Negative response denotes an increase in AP of >25%.
BIDMC patients only; Positive response n = 5; No change n = 7; Negative response n = 1.
Value for n = 1, S.E. not applicable, patient developed gall-bladder cancer.
DISCUSSION
To date there are no effective medical therapies for PSC that result in significant long-term improvements in outcome. Although high dose ursodeoxycholic acid has been used, a large NIH trial was stopped early due to increased complications and is thus no longer recommended.1 Thus, many of the current treatment strategies focus on addressing the secondary complications of the disease rather than targeting the excessive inflammation and subsequent biliary tract fibrosis that develops. Furthermore, some medical strategies and treatments for secondary complications are invasive and may have detrimental side effects.4 In this pilot study we chose serum alkaline phosphatase as the primary outcome measure. This is based on our recent demonstration that normalisation of alkaline phosphatase in PSC patients is associated with a reduction in disease progression from 33 to 14% (P < 0.02).19
We demonstrated in this open-labelled, pilot trial that oral DHA supplementation may be an efficacious treatment for PSC. In our cohort, 22% of the patients had a ‘highly positive’ response to treatment, such that the serum AP levels were decreased by over 50% of baseline levels. A ‘positive’ response, a reduction in serum AP levels by at least 25%, occurred in 35%. Perhaps just as important, 56% had no change, or in other words, no significant deterioration in their serum AP levels while on oral DHA supplementation. With 91% of the cohort with improved or no change in their serum AP levels, the results of the study support an acceptable safety pro-file of oral DHA supplementation at this dosing regimen. Only two patients experienced a worsening of their serum AP levels, one of whom developed cholangiocarcinoma while enrolled in the study.
Indeed, DHA therapy has been used in many clinical trials with no reported significant side effects. In addition, our data suggest that patients who responded most favourably had less severe disease at baseline, supporting earlier treatment during the course of the disease; however, the limited sample size and lack of a placebo group in this pilot trial prevent conclusive treatment recommendations or efficacy conclusions. A higher dose or a longer treatment period may have been more effective.
We have previously reported the important role of altered fatty acid metabolism in disease expression in patients homozygous for the CFTR mutation and in cftr−/−knockout mice.12, 13 However, we have also shown that single allelic CFTR mutations (heterozygotes) in both human studies and animal models also exhibit abnormalities in fatty acid metabolism and altered immunity and regulation of inflammation. Our group has shown that this is not only seen in CFTR expressing epithelial cells but is also present in peripheral blood monocytes from cystic fibrosis (CF) patients as well as healthy obligate heterozygotes.11 This is based on the finding that the half maximal effective concentration (EC50) for lipopolysaccharide (LPS)-induced interleukin (IL)-8 secretion was 100-fold lower in monocytes from CF patients and healthy obligate heterozygotes than that observed from healthy control monocytes. Hence, a single CFTR allele is sufficient to lower the threshold for inflammation. It has now become clear from multiple investigators that CFTR dysfunction leads to an excessive host inmmune response characterised by increased levels of pro-inflammatory cytokines such as IL-1, 6 and 8 and decreased IL-10.9–11, 20 This is of particular importance in this study, as our laboratory has shown that there is an increased prevalence of CFTR abnormalities in adults and children with PSC as demonstrated by genotype and phenotype analyses.5, 6
In addition to an overall improvement in AP levels, we did observe a minor increase in total bilirubin levels. It is possible that the change over time, although still in the normal range, might represent the natural progression of the disease. The fact that when we stratified by whether baseline bilirubin was normal or abnormal, the subjects with normal baseline bilirubin values remained in the normal range at the end of the study, indicating that this interval change is likely to be clinical insignificant and did not represent an adverse effect of DHA. However, future randomised controlled trials of DHA in PSC will need to monitor bilirubin levels in addition to other safety parameters.
The DHA is an important regulator of inflammation. DHA-derived metabolites such as resolvins and docosatrienes decrease neutrophil infiltration, enhance macrophage phagocytosis,21 and down-regulate NFκB activity in cells directly or by activation of PPAR reducing cytokine release.22, 23 Each of these pathways plays an important role in terminating the host’s inflammatory response.10, 17 Our group has previously shown that decreased expression in kupffer cells may play a role in the excessive inflammatory response associated with CFTR dysfunction in cftr−/−mice.20 Furthermore, cftr−/−mice following induction of colitis exhibit a defect in PPARα translocation to the nucleus and subsequent activation when compared to wild-type mice.16 DHA treatment reverses this process resulting in a several-fold increase in PPARα mRNA expression in CF mice as well as increased nuclear translocation of PPARα. We therefore postulate that the mechanism of action of DHA may be through its role as a PPARα agonist.
The positive results of our pilot study support the need for a larger, multicentre, placebo controlled trial evaluating the effectiveness of oral DHA supplementation as a treatment for PSC. In addition to the assessment of the efficacy of oral DHA, additional mechanistic and genotypic analyses may provide new insights into the pathophysiology of PSC, the role of the innate immune system, identify early markers of the disease (fatty acid abnormality) and determine potential modifier genes.
Acknowledgments
Declaration of funding interests: The work performed by C. R. M. was supported by the Programme for Faculty Development and Diversity and the study was conducted in the Harvard-Thorndike Clinical Research Center, both supported by Harvard Catalyst | The Harvard Clinical and Translational Science Center, from the National Center for Research Resources (Award #UL1 RR 025758 and financial contributions from Harvard University and its affiliated academic health care centres). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centres, the National Center for Research Resources or the National Institutes of Health. The work performed by P. G. B. was supported by the Division of Gastroenterology, Beth Israel Deaconess Medical Center, and the Clinical Investigator Training Programme: Beth Israel Deaconess Medical Center – Harvard/MIT Health Sciences and Technology, in collaboration with Pfizer Inc. and Merck & Co. This work was also supported by funding provided by the Morgan Foundation for the Study of PSC. None of the funding bodies had any role in the study design or conduct; data collection, management, analysis or interpretation; or preparation, review, or approval of the manuscript.
Footnotes
Declaration of personal interests: N. H. Afdhal has served as a speaker, a consultant and an advisory board member for Gilead, Echosens, Biogen, GlaxoSmithKline, Vertex, Novartis, Idera Pharmaceuticals, Boehringer Ingelheim, Human Genome Sciences, Biolex, Fibrogen, Ligand, Springbank and Schering/Merck. He has received research funding from Schering Plough/Merck, Novartis, GlaxoSmithKline, Echosens, Vertex, Gilead, Quest, Pharmasett and Abbott. N. H. Afdhal has an academic faculty appointment at Beth Israel Deaconess Medical Center/Harvard Medical School and is not employed by any pharmaceutical or biotechnology company and does not own stock, shares or other equity options in any pharmaceutical or biotechnology companies. N. H. Afdhal does not own any patents or other intellectual property rights. K. D. Lindor has served as a speaker, a consultant and an advisory board member for BioMarin Pharmaceutical, Inc., Biotie Therapies Corporation, Centocor, Gilead Sciences, Inc., Martek Biosciences Corporation, Merck Sharp & Dohme Corp., Sanofi-Aventis and Pfizer, Inc. K. D. Lindor has received research funding from Intercept Pharmaceuticals and NIH. K. D. Lindor is not employed by any pharmaceutical or biotechnology company and does not own stock, shares or other equity options in any pharmaceutical or biotechnology companies. K. D. Lindor has a patent issued on the use of ursodeoxycholic acid to treat nonalcoholic steatohepatitis (October 2, 2001). S. D. Freedman has served as a presenter to the FDA advisory panel on behalf of Alnara/Lilly in January 2011 on Liprotamase, a pancreatic enzyme replacement medication for Cystic Fibrosis. S. D. Freedman has received research funding for the studies described in this manuscript from NIH and the Morgan Foundation for the Study of PSC. Other funding sources in the past two years include NIH, John W. Alden Trust, Charles H. Hood Foundation, Cystic Fibrosis Foundation, and Martek. S. D. Freedman has an academic faculty appointment at Beth Israel Deaconess Medical Center/Harvard Medical School and is not employed by any pharmaceutical or biotechnology company and does not own stock, shares or other equity options in any pharmaceutical or biotechnology companies. S. D. Freedman has the following patents: a patent issued on a method for retrieving pancreatic juice from patients; a patent issued on Jan 30, 2011 for the use of DHA in the treatment of diseases related to Cystic Fibrosis but does not cover PSC; a patent issued on July 19 2011 on Methods for modulating PPAR biologic activity for the treatment of diseases caused by mutations in the CFTR gene.
References
- 1.Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808–14. doi: 10.1002/hep.23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El Fouly A, Dechene A, Gerken G. Surveillance and screening of primary sclerosing cholangitis. Dig Dis. 2009;27:526–35. doi: 10.1159/000233293. [DOI] [PubMed] [Google Scholar]
- 3.Franco J, Saeian K. Biliary tract inflammatory disorders: primary sclerosing cholangitis and primary biliary cirrhosis. Curr Gastroenterol Rep. 1999;1:95–101. doi: 10.1007/s11894-996-0006-8. [DOI] [PubMed] [Google Scholar]
- 4.McLoughlin M, Enns R. Endoscopy in the management of primary sclerosing cholangitis. Curr Gastroenterol Rep. 2008;10:177–85. doi: 10.1007/s11894-008-0040-9. [DOI] [PubMed] [Google Scholar]
- 5.Sheth S, Shea JC, Bishop MD, et al. Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Hum Genet. 2003;113:286–92. doi: 10.1007/s00439-003-0963-z. [DOI] [PubMed] [Google Scholar]
- 6.Pall H, Zielenski J, Jonas MM, et al. Primary sclerosing cholangitis in childhood is associated with abnormalities in cystic fibrosis-mediated chloride channel function. J Pediatr. 2007;151:255–9. doi: 10.1016/j.jpeds.2007.03.062. [DOI] [PubMed] [Google Scholar]
- 7.Cohn JA, Strong TV, Picciotto MR, Nairn AC, Collins FS, Fitz JG. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterology. 1993;105:1857–64. doi: 10.1016/0016-5085(93)91085-v. [DOI] [PubMed] [Google Scholar]
- 8.Gaskin KJ, Waters DL, Howman-Giles R, et al. Liver disease and common-bile-duct stenosis in cystic fibrosis. N Engl J Med. 1988;318:340–6. doi: 10.1056/NEJM198802113180602. [DOI] [PubMed] [Google Scholar]
- 9.Freedman SD, Weinstein D, Blanco PG, et al. Characterization of LPS-induced lung inflammation in cftr−/− mice and the effect of docosahexaenoic acid. J Appl Physiol. 2002;92:2169–76. doi: 10.1152/japplphysiol.00927.2001. [DOI] [PubMed] [Google Scholar]
- 10.Karp CL, Flick LM, Park KW, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol. 2004;5:388–92. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- 11.Zaman MM, Gelrud A, Junaidi O, et al. Interleukin 8 secretion from monocytes of subjects heterozygous for the deltaF508 cystic fibrosis transmembrane conductance regulator gene mutation is altered. Clin Diagn Lab Immunol. 2004;11:819–24. doi: 10.1128/CDLI.11.5.819-824.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freedman SD, Blanco PG, Zaman MM, et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med. 2004;350:560–9. doi: 10.1056/NEJMoa021218. [DOI] [PubMed] [Google Scholar]
- 13.Freedman SD, Katz MH, Parker EM, Laposata M, Urman MY, Alvarez JG. A membrane lipid imbalance plays a role in the phenotypic expression of cystic fibrosis in cftr(−/−) mice. Proc Natl Acad Sci U S A. 1999;96:13995–4000. doi: 10.1073/pnas.96.24.13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beharry S, Ackerley C, Corey M, et al. Long-term docosahexaenoic acid therapy in a congenic murine model of cystic fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G839–48. doi: 10.1152/ajpgi.00582.2005. [DOI] [PubMed] [Google Scholar]
- 15.Blanco PG, Zaman MM, Junaidi O, et al. Induction of colitis in cftr−/− mice results in bile duct injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G491–6. doi: 10.1152/ajpgi.00452.2003. [DOI] [PubMed] [Google Scholar]
- 16.Pall H, Zaman MM, Andersson C, Freedman SD. Decreased peroxisome proliferator activated receptor alpha is associated with bile duct injury in cystic fibrosis transmembrane conductance regulator−/− mice. J Pediatr Gastroenterol Nutr. 2006;42:275–81. doi: 10.1097/01.mpg.0000189368.37535.42. [DOI] [PubMed] [Google Scholar]
- 17.Serhan CN, Arita M, Hong S, Gotlinger K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids. 2004;39:1125–32. doi: 10.1007/s11745-004-1339-7. [DOI] [PubMed] [Google Scholar]
- 18.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis) Virchows Arch A Pathol Anat Histol. 1978;379:103–12. doi: 10.1007/BF00432479. [DOI] [PubMed] [Google Scholar]
- 19.Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis. 2011;43:309–13. doi: 10.1016/j.dld.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ollero M, Junaidi O, Zaman MM, et al. Decreased expression of peroxisome proliferator activated receptor gamma in cftr−/− mice. J Cell Physiol. 2004;200:235–44. doi: 10.1002/jcp.20020. [DOI] [PubMed] [Google Scholar]
- 21.Seki H, Sasaki T, Ueda T, Arita M. Resolvins as regulators of the immune system. ScientificWorldJournal. 2010;10:818–31. doi: 10.1100/tsw.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calder PC. Polyunsaturated fatty acids, inflammation, and immunity. Lipids. 2001;36:1007–24. doi: 10.1007/s11745-001-0812-7. [DOI] [PubMed] [Google Scholar]
- 23.Wall R, Ross RP, Fitzgerald GF, Stanton C. Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev. 2010;68:280–9. doi: 10.1111/j.1753-4887.2010.00287.x. [DOI] [PubMed] [Google Scholar]