

Abstract
Polyamines are small essential polycations involved in many biological processes. Enzymes of polyamine metabolism have been extensively studied and are attractive drug targets. Nevertheless, the reversible acetylation of polyamines remains poorly understood. Although eukaryotic N8-acetylspermidine deacetylase activity has already been detected and studied, the specific enzyme responsible for this activity has not yet been identified. However, a zinc deacetylase from Mycoplana ramosa, acetylpolyamine amidohydrolase (APAH), has been reported to use various acetylpolyamines as substrates. The recently solved crystal structure of this polyamine deacetylase revealed the formation of an “L”-shaped active site tunnel at the dimer interface, with ideal dimensions and electrostatic properties for accommodating narrow, flexible, cationic polyamine substrates. Here, we report the design, synthesis, and evaluation of N8-acetylspermidine analogues bearing different zinc binding groups as potential inhibitors of APAH. Most of the synthesized compounds exhibit modest potency, with IC50 values in the mid-micromolar range, but compounds bearing hydroxamate or trifluoromethylketone zinc binding groups exhibit enhanced inhibitory potency in the mid-nanomolar range. These inhibitors will enable future explorations of acetylpolyamine function in both prokaryotes and eukaryotes.
Keywords: Metalloenzyme, Polyamine deacetylase, Enzyme inhibitor, Polyamine analogues
1. Introduction
Polyamines such as putrescine, spermidine, and spermine are ubiquitous in living organisms and implicated in numerous essential biological processes.1 For instance, polyamine concentrations affect the cell cycle progression through tightly regulated biosynthetic pathways.1, 2 At the molecular level, since polyamines are polycations, they can bind to nucleic acids and modulate DNA-protein interactions.2 Given the importance of polyamines in different cellular processes, various enzymes of polyamine metabolism have been studied as potential drug targets.3, 4 For example, since upregulation of polyamine biosynthesis is a hallmark of certain cancers,5, 6 inhibitors of ornithine decarboxylase (ODC), S-adenosylmethionine decarboxylase, spermidine synthase, and spermine synthase have been evaluated in approaches to cancer chemotherapy. Depletion of putrescine and spermidine was achieved in vitro by cell treatment with the irreversible ODC inhibitor α-difluoromethylornithine (DFMO),7 leading to inhibition of cell growth.8 Although DFMO failed in clinical trials as a cancer chemotherapeutic agent, it was approved by the FDA for the treatment of parasitic infections such as African sleeping sickness.9
While most of the enzymes of polyamine metabolism have been extensively studied, enzymes involved in the reversible acetylation of polyamines are less well understood. The acetylation of polyamines decreases their overall charge, which is believed to regulate their function in vivo. Indeed, acetylated polyamines destabilize nucleosome structure, whereas the corresponding free polyamines bind to DNA and facilitate condensation.10, 11 In eukaryotes, spermidine can be either N1- or N8-acetylated by two distinct enzymes: a cytoplasmic spermidine/spermine N1-acetyltransferase12 or a nuclear spermidine N8-acetyltransferase.13 Despite similar structures, N1- and N8-acetylspermidine are metabolized differently once formed: N1-acetylspermidine is catabolized to putrescine by a cytosolic polyamine oxidase,14 and N8-acetylspermidine is hydrolyzed to generate spermidine by a specific cytosolic N8-acetylspermidine deacetylase that is unable to use N1-acetylspermidine as a substrate.15
Even though the N8-acetylspermidine deacetylase activity has been characterized and studied in vivo and in subcellular fractions, no eukaryotic polyamine deacetylase has been identified to date. However, a prokaryotic polyamine deacetylase has been reported: acetylpolyamine amidohydrolase (APAH) from Mycoplana ramosa.16 Notably, APAH has broader substrate specificity in comparison with the mammalian enzyme. As shown in Figure 1, APAH substrates include both small and large acetylpolyamines such as acetylputrescine, acetylcadaverine, N1- and N8-acetylspermidine, and N1-acetylspermine.16, 17 Bacterial APAH is a dimeric zinc-dependent hydrolase16, 17 as recently confirmed in crystal structure determinations of APAH from M. ramosa18 and from B. pseudomallei.19 As previously proposed,20 APAH adopts the α/β fold first observed for the binuclear manganese metalloenzyme arginase21 and also shared by the histone deacetylases (HDACs).22–24 Key active site residues required for the chemical mechanism of deacetylation are conserved between APAH and HDACs.25 In contrast with the HDACs, the dimerization of APAH results in the formation of a narrow “L”-shaped active site at the dimer interface,18 conferring specificity for slender, flexible substrates rather than the large peptide substrates typically processed by HDACs. This structural feature now guides the design of specific inhibitors of APAH.
Figure 1.

Substrates of APAH from Mycoplana ramosa: (A) acetylputrescine, (B) acetylcadaverine, (C) N1-acetylspermidine, (D) N8-acetylspermidine, and (E) N1-acetylspermine.
Inhibitors of APAH will be helpful tools for the exploration of both prokaryotic and eukaryotic acetylpolyamine function in vivo. For instance, inhibitors of the mammalian N8-acetylspermidine deacetylase have been described earlier, some of which exhibit low nanomolar inhibitory activity.26, 27 So far, the HDAC inhibitor M34428 and the trifluoromethylketone analogue of l-arginine29 are the only compounds reported to inhibit the bacterial polyamine deacetylase in the low and mid-micromolar range, respectively.18, 29 Here, we report the synthesis of new polyamine derivatives as well as new synthetic routes for some of the previously described mammalian N8-acetylspermidine deacetylase inhibitors.26, 27 All compounds synthesized in the current study are analogues of N8-acetylspermidine bearing different functional groups targeting Zn2+ coordination. We also report the inhibitory potency of these compounds against M. ramosa APAH.
2. Results and discussion
2.1. Inhibitor design
The X-ray crystal structure of inactive H159A APAH complexed with N8-acetylspermidine (PDB accession code 3Q9C) illustrates the molecular details of substrate recognition in the enzyme active site.18 Key interactions are made by the N4 secondary amino group of N8-acetylspermidine, which donates a hydrogen bond to E117 and makes a cation-π interaction with F225; the N1 primary amino group, which donates a hydrogen bond to E106 in the other monomer of the homodimer; the amide NH group, which donates a hydrogen bond to the backbone carbonyl of G167; and the amide carbonyl group, which coordinates to the Zn2+ ion and accepts a hydrogen bond from Y323. Based on the mechanism of catalysis by the related HDACs,30–32 a nucleophilic Zn2+-bound water molecule is activated by metal coordination and general base H159 (Figure 2). Nucleophilic attack at the scissile carbonyl group of the substrate results in the formation of a tetrahedral intermediate stabilized by metal coordination and hydrogen bond interactions with surrounding residues. The collapse of this intermediate is enabled by H159, which serves as a general acid catalyst in this step of the mechanism,31 leading to the formation of products spermidine and acetate.25
Figure 2.
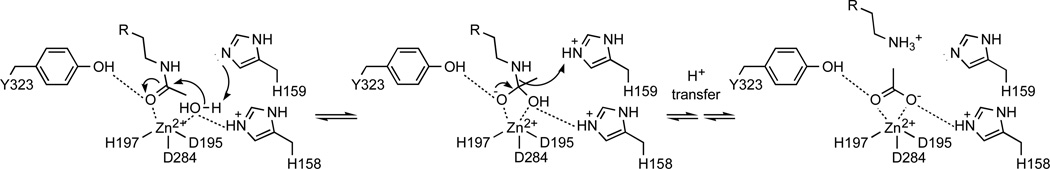
Mechanism of APAH.
Based on the structural features important for substrate recognition and catalysis, including the tetrahedral structure of the transition state flanking the tetrahedral intermediate, we designed and synthesized potential APAH inhibitors based on the N8-acetylspermidine substrate-like scaffold. As shown in Figure 3, compounds I–X all share a common 1,3-diaminopropane moiety to preserve key enzyme-substrate hydrogen bond interactions with the polyamine N1 and N4 groups (Figure 2). However, each compound differs in the nature of its head group designed to mimic substrate or tetrahedral transition state binding to the active site Zn2+ ion. Most of these Zn2+-binding groups have been successfully incorporated into effective inhibitors of HDACs29, 33–39 and other metallohydrolases.40–43
Figure 3.

Structures of target compounds as potential APAH inhibitors. These compounds are analogues of the substrate N8-acetylspermidine (Figure 1D).
2.2. Chemistry
Syntheses of compounds I–X are summarized in Schemes 1–4. Compounds I–X were each synthesized from key intermediates 3 or 4. As shown in Scheme 1, 3 and 4 were obtained in two steps. In the first step, 1,3-diaminopropane was N-alkylated with an alkylbromide, either 5-bromopent-1-ene or 7-bromohept-1-ene, to yield alkylamines 1 and 2, respectively. This reaction was performed with an excess of 1,3-diaminopropane (10 equivalents, neat) to favor the monoalkylation of the unprotected diamine over polyalkylation. Monoalkylamines 1 and 2 were then quantitatively N-protected with tert-butoxycarbonyl (Boc) groups with di-tert-butyl pyrocarbonate (Boc2O).
Scheme 1.
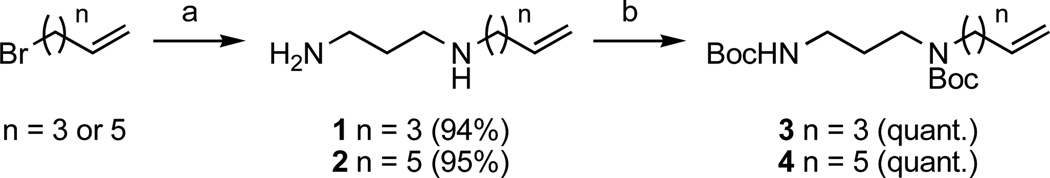
Synthesis of intermediates 3 and 4. Reagents and conditions: (a) 1,3-diaminopropane (neat, 10 eq), 0°C (1 h) then room temperature (RT) (2 h); (b) Boc2O (3 eq) in CH2Cl2, 0°C then RT (overnight).
Scheme 4.

Synthesis of compounds VII–X. Reagents and conditions: (a) 9-BBN (2.5 eq) in THF, 0°C then RT (20 h), then NaOH, H2O2, 0°C then RT (30 min); (b) CBr4 (2 eq), PPh3 (2 eq) in THF, 0°C then RT (overnight); (c) KSAc (6 eq) in MeCN, RT (overnight); (d) anhydrous HCl (4 N) in dioxane, RT (2 h); (e) NaOMe (2 eq) in MeOH, RT (2 h); (f) anhydrous HCl (1 N) in AcOEt, RT (5 h); (g) NaSMe (6 eq) in EtOH, 60°C (overnight); (h) m-CPBA (3 eq) in CH2Cl2, 0°C then RT (2 h); (i) [Ir(cod)Cl]2 (5 mol %), dppm (10 mol %), pinacolborane (1.9 eq), RT (24 h); (j) aqueous HCl (6 N), reflux (24 h).
We developed an alternative route for the synthesis of compounds I, II, and III compared with that previously published.26, 27 The new synthetic route allows for more flexibility in generating additional compounds from intermediate 4. As depicted in Scheme 2, compounds I, II, and III were synthesized from carboxylic acid 5. Oxidative cleavage of alkene 4 into 5 was performed by the Sharpless method with ruthenium chloride as catalyst and sodium periodate as oxidant44, 45 in solvent system H2O/AcOEt/MeCN (3/2/2).46 N,N’-carbonyldiimidazole (CDI) mediated coupling of carboxylic acid 5 with N-O-dimethylamine quantitatively led to Weinreb amide 6. N-methoxy-N-methylamides are well-known reagents for the synthesis of ketones from carboxylic acids in the presence of Grignard or organolithium reagents.47 Using this strategy, methylketone 7 was synthesized from Weinreb amide 6 with an excess of methylmagnesium bromide (5 equivalents). CDI-activated carboxylic acid 5 was reacted with unprotected hydroxylamine to form the corresponding hydroxamic acid 8.48 Deprotection of compounds 5, 7, and 8 with anhydrous HCl (4 N) in dioxane at room temperature led to target compounds I, II, and III as dihydrochloride salts.
Scheme 2.

Synthesis of compounds I–III. Reagents and conditions: (a) RuCl3 (4 mol %), NaIO4 (4.5 eq) in H2O/AcOEt/MeCN (3/2/2), RT (3 h); (b) anhydrous HCl (4 N) in dioxane, RT (2 h); (c) CDI (2 eq) in CH2Cl2, RT (1 h), then MeNHOMe•HCl (2 eq), RT (overnight); (d) MeMgBr (5 eq) in THF, 0°C (2 h) then RT (1 h); (e) CDI (1.5 eq) in THF, RT (1 h), then NH2OH•HCl (2 eq), RT (overnight).
Alkene 4 also served as a key common intermediate for the synthesis of compounds IV, V, and VI as shown in Scheme 3. As for the synthesis of carboxylic acid 5, introduction of the aldehyde functionality was achieved by an oxidative cleavage of alkene 4. This was done sequentially by first oxidizing 4 to the corresponding cis-diol with osmium tetroxide as catalyst in the presence of N-methylmorpholine N-oxide (NMO) in dioxane/H2O (4/1) as solvent49 until completion of the reaction (3 hours). The cis-diol derived from 4 was not isolated, but directly cleaved to aldehyde 9 by reaction with sodium periodate. Nucleophilic trifluoromethylation of aldehyde 9 was achieved using Ruppert’s reagent (trifluoromethyltrimethylsilane, TMSCF3)50 and a source of fluoride, tetra-n-butylammonium fluoride (TBAF), as initiator under usual conditions.51, 52 Trifluoromethyl carbinol 10 was then easily oxidized to the corresponding trifluoromethyl ketone 11 by Dess-Martin periodinane (DMP).53, 54 Alkene 4 was quantitatively oxidized to epoxide 12 using m-chloroperbenzoic acid (m-CPBA). Regioselective ring opening of unsymetrically substituted epoxide 12 was achieved using lithium bromide and acetic acid yielding α-bromohydrin 13.55 Subsequent alcohol oxidation of 13 with DMP led to the formation of α-bromoketone 14.53 Bromide 14 was then treated with potassium thioacetate to afford thioester 15. Oxirane 12 also served as a precursor for the synthesis of α-methoxyketone derivative VI. Ring opening of its epoxide moiety with sodium methoxide in MeOH regioselectively formed the corresponding α-methoxyalcohol 16, after which oxidation with DMP afforded α-methoxyketone 17.53 Deprotection of compounds 11, 15, and 17 with anhydrous HCl (4 N in dioxane, or 1 N in AcOEt) at room temperature led to target compounds IV, V, and VI as dihydrochloride salts. Synthesis of the α-mercaptoketone from thioester 15 was attempted. Successful thioacetate group alcoholysis in MeOH under basic conditions (sodium methoxide) gave the corresponding N-Boc protected α-mercaptoketone, but no pure deprotected α-mercaptoketone could be isolated after deprotection under acidic conditions.
Scheme 3.

Synthesis of compounds IV–VI. Reagents and conditions: (a) NMO (2.5 eq), OsO4 (2.5 mol %) in dioxane/H2O (4/1), RT (3 h), then NaIO4 (2.5 eq), RT (20 min); (b) TMSCF3 (3 eq), TBAF (10 mol %) in THF, RT (2 h), then TBAF (in THF containing ca. 5% H2O, 1.5 eq), RT (45 min); (c) DMP (4 eq) in CH2Cl2, RT (overnight); (d) anhydrous HCl (4 N) in dioxane, RT (2 h); (e) m-CPBA (2 eq) in CH2Cl2, 0°C then RT (21 h); (f) LiBr (3.2 eq), AcOH (3 eq) in THF, RT (overnight); (g) DMP (3 eq), RT (3 h); (h) KSAc (6 eq) in MeCN, RT (overnight); (i) NaOMe (6 eq) in MeOH, RT (24 h), (j) DMP (6 eq) in CH2Cl2, RT (24 h); (k) anhydrous HCl (1 N) in AcOEt, RT (6 h).
As shown in Scheme 4, alkene 3 was used as precursor for compounds VII–X. Alkene 3 hydroboration with 9-borabicyclo[3.3.1]nonane (9-BBN)56 followed by oxidation with H2O2 under basic conditions (NaOH) quantitatively afforded alcohol 18. Subsequent bromination of 18 was achieved using carbon tetrabromide (CBr4) in the presence of triphenylphosphine (PPh3). Corresponding alkyl bromide 19 was then treated with potassium thioacetate to afford 20, as for the synthesis of thioesther 15. Thioacetate group alcoholysis in MeOH under basic conditions (sodium methoxide) gave thiol 21. Alkyl bromide 19 also served a precursor for the synthesis of sulfone derivative IX. Nucleophilic substitution of bromine by sodium thiomethoxide in EtOH afforded thioether 22, after which oxidation with m-CPBA led to sulfone 23. Hydroboration of alkene 3 with pinacolborane using [Ir(cod)Cl]2 as catalyst and 1,1-bis(diphenylphosphino)methane (dppm) as ligand under usual conditions selectively gave terminal boronic ester 24.57 Complete deprotection of 24 was achieved in aqueous HCl (6 N) under reflux affording compound X as a dihydrochloride salt. Deprotection of compounds 20, 21, and 23 with anhydrous HCl (4 N in dioxane, or 1 N in AcOEt) at room temperature led to target compounds VII, VIII, and IX as dihydrochloride salts.
2.3. Enzyme inhibition
All N8-acetylspermidine analogues were tested in vitro for APAH inhibition. Results are summarized in Table 1. Three compounds exhibit very poor inhibitory potency against APAH: carboxylic acid I, thioester VII, and sulfone IX, with IC50 values in the millimolar range. In contrast, sulfone and thioester analogues of SAHA are effective inhibitors of HDAC in the mid- or low micromolar range, respectively,33, 58 and carboxylic acid I was previously shown to be a potent inhibitor of the mammalian N8-acetylspermidine deacetylase in the low micromolar range.27
Table 1.
Inhibitory potency of compounds I–X against M. ramosa APAH
| Compound | Head-group | IC50 (µM) |
|---|---|---|
| I | -COOH | 1800 ± 200 |
| II | -COCH3 | 160 ± 10 |
| III | -CONHOH | 0.39 ± 0.03 |
| IV | -COCF3 | 0.27 ± 0.03 38 ± 6 |
| V | -COCH2SAc | 39 ± 10 4000 ± 1000 |
| VI | -COCH2OCH3 | 380 ± 50 |
| VII | -SAc | 1900 ± 200 |
| VIII | -SH | 26 ± 3 |
| IX | -SO2CH3 | 10000 ± 3000 |
| X | -B(OH)2 | 230 ± 40 |
A second set of compounds are modest inhibitors of APAH, with inhibitory potencies in the mid-micromolar range. In increasing order of potency, these compounds are: α-methoxyketone VI, boronic acid X, ketone II, thioester V, and thiol VIII. Ketone II is a modest inhibitor or the bacterial polyamine deacetylase but a mid-nanomolar selective inhibitor of the mammalian N8-acetylspermidine deacetylase.26 The efficacy and specificity of ketone I have also been demonstrated in vivo, and this compound was used to probe the function of the mammalian polyamine deacetylase.59 In general, the incorporation of each of these functional groups in the design of HDAC inhibitors resulted in the generation of highly potent compounds with inhibitory potency in the nanomolar range.33, 35–39 However, in contrast to our N8-acetylspermidine analogues, HDAC inhibitors are designed based on the combination of a metal-binding group, a linker, and an active site-capping group. For a given Zn2+-binding group, optimization of the linker and the capping group to optimize interactions with the mouth of the active site cleft is usually required to achieve exceptional inhibitory potency.
The best two APAH inhibitors showing potency in the nanomolar range are hydroxamate III and trifluoromethylketone IV (Table 1). Hydroxamates have been extensively studied as metal-coordinating groups for the design of metalloenzyme inhibitors, such as the FDA-approved HDAC inhibitor SAHA for anti-cancer chemotherapy.60 Hydroxamates form a stable bidentate five-membered ring complex with the catalytic Zn2+ ion that contributes to high affinity. Hydroxamate III exhibits an IC50 value of 390 nM against APAH. This compound is also a potent inhibitor of mammalian N8-acetylspermidine deacetylase activity, with an apparent Ki of 1 nM measured with subcellular extracts.27
Trifluoromethylketones are well-known to exist as gem-diol hydrate in aqueous solution. This was further demonstrated for compound IV with 13C and 19F NMR in D2O. Indeed, a single peak at −85.1 ppm for 19F NMR and a quadruplet at 93.6 ppm clearly indicates that the trifluoromethylketone group is hydrated in water. This gem-diol form mimics the tetrahedral intermediate in zinc metalloenzyme-catalyzed hydrolytic reactions, as first demonstrated for carboxypeptidase A.61, 62 Surprisingly, as shown in Figure 4, the data obtained for compound IV fit better with a two-site binding model, whereas the concentration-response curve of most of the other compounds were typical of a one-site binding model as for hydroxamate III. The biphasic curve obtained for inhibition by trifluoromethylketone IV yields IC50 values of 270 nM and 38 µM. Similar behavior is also observed for compound V, with IC50 values of 39 µM and 4 mM. While such biphasic inhibition is also observed in other systems, e.g., the inhibition of smooth muscle endothelin-converting enzyme by the metalloprotease inhibitor phosphoramidon,63 the reason for this uncommon inhibition mode against APAH remains unclear. This type of dose-response curve is usually observed when a ligand binds to a receptor existing in two different affinity states,64 or to a receptor or a transporter in two different sites.65 However, the X-ray crystal structure of the APAH-IV complex reveals the binding of the gem-diol form of the inhibitor solely in the enzyme active site (study in progress), and we have not observed any other notable features, e.g., time-dependent inhibition, in our measurements. Thus, it is possible that monomers A and B of APAH exist in two different affinity states with regard to the binding of IV.
Figure 4.

Inhibition of APAH by: (A) hydroxamate III and (B) trifluoromethylketone IV.
3. Conclusions
In summary, we have designed and synthesized a series of N8-acetylspermidine analogues bearing different functional groups targeting Zn2+ coordination interactions in the active site of APAH. Most analogues studied are modest inhibitors, but two – compounds III and IV, bearing hydroxamate and trifluoromethylketone groups, respectively – exhibit nanomolar inhibitory potency. Future work on the optimization of these leads may facilitate the development of even better APAH inhibitors. Moreover, compounds III and IV may also be useful tools for probing the function of acetylpolyamines in both eukaryotes and prokaryotes, and in searching for the as-yet unidentified mammalian N8-acetylspermidine deacetylase.
4. Experimental Section
4.1. Chemistry
4.1.1. General Procedures
All reagents were of at least 95% purity and purchased from Fisher Scientific, Alfa Aesar or Sigma Aldrich. All solvents were of HPLC grade and purchased from Fisher Scientific or Sigma Aldrich. For reactions requiring anhydrous conditions, solvents (THF, MeCN, and MeOH) were purchased as anhydrous grade from Fisher Scientific (except CH2Cl2, which was freshly distilled under N2 from P2O5). Reactions were monitored by TLC with Sigma Aldrich aluminum plates (silica gel with fluorescent indicator, 60 Å, 200 µm) and visualized by staining with a ninhydrin solution or under UV light when necessary. Flash column chromatography was performed using Fisher Scientific silica gel 60 (230–400 mesh). Melting points were determined using a Mel-Temp Electrothermal apparatus and were uncorrected. High-resolution mass spectrometry (HRMS) was performed on a Waters LC-TOF mass spectrometer (model LCT-XE Premier) using electrospray ionization (ESI) in positive mode. For compound X, the boronic acid moiety was derivatized by adding (+)-pinanediol to enable analysis by mass spectrometry.
1H and 13C NMR spectra were recorded on Bruker DMX 360 and DRX 500 spectrometers operating at 360 and 500 MHz, respectively, for 1H NMR and at 90.6 and 125.6 MHz, respectively, for 13C NMR. 19F NMR spectra were recorded at 282.4 MHz on a Bruker DMX 360 spectrometer, and 11B NMR spectra at 128 MHz on a Bruker DMX 400 spectrometer. 1H and 13C NMR chemical shifts (δ) are reported in ppm relative to the residual solvent peak. NMR coupling constants (J) are reported in Hz, and multiplicities are denoted as follows: s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quadruplet; and m, multiplet.
4.1.2. N1-(pent-4-enyl)propane-1,3-diamine (1)
To 35.2 mL of 1,3-diaminopropane (422 mmol) at 0°C and under argon was added dropwise 5-bromopent-1-ene (5.0 mL, 42.2 mmol). The solution was stirred at 0°C one hour, and two additional hours at room temperature. The reaction mixture was then partitioned between AcOEt (250 mL), brine (40 mL), saturated aqueous NaHCO3 (40 mL), and H2O (40 mL). The aqueous layer was extracted with AcOEt and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by flash column chromatography on SiO2 with CHCl3/MeOH/NH4OH gradients afforded pure alkyl diamine 1 (5.64 g, 94%) as a slightly yellow oil. 1H NMR (360 MHz, CDCl3) δ: 5.81-5.70 (m, 1H), 4.96 (dd, J = 1.8 Hz, J = 17.3 Hz, 1H), 4.90 (dd, J = 1.8 Hz, J = 10.1 Hz, 1H), 2.72 (t, J = 6.6 Hz, 2H), 2.63 (t, J = 6.8 Hz, 2H), 2.56 (t, J = 7.2 Hz, 2H), 2.06-2.00 (m, 2H), 1.63-1.41 (m, 7H). 13C NMR (125.6 MHz, CDCl3) δ: 138.2, 114.4, 49.3, 47.7, 40.3, 33.5, 31.3, 29.0. HRMS (ESI) calcd for C8H19N2 [M + H]+ 143.1548, found 143.1549.
4.1.3. N1-(hept-6-enyl)propane-1,3-diamine (2)
Alkylation of 1,3-diaminopropane (27.4 mL, 328 mmol) with 7-bromohept-1-ene (5 mL, 32.8 mmol) was performed under the same conditions as for 1, and afforded after purification alkyl diamine 2 as a slightly yellow oil (5.31 g, 95%). 1H NMR (360 MHz, CDCl3) δ: 5.79-5.68 (m, 1H), 4.93 (dd, J = 1.1 Hz, J = 17.3 Hz, 1H), 4.87 (dd, J = 1.1 Hz, J = 10.1 Hz, 1H), 2.70 (t, J = 6.8 Hz, 2H), 2.60 (t, J = 6.8 Hz, 2H), 2.53 (t, J = 6.8 Hz, 2H), 1.99 (m, 2H), 1.57 (m, 2H), 1.45 (m, 2H), 1.36-1.24 (m, 4H), 1.15 (s, 3H). 13C NMR (90.6 MHz, CDCl3) δ: 139.0, 114.3, 50.2, 48.0, 40.7, 34.0, 33.7, 30.1, 28.9, 26.9. HRMS (ESI) calcd for C10H23N2 [M + H]+ 171.1861, found 171.1853.
4.1.4. N1,N3-Bis(tert-butoxycarbonyl)-N1-(pent-4-enyl)propane-1,3-diamine (3)
To a solution of alkyl diamine 1 (5.40 g, 38.0 mmol) in dry CH2Cl2 (150 mL) at 0°C and under argon was added dropwise Boc2O (26.2 mL, 114 mmol). The solution was allowed to warm gradually to room temperature and was stirred overnight. The reaction mixture was concentrated in vacuo and the resulting residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford pure 3 as a colorless oil (12.7 g, quantitative). 1H NMR (500 MHz, CDCl3) δ: 5.78-5.70 (m, 1H), 5.10 (bs, 1H), 4.96 (dd, J = 2.0 Hz, J = 17.3 Hz, 1H), 4.91 (dd, J = 2.0 Hz, J = 10.0 Hz, 1H), 3.19 (t, J = 6.3 Hz, 2H), 3.08 (t, J = 7.5 Hz, 2H), 3.04 (t, J = 6.5 Hz, 2H), 1.97 (m, 2H), 1.62-1.52 (m, 4H), 1.40 (s, 9H), 1.38 (s, 9H). 13C NMR (125.6 MHz, CDCl3) δ: 156.1 (2C), 137.9, 115.0, 79.5, 79.0, 46.5, 44.0, 37.7, 31.0, 28.5 (7C), 27.6. HRMS (ESI) calcd for C18H34N2NaO4 [M + Na]+ 365.2416, found 365.2410.
4.1.5. N1,N3-Bis(tert-butoxycarbonyl)-N1-(hept-6-enyl)propane-1,3-diamine (4)
Boc-protection of alkyl diamine 2 (5.1 g, 29.9 mmol) with Boc2O (20.6 mL, 89.7 mmol) was performed under the same conditions as for 3, and afforded 4 as a colorless oil (11.1 g, quantitative). 1H NMR (500 MHz, CDCl3) δ: 5.63 (m, 1H), 5.26 (bs, 1H), 4.84 (dd, J = 1.6 Hz, J = 17.2 Hz, 1H), 4.79 (dd, J = 1.6 Hz, J = 10.1 Hz, 1H), 3.11 (t, J = 6.0 Hz, 2H), 3.00 (t, J = 6.0 Hz, 2H), 2.95 (t, J = 6.3 Hz, 2H), 1.91 (apparent q (dt), J = 7.2 Hz, 2H), 1.55-1.50 (m, 2H), 1.42-1.33 (m, 2H), 1.32 (s, 9H), 1.30 (s, 9H), 1.29-1.24 (m, 2H), 1.19-1.11 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 155.9 (2C), 138.5, 114.3, 79.2, 78.6, 46.8, 43.7, 37.6, 33.5, 28.4, 28.8 (7C), 28.2, 26.2. HRMS (ESI) calcd for C20H38N2NaO4 [M + Na]+ 393.2729, found 393.2727.
4.1.6. N,N’-Bis(tert-butoxycarbonyl)-6-[(3-aminopropyl)amino]hexanoic acid (5)
To a solution of alkene 4 (4.00 g, 10.8 mmol) in 200 mL of a H2O/AcOEt/MeCN (3/2/2) mixture were added successively ruthenium(III) chloride hydrate (35%, 256 mg, 432 µmol) and sodium periodate (10.4 g, 48.6 mmol). After stirring at room temperature for 3 hours, the reaction mixture was quenched by the addition of isopropanol (80 mL), the suspension filtered over a celite pad, and the filtrate concentrated in vacuo. The aqueous phase was extracted with AcOEt and the combined organic extracts were washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was further purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford carboxylic acid 5 as a colorless oil (4.11 g, 98%). 1H NMR (500 MHz, CDCl3) δ: 8.79 (bs, 1H), 5.37 (bs, 1H), 3.21 (t, J = 6.3 Hz, 2H), 3.12 (t, J = 6.6 Hz, 2H), 3.07 (t, J = 6.0 Hz, 2H), 2.32 (t, J = 7.4 Hz, 2H), 1.66-1.60 (m, 4H), 1.54-1.48 (m, 2H), 1.43 (s, 9H), 1.42 (s, 9H), 1.33-1.27 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 178.6, 156.5 (2C), 79.7, 79.4, 46.9, 44.2, 38.0, 34.0, 28.6 (7C), 28.2, 26.5, 24.5. HRMS (ESI) calcd for C19H36N2NaO6 [M + Na]+ 411.2471, found 411.2473.
4.1.7. N’,N’’-Bis(tert-butoxycarbonyl)-6-[(3-aminopropyl)amino]-N-methoxy-N-methylhexanamide (6)
To a solution of carboxylic acid 5 (2.08 g, 5.35 mmol) in dry CH2Cl2 (80 mL) under argon was added CDI (1.74 g, 10.7 mmol). After one hour at room temperature, N,O-dimethylhydroxylamine hydrochloride (1.04 g, 10.7 mmol) was added to the solution. After stirring overnight, the reaction mixture was diluted CH2Cl2 (100 mL), washed with 0.1 M HCl (3 × 50 mL), water, and brine. The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was further purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford Weinreb amide 6 (2.31 g, quantitative) as a colorless oil. 1H NMR (360 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.65 (s, 3H), 3.22 (t, J = 6.1 Hz, 2H), 3.15 (s, 3H), 3.14-3.03 (m, 4H), 2.39 (t, J = 7.2 Hz, 2H), 1.67-1.58 (m, 4H), 1.56-1.46 (m, 2H), 1.43 (s, 9H), 1.41 (s, 9H), 1.34-1.26 (m, 2H). 13C NMR (90.6 MHz, CDCl3) δ: 174.6, 156.1 (2C), 79.6, 79.1, 61.3, 46.9, 44.1, 37.8, 32.3, 31.9, 28.6 (7C), 28.3, 26.8, 24.5. HRMS (ESI) calcd for C21H42N3O6 [M + H]+ 432.3074, found 432.3091.
4.1.8. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]heptan-2-one (7)
To a solution of Weinreb amide 6 (1.36 g, 3.15 mmol) in dry THF (25 mL) at 0°C and under argon was added dropwise methylmagnesium bromide (1.0 M in THF, 15.8 mL, 15.8 mmol). The solution was stirred 2 hours at 0°C and then one hour at room temperature. The reaction mixture was cooled down to 0°C, quenched by the addition of saturated aqueous NH4Cl (20 mL), diluted with AcOEt (100 mL), and the aqueous phase was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was further purified by flash column chromatography with hexanes/AcOEt gradients to afford ketone 7 as a colorless oil (1.12 g, 92%). 1H NMR (500 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.17 (t, J = 6.5 Hz, 2H), 3.06 (t, J = 6.5 Hz, 2H), 3.03 (t, J = 6.5 Hz, 2H), 2.36 (t, J = 7.5 Hz, 2H), 2.06 (s, 3H), 1.61-1.56 (m, 2H), 1.55-1.49 (m, 2H), 1.48-1.41 (m, 2H), 1.39 (s, 9H), 1.37 (s, 9H), 1.22-1.16 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 208.9, 156.1 (2C), 79.5, 79.0, 46.8, 43.9, 43.6, 37.7, 29.9, 28.5 (7C), 28.3, 26.4, 23.5. HRMS (ESI) calcd for C20H38N2O5Na [M + Na]+ 409.2678, found 409.2670.
4.1.9. N’,N’’-Bis(tert-butoxycarbonyl)-6-[(3-aminopropyl)amino]-N-hydroxyhexanamide (8)
To a solution of carboxylic acid 5 (600 mg, 1.54 mmol) in dry THF (20 mL) under argon was added CDI (376 mg, 2.32 mmol). After one hour at room temperature, hydroxylamine hydrochloride (215 mg, 3.09 mmol) was added to the solution. After stirring overnight, the reaction mixture was diluted with a 5% KHSO4 aqueous solution (60 mL) and extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The residue was further purified by flash column chromatography on SiO2 with AcOEt/MeOH gradients to afford hydroxamic acid 8 (550 mg, 88%) as a slightly yellow oil. 1H NMR (500 MHz, DMSO-d6) δ: 10.29 (bs, 1H), 8.61 (bs, 1H), 6.72 (bs, 1H), 3.08 (apparent q (2t), J = 7.5 Hz, 4H), 2.88 (apparent q (dt), J = 6.5 Hz, 2H), 1.93 (t, J = 7.4 Hz, 2H), 1.58-1.54 (m, 2H), 1.52-1.46 (m, 2H), 1.44-1.39 (m, 2H), 1.38 (s, 9H), 1.37 (s, 9H), 1.21-1.15 (m, 2H). 13C NMR (125.6 MHz, DMSO-d6) δ: 169.0, 155.6, 154.6, 78.2, 77.4, 46.3, 44.3, 37.6, 32.2, 28.2, 28.1 (6C), 27.6, 25.9, 24.9. HRMS (ESI) calcd for C19H37N3O6Na [M + Na]+ 426.2580, found 426.2576.
4.1.10. N,N’-Bis(tert-butoxycarbonyl)-6-[(3-aminopropyl)amino]hexanal (9)
To a solution of alkene 4 (2.71 g, 7.31 mmol) in 150 mL of a dioxane/H2O (4/1) mixture were successively added NMO monohydrate (2.47 g, 18.3 mmol) dissolved in 6 mL of water, and osmium tetroxide (47 mg, 185 µmol). TLC monitoring showed completion after 3 hours at room temperature. Sodium periodate (3.91 g, 18.3 mmol) was added to the solution. After stirring for 20 min, the reaction mixture was quenched with isopropanol (50 mL), the suspension filtered over a celite pad, and the filtrate concentrated in vacuo. The aqueous phase was extracted with AcOEt and the combined organic extracts were washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was further purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford aldehyde 9 as a colorless oil (2.64 g, 97%). 1H NMR (500 MHz, CDCl3) δ: 9.63 (s, 1H), 5.25 (bs, 1H), 3.11 (t, J = 7.0 Hz, 2H), 3.02 (t, J = 6.7 Hz, 2H), 2.96 (t, J = 7.4 Hz, 2H), 2.31 (t, J = 6.9 Hz, 2H), 1.55-1.49 (m, 4H), 1.42-1.37 (m, 2H), 1.32 (s, 9H), 1.30 (s, 9H), 1.21-1.15 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 202.1, 155.9 (2C), 79.3, 78.7, 46.6, 43.8, 43.6, 37.5, 28.3 (7C), 27.8, 26.2, 21.6. HRMS (ESI) calcd for C19H36N2O5Na [M + Na]+ 395.2522, found 395.2509.
4.1.11. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-1,1,1-trifluoroheptan-2-ol (10)
To a solution of aldehyde 9 (1.53 g, 4.11 mmol) in dry THF (20 mL) at room temperature and under argon were added successively TMSCF3 (1.82 mL, 12.3 mmol) and anhydrous TBAF (1.0 M in THF, 410 µL, 410 µmol). After 2 hours at room temperature, TBAF (1.0 M in THF containing ca. 5% H2O, 6.2 mL, 6.20 mmol) was added dropwise and the solution was stirred for 45 min and concentrated in vacuo. The corresponding residue was partitioned between AcOEt (100 mL) and saturated aqueous NaHCO3 (50 mL) and the aqueous phase was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. Purification by flash column chromatography on SiO2 with hexanes/AcOEt gradients afforded trifluoromethylalcohol 10 as a colorless oil (1.49 g, 82%). 1H NMR (360 MHz, CDCl3) δ: 5.32 (bs, 1H), 4.46 (bs, 1H), 3.79-3.73 (m, 1H), 3.11 (t, J = 6.5 Hz, 2H), 3.03 (t, J = 6.5 Hz, 2H), 2.97 (t, J = 6.5 Hz, 2H), 1.58-1.48 (m, 6H), 1.46-1.38 (m, 2H), 1.34 (s, 9H), 1.32 (s, 9H), 1.25-1.14 (m, 2H). 13C NMR (90.6 MHz, CDCl3) δ: 156.3 (2C), 125.5 (q, J = 281.8 Hz), 79.6, 79.1, 69.7 (q, J = 29.0 Hz), 46.9, 43.9, 37.6, 29.5, 28.3 (7C), 28.1, 26.4, 24.6. 19F NMR (338.8 MHz) δ: −79.8. HRMS (ESI) calcd for C20H37N2NaO5F3 [M + Na]+ 465.2552, found 465.2553.
4.1.12. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-1,1,1-trifluoroheptan-2-one (11)
To a solution of trifluoromethylalcohol 10 (1.40 g, 3.16 mmol) in dry CH2Cl2 (50 mL) at room temperature and under argon was added DMP (5.37 g, 12.7 mmol). The reaction mixture was stirred overnight at room temperature, then cooled down to 0°C, quenched with the addition of an aqueous sodium thiosulfate solution (0.5 M) saturated with NaHCO3 (150 mL), and the aqueous layer was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford trifluoromethylketone 11 as a slightly yellow oil (1.32 g, 95%). 1H NMR (500 MHz, CDCl3) (ketone/hydrate : 1/0) δ: 5.26 (bs, 1H), 3.11 (t, J = 6.7 Hz, 2H), 3.03 (t, J = 6.5 Hz, 2H), 2.96 (t, J = 6.0 Hz, 2H), 2.60 (t, J = 7.1 Hz, 2H), 1.60-1.53 (m, 4H), 1.45-1.39 (m, 2H), 1.33 (s, 9H), 1.30 (s, 9H), 1.23-1.17 (m, 2H). 13C NMR (500 MHz, CDCl3) δ: 191.2 (q, J = 35.2 Hz), 156.1 (2C), 115.5 (q, J = 291.4 Hz), 79.5, 78.8, 46.6, 43.4, 37.6, 36.1, 28.3 (7C), 28.1, 25.9, 22.0. 19F NMR (338.8 MHz) δ: −79.4. HRMS (ESI) calcd for C20H35N2O5F3Na [M + Na]+ 463.2396, found 463.2397.
4.1.13. N1,N3-Bis(tert-butoxycarbonyl)-N1-[5-(oxiran-2-yl)pentyl]propane-1,3-diamine (12)
To a solution of alkene 4 (2.89 g, 7.80 mmol) in dry CH2Cl2 (150 mL) under argon at 0°C was added dropwise m-CPBA (77%, 3.50 g, 15.6 mmol) in 40 mL dry CH2Cl2. The solution was allowed to reach room temperature and stirred until completion of the reaction (21 hours) as shown by TLC. Saturated aqueous NaHCO3 (50 mL) was added to the reaction mixture and the aqueous layer was extracted with AcOEt. Combined organic extracts were dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford epoxide 12 (3.01 g, quantitative) as a colorless oil. 1H NMR (360 MHz, CDCl3) δ: 5.28 (bs, 1H), 3.05 (t, J = 7.2 Hz, 2H), 2.97-2.88 (m, 4H), 2.71-2.67 (m, 1H), 2.54-2.52 (m, 1H), 2.26-2.24 (m, 1H), 1.51-1.46 (m, 2H), 1.38-1.26 (m, 6H), 1.27 (s, 9H), 1.24 (s, 9H), 1.18-1.14 (m, 2H). 13C NMR (90.6 MHz, CDCl3) δ: 155.8 (2C), 79.0, 78.4, 51.8, 46.6, 46.5, 43.6, 37.3, 32.1, 28.3, 28.2 (7C), 26.4, 25.5. HRMS (ESI) calcd for C20H39N2O5 [M + H]+ 387.2859, found 387.2854.
4.1.14. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-1-bromoheptan-2-ol (13)
To a solution of epoxide 12 (2.10 g, 5.43 mmol) in dry THF (25 mL) under argon were added successively lithium bromide (1.51 g, 17.4 mmol) and glacial acetic acid (930 µL, 16.2 mmol) dropwise. After stirring at room temperature overnight, saturated aqueous NaHCO3 (30 mL) was added to the reaction mixture. The aqueous layer was extracted with AcOEt and the combined organic extracts were dried over Na2SO4, filtered and concentrated in vacuo. Purification by flash column chromatography on SiO2 with hexanes/AcOEt gradients afforded α-bromohydrin 13 (2.31 g, 91%) as a colorless oil. 1H NMR (360 MHz, CDCl3) δ: 5.26 (bs, 1H), 3.71-3.64 (m, 1H), 3.38 (dd, J = 4.0 Hz, J = 10.1 Hz, 1H), 3.30 (dd, J = 6.5 Hz, J = 10.1 Hz, 1H), 3.13 (t, J = 6.5 Hz, 2H), 3.05-2.96 (m, 5H), 1.58-1.53 (m, 2H), 1.48-1.39 (m, 4H), 1.37-1.31 (m, 2H), 1.36 (s, 9H), 1.34 (s, 9H), 1.22-1.15 (m, 2H). 13C NMR (90.6 MHz, CDCl3) δ: 156.0 (2C), 79.4, 78.9, 70.7, 46.9, 43.9, 39.8, 37.6, 34.9, 28.4 (7C), 27.9, 26.6, 25.2. HRMS (ESI) calcd for C20H39N2O5BrNa [M + Na]+ 489.1940, found 489.1931.
4.1.15. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-1-bromoheptan-2-one (14)
To a solution of α-bromohydrin 13 (2.10 g, 4.49 mmol) in dry CH2Cl2 (50 mL) at room temperature and under argon was added DMP (5.72 g, 13.5 mmol). After 3 hours, the reaction mixture was cooled down to 0°C, quenched with the addition of an aqueous sodium thiosulfate solution (0.5 M) saturated with NaHCO3 (100 mL), and the aqueous layer was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford α-bromoketone 14 as a colorless oil (2.03 g, 97%). 1H NMR (360 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.78 (s, 2H), 3.12 (t, J = 6.5 Hz, 2H), 3.04-2.95 (m, 4H), 2.54 (t, J = 7.2 Hz, 2H), 1.56-1.47 (m, 4H), 1.45-1.36 (m, 2H), 1.33 (s, 9H), 1.31 (s, 9H), 1.21-1.12 (m, 2H). 13C NMR (90.6 MHz, CDCl3) δ: 201.7, 155.9 (2C), 79.3, 78.7, 46.6, 43.8, 39.5, 37.6, 34.2, 28.3, 27.8 (7C), 26.1, 23.4. HRMS (ESI) calcd for C20H37N2O5BrNa [M + Na]+ 487.1784, found 487.1794.
4.1.16. S-{N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-2-oxoheptyl}thioacetate (15)
To a solution of α-bromoketone 14 (1.72 g, 3.70 mmol) in dry MeCN (25 mL) at room temperature under argon was added potassium thioacetate (2.54 g, 22.2 mmol). After stirring overnight, the reaction mixture was diluted with water and the aqueous layer was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by flash column chromatography on SiO2 with hexanes/AcOEt gradients afforded thioester 15 (1.65 g, 97%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.68 (s, 2H), 3.18 (t, J = 6.5 Hz, 2H), 3.08-3.02 (m, 4H), 2.50 (t, J = 7.5 Hz, 2H), 2.32 (s, 3H), 1.62-1.54 (m, 4H), 1.49-1.43 (m, 2H), 1.40 (s, 9H), 1.38 (s, 9H), 1.25-1.18 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 203.8, 194.3, 156.1 (2C), 79.5, 79.0, 46.7, 43.9, 41.5, 39.0, 37.7, 30.2, 28.5 (7C), 28.4, 26.3, 23.4. HRMS (ESI) calcd for C22H40N2O6SNa [M + Na]+ 483.2505, found 483.2517.
4.1.17. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-1-methoxyheptan-2-ol (16)
Epoxide 12 (1.33 g, 3.44 mmol) was dissolved in a 0.5 M solution of sodium methoxide in MeOH (41.2 mL, 20.6 mmol). The reaction mixture was stirred under argon at room temperature until completion of the reaction (24 hours) as shown by TLC. After removal of the solvent in vacuo, the residue was partitioned between AcOEt (100 mL) and water (25 mL), and the aqueous layer was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. Purification by flash column chromatography on SiO2 with hexanes/AcOEt gradients afforded alcohol 16 (1.34 g, 93%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 5.33 (bs, 1H), 3.66-3.60 (m, 1H), 3.27-3.24 (m, 4H), 3.15-3.10 (m, 3H), 3.02-2.96 (m, 4H), 2.72 (bs, 1H), 1.55-1.50 (m, 2H), 1.43-1.35 (m, 4H), 1.33 (s, 9H), 1.31 (s, 9H), 1.29-1.23 (m, 2H), 1.20-1.14 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 156.0 (2C), 79.2, 78.7, 77.1, 69.9, 58.8, 46.9, 43.7, 37.4, 33.1, 28.3 (7C), 28.2, 26.8, 25.2. HRMS (ESI) calcd for C21H43N2O6 [M + H]+ 419.3121, found 419.3124.
4.1.18. N,N’-Bis(tert-butoxycarbonyl)-7-[(3-aminopropyl)amino]-1-methoxyheptan-2-one (17)
To a solution of alcohol 16 (1.19 g, 2.84 mmol) in dry CH2Cl2 (50 mL) at room temperature and under argon was added DMP (7.24 g, 17.1 mmol). After 24 hours, the reaction mixture was cooled down to 0°C, quenched with the addition of an aqueous sodium thiosulfate solution (0.5 M) saturated with NaHCO3 (150 mL), and the aqueous layer was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford α-methoxyketone 17 as a colorless oil (1.08 g, 91%). 1H NMR (500 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.94 (s, 2H), 3.35 (s, 3H), 3.18 (t, J = 6.5 Hz, 2H), 3.08-3.02 (m, 4H), 2.38 (t. J = 7.5 Hz, 2H), 1.61-1.52 (m, 4H), 1.49-1.43 (m, 2H), 1.39 (s, 9H), 1.37 (s, 9H), 1.29-1.23 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 208.5, 156.24 (2C), 79.5, 79.0, 77.6, 59.3, 46.8, 43.9, 38.6, 37.7, 28.4 (7C), 28.3, 26.4, 23.0. HRMS (ESI) calcd for C21H40N2O6Na [M + Na]+ 439.2784, found 439.2772.
4.1.19. N,N’-Bis(tert-butoxycarbonyl)-5-[(3-aminopropyl)amino]pentan-1-ol (18)
To a solution of alkene 3 (3.00 g, 8.76 mmol) in dry THF (150 mL) at 0°C and under argon was added dropwise 9-BBN (0.5 M in THF, 43.8 mL, 21.9 mmol). The solution was allowed to warm up to room temperature and stirred for 20 hours. The reaction mixture was then cooled down to 0°C, and aqueous NaOH (6 M, 30 mL) was added, followed by the dropwise addition of H2O2 (30%, 15 mL). After stirring 30 min at room temperature, the solution was concentrated in vacuo. The aqueous phase was diluted with 50 mL of water, and extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford alcohol 18 as a colorless oil (3.15 g, quantitative). 1H NMR (500 MHz, CDCl3) δ: 5.35 (bs, 1H), 3.54 (t, J = 6.5 Hz, 2H), 3.16 (t, J = 6.5 Hz, 2H), 3.06 (t, J = 6.5 Hz, 2H), 3.01 (t, J = 6.5 Hz, 2H), 2.74 (bs, 1H), 1.61-1.55 (m, 2H), 1.53-1.43 (m, 4H), 1.38 (s, 9H), 1.36 (s, 9H), 1.29-1.23 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ:156.2 (2C), 79.5, 79.0, 62.4, 47.0, 43.9, 37.6, 32.3, 28.4 (7C), 27.8, 23.0. HRMS (ESI) calcd for C18H36N2O5Na [M + Na]+ 383.2522, found 383.2508.
4.1.20. N1,N3-Bis(tert-butoxycarbonyl)-N1-(5-bromopentyl)propane-1,3-diamine (19)
To a solution of alcohol 18 (3.00 g, 8.32 mmol) in dry THF (100 mL) at 0°C were added successively CBr4 (5.52 g, 16.6 mmol) and PPh3 (4.37 g, 16.7 mmol). The reaction mixture was allowed to reach room temperature and stirred overnight. After removal of the solvent in vacuo, purification of the residue by flash column chromatography on SiO2 with hexanes/AcOEt gradients afforded alkyl bromide 19 (3.38 g, 96%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.23 (t, J = 6.5 Hz, 2H), 3.17 (t, J = 6.5 Hz, 2H), 3.08 (t, J = 6.5 Hz, 2H), 3.02 (t, J = 6.5 Hz, 2H), 1.83-1.77 (m, 2H), 1.61-1.56 (m, 2H), 1.51-1.44 (m, 2H), 1.39 (s, 9H), 1.37 (s, 9H), 1.38-1.31 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 156.0 (2C), 79.5, 78.9, 46.7, 43.8, 37.7, 33.6, 32.4, 28.4 (7C), 27.7, 25.4. HRMS (ESI) calcd for C18H36N2O4Br [M + H]+ 423.1858, found 423.1866.
4.1.21. S-{N,N’-Bis(tert-butoxycarbonyl)-5-[(3-aminopropyl)amino]pentyl} thioacetate (20)
Reaction of alkyl bromide 19 (1.70 g, 4.02 mmol) and potassium thioacetate (2.75 g, 24.1 mmol) under the same conditions as for compound 15 afforded after purification thioester 20 (1.58 g, 94%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.07 (t, J = 6.5 Hz, 2H), 2.96 (t, J = 6.5 Hz, 2H), 2.91 (t, J = 6.0 Hz, 2H), 2.68 (t, J = 7.0 Hz, 2H), 2.14 (s, 3H), 1.51-1.47 (m, 2H), 1.44-1.38 (m, 2H), 1.38-1.32 (m, 2H), 1.28 (s, 9H), 1.26 (s, 9H), 1.19-1.13 (m, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 195.3, 155.8 (2C), 79.1, 78.5, 46.5, 43.6, 37.5, 30.3, 29.0, 28.7, 28.2 (7C), 27.8, 25.7. HRMS (ESI) calcd for C20H38N2O5SNa [M + Na]+ 441.2399, found 441.2406.
4.1.22. N,N’-Bis(tert-butoxycarbonyl)-5-[(3-aminopropyl)amino]pentane-1-thiol (21)
To a solution of thioester 20 (550 mg, 1.31 mmol) in dry MeOH (10 mL) under argon was added sodium methoxide (0.5 M solution in MeOH, 5.3 mL, 2.65 mml). The solution was stirred at room temperature for 2 hours, quenched by the addition of 10% aqueous citric acid (100 mL), and the aqueous phase was extracted with AcOEt. Combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford thiol 21 as a colorless oil (485 mg, 98%). 1H NMR (500 MHz, CDCl3) δ: 5.19 (bs, 1H), 3.07 (t, J = 6.0 Hz, 2H), 2.98 (t, J = 6.3 Hz, 2H), 2.92 (t, J = 6.3 Hz, 2H), 2.35 (apparent q (dt), J = 7.3 Hz, 2H), 1.51-1.41 (m, 4H), 1.37-1.32 (m, 2H), 1.29 (s, 9H), 1.26 (s, 9H), 1.23-1.17 (m, 2H), 1.16 (t, J = 7.3 Hz, 1H). 13C NMR (125.6 MHz, CDCl3) δ: 155.8 (2C), 79.1, 78.5, 46.6, 43.7, 37.5, 33.4, 28.2 (7C), 27.7, 25.3, 24.2. HRMS (ESI) calcd for C18H36N2O4SNa [M + Na]+ 399.2293, found 399.2293.
4.1.23. N1,N3-Bis(tert-butoxycarbonyl)-N1-[5-(methylthio)pentyl]propane-1,3-diamine (22)
To a solution of alkyl bromide 19 (1.22 g, 2.88 mmol) in EtOH (15 mL) was added sodium thiomethoxide (1.21 g, 17.3 mmol). The reaction mixture was stirred overnight at 60°C under argon. After removal of the solvent under vacuo, the residue was partitioned between CH2Cl2 (50 mL) and saturated aqueous NaHCO3 (25 mL), and the aqueous layer was extracted with CH2Cl2. Combined organic extracts were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by flash column chromatography on SiO2 with hexanes/AcOEt gradients afforded thioether 22 (1.10 g, 98%) as a colorless oil. 1H NMR (360 MHz, CDCl3) δ: 5.21 (bs, 1H), 3.20 (t, J = 6.5 Hz, 2H), 3.13-3.05 (m, 4H), 2.46 (t, J = 7.4 Hz, 2H), 2.06 (s, 3H), 1.66-1.54 (m, 4H), 1.53-1.46 (m, 2H), 1.43 (s, 9H), 1.41 (s, 9H), 1.38-1.29 (m, 2H). 13C NMR (90.6 MHz, CDCl3) δ: 156.1 (2C), 79.6, 79.1, 46.9, 44.1, 37.8, 34.3, 29.0, 28.5 (7C), 28.2, 26.1, 15.6. HRMS (ESI) calcd for C19H39N2O4S [M + H]+ 391.2631, found 391.2633.
4.1.24. N1,N3-Bis(tert-butoxycarbonyl)-N1-[5-(methylsulfonyl)pentyl]propane-1,3-diamine (23)
To a solution of thioether 22 (500 mg, 1.28 mmol) in dry CH2Cl2 (10 mL) under argon at 0°C was added dropwise m-CPBA (77%, 861 mg, 3.84 mmol) in 10 mL dry CH2Cl2. The solution was allowed to reach room temperature and stirred until completion of the reaction (2 hours) as shown by TLC. Saturated aqueous NaHCO3 (10 mL) was added to the reaction mixture and the aqueous layer was extracted with AcOEt. Combined organic extracts were dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford sulfone 23 (524 mg, 97%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ: 5.14 (bs, 1H), 3.07 (t, J = 7.0 Hz, 2H), 3.01 (t, J = 6.5 Hz, 2H), 2.92 (t, J = 6.5 Hz, 2H), 2.87 (t, J = 7.0 Hz, 2H), 2.75 (s, 3H), 1.73-1.67 (m, 2H), 1.53-1.48 (m, 2H), 1.44-1.40 (m, 2H), 1.31-1.26 (m, 2H), 1.30 (s, 9H), 1.27 (s, 9H). 13C NMR (125.6 MHz, CDCl3) δ: 155.9 (2C), 79.3, 78.6, 54.3, 46.3, 43.6, 40.2, 37.5, 28.2 (7C), 28.0, 25.4, 22.0. HRMS (ESI) calcd for C19H38N2O6SNa [M + Na]+ 445.2348, found 445.2344.
4.1.25. N1,N3-Bis(tert-butoxycarbonyl)-N1-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pentyl]propane-1,3-diamine (24)
To a solution of [Ir(cod)Cl]2 (196 mg, 292 µmol) and dppm (225 mg, 585 µmol) in dry CH2Cl2 (150 mL) under argon was added pinacolborane (1.65 mL, 11.3 mmol) and alkene 3 (2.00 g, 5.84 mmol) in 30 mL dry CH2Cl2. After stirring 24 hours at room temperature, the reaction mixture was cooled to 0°C, quenched with the addition of 60 mL of water, and the aqueous phase extracted with Et2O. Combined organic extracts were dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 with hexanes/AcOEt gradients to afford 24 as a colorless oil (2.25 g, 82%). 1H NMR (500 MHz, CDCl3) δ: 5.24 ppm (bs, 1H), 3.16 (t, J = 6.6 Hz, 2H), 3.04-2.99 (m, 4H), 1.59-1.54 (m, 2H), 1.45-1.39 (m, 2H), 1.37 (s, 9H), 1.35 (s, 9H), 1.35-1.30 (m, 2H), 1.21-1.15 (m, 2H), 1.15 (s, 12H), 0.68 (t, J = 7.8 Hz, 2H). 13C NMR (125.6 MHz, CDCl3) δ: 156.0 (2C), 82.6 (2C), 79.3, 78.8, 47.0, 43.8, 37.7, 29.5, 28.5 (7C), 28.3, 24.8 (4C), 23.8, 11.2 (bs). 11B NMR (128 MHz, CDCl3) δ: 33.0 ppm. HRMS (ESI) calcd for C24H48N2O5B [M + H]+ 471.3605, found 471.3615.
4.1.26. 6-[(3-aminopropyl)amino]hexanoic acid dihydrochloride (I)
A solution of compound 5 (300 mg, 772 µmol) in anhydrous 4 N HCl in dioxane (20 mL) was stirred at room temperature under argon for 2 hours. The reaction mixture was cooled down to 0°C and, diluted with Et2O (20 mL). The precipitate was filtered, washed with cold Et2O, and dried in vacuo to afford carboxylic acid I (dihydrochloride salt) as a white solid (188 mg, 93%). mp: 172-174 °C. 1H NMR (500 MHz, D2O) δ: 3.20 (t, J = 8.0 Hz, 2H), 3.17-3.11 (m, 4H), 2.45 (t, J = 7.4 Hz, 2H), 2.17-2.11 (m, 2H), 1.79-1.72 (m, 2H), 1.71-1.65 (m, 2H), 1.49-1.43 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 178.8, 47.6, 44.4, 36.6, 33.5, 25.2, 25.1, 23.8, 23.7. HRMS (ESI) calcd for C9H21N2O2 [M + H]+ 189.1603, found 189.1604.
4.1.27. 7-[(3-aminopropyl)amino]heptan-2-one dihydrochloride (II)
Deprotection of compound 7 (320 mg, 828 µmol) was performed under the same conditions as for I to afford ketone II (dihydrochloride salt) as a white powder (202 mg, 94%). mp: 206-209 °C (dec.). 1H NMR (500 MHz, D2O) δ: 3.14 (t, J = 8.0 Hz, 2H), 3.10 (t, J = 8.0 Hz, 2H), 3.06 (t, J = 7.8 Hz, 2H), 2.58 (t, J = 7.5 Hz, 2H), 2.19 (s, 3H), 2.11-2.05 (m, 2H), 1.71-1.65 (m, 2H), 1.60-1.54 (m, 2H), 1.38-1.31 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 216.9, 47.5, 44.3, 42.7, 36.5, 29.2, 25.2, 25.0, 23.6, 22.5. HRMS (ESI) calcd for C10H23N2O [M + H]+ 187.1810, found 187.1813.
4.1.28. 6-[(3-aminopropyl)amino]-N-hydroxyhexanamide dihydrochloride (III)
Deprotection of compound 8 (350 mg, 867 µmol) was performed under the same conditions as for I to afford hydroxamic acid III (dihydrochloride salt) as a white powder (230 mg, 96%). mp: 135-139 °C. 1H NMR (500 MHz, D2O) δ: 3.19 (t, J = 8.0 Hz, 2H), 3.16-3.10 (m, 4H), 2.23 (t, J = 7.3 Hz, 2H), 2.16-2.10 (m, 2H), 1.77-1.71 (m, 2H), 1.70-1.64 (m, 2H), 1.45-1.39 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 173.1, 47.6, 44.4, 36.6, 32.0, 25.2, 25.0, 24.3, 23.7. HRMS (ESI) calcd for C9H22N3O2 [M + H]+ 204.1712, found 204.1722.
4.1.29. 7-[(3-aminopropyl)amino]-1,1,1-trifluoroheptan-2-one dihydrochloride (IV)
Deprotection of compound 11 (350 mg, 795 µmol) was performed under the same conditions as for I to afford trifluoromethylketone IV (dihydrochloride salt) as a white powder (234 mg, 94%). mp: 228–231 °C (dec.). 1H NMR (500 MHz, D2O) (ketone/hydrate : 0/1) δ: 3.20 (t, J = 8.0 Hz, 2H), 3.17-3.12 (m, 4H), 2.18-2.11 (m, 2H), 1.88 (t, J = 8.2 Hz, 2H), 1.80-1.74 (m, 2H), 1.62-1.55 (m, 2H), 1.51-1.45 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 123.6 (q, J = 286.4 Hz), 93.6 (q, J = 31.4 Hz), 47.7, 44.4, 36.6, 33.7, 25.7, 25.3, 23.7, 20.7. 19F NMR (338.8 MHz) δ: −85.1. HRMS (ESI) calcd for C10H20N2OF3 [M + H]+ 241.1528, found 241.1534.
4.1.30. 7-{[(3-aminopropyl)amino]-2-oxoheptyl} thioacetate dihydrochloride (V)
Deprotection of compound 15 (300 mg, 651 µmol) was performed under the same conditions as for I to afford thioacetate derivative V (dihydrochloride salt) as a white powder (210 mg, 97%). mp: 211–213 °C (dec.). 1H NMR (500 MHz, D2O) δ: 4.00 (s, 2H), 3.23 (t, J = 8.0 Hz, 2H), 3.19 (t, J = 7.8 Hz, 2H), 3.16 (t, J = 7.8 Hz, 2H), 2.81 (t, J = 7.2 Hz, 2H), 2.49 (s, 3H), 2.21-2.15 (m, 2H), 1.82-1.76 (m, 2H), 1.74-1.68 (m, 2H), 1.50-1.43 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 209.8, 200.1, 47.7, 44.5, 41.1, 39.3, 36.7, 29.5, 25.3, 25.1, 23.8, 22.7. HRMS (ESI) calcd for C12H25N2O2S [M + H]+ 261.1637, found 261.1626.
4.1.31. 7-[(3-aminopropyl)amino]-1-methoxyheptan-2-one dihydrochloride (VI)
A solution of compound 17 (240 mg, 576 µmol) in anhydrous 1 N HCl in AcOEt (30 mL) was stirred at room temperature under argon for 6 hours. The suspension was diluted with hexanes (20 mL). The precipitate was filtered, washed with AcOEt and hexanes, and dried in vacuo to afford α-methoxyketone VI (dihydrochloride salt) as an off-white solid (151 mg, 91 %). mp: 216–218 °C (dec.). 1H NMR (500 MHz, D2O) δ: 4.35 (s, 2H), 3.44 (s, 3H), 3.19 (t, J = 8.0 Hz, 2H), 3.15 (t, J = 7.9 Hz, 2H), 3.11 (t, J = 7.8 Hz, 2H), 2.55 (t, J = 7.3 Hz, 2H), 2.16-2.10 (m, 2H), 1.77-1.71 (m, 2H), 1.68-1.62 (m, 2H), 1.45-1.39 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 212.8, 76.5, 58.7, 47.6, 44.4, 37.8, 36.6, 25.3, 25.2, 23.7, 22.3. HRMS (ESI) calcd for C11H25N2O2 [M + H]+ 217.1916, found 217.1925.
4.1.32. S-{5-[(3-aminopropyl)amino]pentyl} thioacetate dihydrochloride (VII)
Deprotection of compound 20 (300 mg, 717 µmol) was performed under the same conditions as for I to afford thioacetate derivative VII (dihydrochloride salt) as a white powder (196 mg, 94%). mp: 260–262 °C (dec.). 1H NMR (500 MHz, D2O) δ: 3.19 (t, J = 8.0 Hz, 2H), 3.15 (t, J = 7.9 Hz, 2H), 3.11 (t, J = 7.8 Hz, 2H), 2.94 (t, J = 7.2 Hz, 2H), 2.41 (s, 3H), 2.16-2.10 (m, 2H), 1.78-1.72 (m, 2H), 1.69-1.64 (m, 2H), 1.52-1.46 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 202.2, 47.6, 44.4, 36.6, 30.1, 28.4, 28.1, 25.0, 24.7, 23.7. HRMS (ESI) calcd for C10H23N2OS [M + H]+ 219.1531, found 219.1532.
4.1.33. 5-[(3-aminopropyl)amino]pentane-1-thiol dihydrochloride (VIII)
A solution of compound 21 (208 mg, 552 µmol) in anhydrous 1 N HCl in AcOEt (40 mL) was stirred at room temperature under argon for 5 hours. The reaction mixture was concentrated in vacuo and resuspended in hexanes (20 mL). The precipitate was filtered, washed with hexanes, and dried in vacuo to afford thiol derivative VIII (dihydrochloride salt) as a white solid (129 mg, 93%). mp: 284–286 °C (dec.). 1H NMR (500 MHz, D2O) δ: 3.24 (t, J = 8.0 Hz, 2H), 3.21-3.15 (m, 4H), 2.65 (t, J = 7.1 Hz, 2H), 2.21-2.15 (m, 2H), 1.82-1.76 (m, 2H), 1.76-1.70 (m, 2H), 1.59-1.52 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 47.8, 44.5, 36.7, 32.4; 25.0, 24.5, 23.8, 23.5. HRMS (ESI) calcd for C8H21N2S [M + H]+ 177.1425, found 177.1424.
4.1.34. N1-[5-(methylsulfonyl)pentyl]propane-1,3-diamine dihydrochloride (IX)
Deprotection of compound 23 (300 mg, 710 µmol) was performed under the same conditions as for I to afford sulfone IX (dihydrochloride salt) as a white powder (202 mg, 97%). mp: 202–204 °C. 1H NMR (500 MHz, D2O) δ: 3.28 (t, J = 7.8 Hz, 2H), 3.15 (t, J = 7.8 Hz, 2H), 3.11-3.07 (m, 7H), 2.12-2.05 (m, 2H), 1.89-1.82 (m, 2H), 1.78-1.71 (m, 2H), 1.58-1.51 (m, 2H). 13C NMR (125.6 MHz, D2O) δ: 53.1, 47.3, 44.3, 39.5, 36.4, 24.9, 24.3, 23.6, 20.9. HRMS (ESI) calcd for C9H23N2O2S [M + H]+ 223.1480, found 223.1483.
4.1.35. 5-[(3-aminopropyl)amino]pentylboronic acid dihydrochloride (X)
The protected compound 24 (300 mg, 638 µmol) was stirred to reflux with 6N aqueous HCl (10 mL) for 24 hours. After cooling to room temperature, the reaction mixture was washed with Et2O (20 mL) and the aqueous phase was concentrated in vacuo to give boronic acid X as an off-white solid (148 mg, 89%). mp: 288–290 °C (dec.). 1H NMR (500 MHz, D2O) δ: 3.18 (t, J = 8.0 Hz, 2H), 3.15 (t, J = 7.9 Hz, 2H), 3.10 (t, J = 7.8 Hz, 2H), 2.16-2.10 (m, 2H), 1.76-1.70 (m, 2H), 1.50-144 (m, 2H), 1.43-1.37 (m, 2H), 0.83 (t, J = 7.6 Hz, 2H). 13C NMR (125.6 MHz, D2O) δ: 47.8, 44.4, 36.6, 28.3, 25.3, 23.7, 23.1, 13.8 (bs). 11B NMR (128 MHz, D2O) δ: 32.4. HRMS (ESI) calcd for C18H36N2O2B [M + (+)-pinanediol - 2H2O + H]+ 323.2870, found 323.2869.
4.2. APAH expression, purification, and activity assay
APAH was expressed in Escherichia coli BL21(DE3) cells (Novagen Inc.) and purified as previously described.18 The inhibition of APAH by the synthesized diamine derivatives was analyzed using a fluorimetric assay, as previously described.18 Activity was measured using the commercially available Fluor-de-Lys deacetylase fluorogenic substrate (BML-KI104, Enzo Life Sciences). Briefly, deacetylation of the substrate molecule allows a protease developer to cleave the peptide bond linking the C-terminal fluorophore, resulting in a fluorescence shift. Assays were run at 25 °C and contained 250 nM enzyme, 150 µM substrate, and 0–100 mM inhibitor in assay buffer (25 mM Tris (pH 8.2), 137 mM NaCl, 2.7 mM KCl and 1 mM MgCl2 (100 µM tris-(2-carboxyethyl)phosphine was added only for the assay of thiol compound VIII) in a final volume of 50 µL. After 30 min, reactions were quenched by adding 100 µM M344 (Sigma Aldrich) and the appropriate Fluor-de-Lys developer (BML-KI105, Enzo Life Sciences, 50 µL). Fluorescence was measured after 45 min using a Fluoroskan II plate reader (excitation = 355 nm, emission = 460 nm). Assays for each concentration of inhibitor were performed in triplicate in separate experiments. IC50 values for each compound were determined using the software Graphpad Prism (2008).
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Health Grant GM49758. The Roy and Diana Vagelos Scholars Program in Molecular Life Sciences supports the research of undergraduate student Christine M. Bowman. Finally, we thank Dr. Rakesh Kohli (University of Pennsylvania, Department of Chemistry) for recording the high resolution mass spectra.
Abbreviations
- APAH
acetylpolyamine amidohydrolase
- dppm
bis(diphenylphosphino)methane
- 9-BBN
9-borabicyclo[3.3.1]nonane
- DMP
Dess-Martin periodinane
- DFMO
α-difluoromethylornithine
- Boc2O
di-tert-butyl pyrocarbonate
- ESI
electrospray ionization
- HRMS
high-resolution mass spectrometry
- HDAC
histone deacetylase
- m-CPBA
m-chloroperbenzoic acid
- NMO
N-methylmorpholine N-oxide
- CDI
N,N’-carbonyldiimidazole
- ODC
ornithine decarboxylase
- RT
room temperature
- Boc
tert-butoxycarbonyl
- TBAF
tetra-n-butylammonium fluoride
- TMSCF3
trifluoromethyltrimethylsilane
- PPh3
triphenylphosphine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
NMR spectra for compounds I–X are provided in the Supplementary Information.
References
- 1.Pegg AE, McCann PP. Am. J. Physiol. 1982;243:C212. doi: 10.1152/ajpcell.1982.243.5.C212. [DOI] [PubMed] [Google Scholar]
- 2.Thomas T, Thomas TJ. Cell. Mol. Life Sci. 2001;58:244. doi: 10.1007/PL00000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace HM, Fraser AV. Amino Acids. 2004;26:353. doi: 10.1007/s00726-004-0092-6. [DOI] [PubMed] [Google Scholar]
- 4.Heby O, Persson L, Rentala M. Amino Acids. 2007;33:359. doi: 10.1007/s00726-007-0537-9. [DOI] [PubMed] [Google Scholar]
- 5.Gerner EW, Meyskens FL., Jr Nat. Rev. Cancer. 2004;4:781. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 6.Casero RA, Jr, Marton LJ. Nat. Rev. Drug Discovery. 2007;6:373. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 7.Bey P, Danzin C, Van Dorsselaer V, Mamont P, Jung M, Tardif C. J. Med. Chem. 1978;21:50. doi: 10.1021/jm00199a009. [DOI] [PubMed] [Google Scholar]
- 8.Mamont PS, Duchesne MC, Grove J, Bey P. Biochem. Biophys. Res. Commun. 1978;81:58. doi: 10.1016/0006-291x(78)91630-3. [DOI] [PubMed] [Google Scholar]
- 9.Bacchi CJ, Nathan HC, Hutner SH, McCann PP, Sjoerdsma A. Science. 1980;210:332. doi: 10.1126/science.6775372. [DOI] [PubMed] [Google Scholar]
- 10.Blankenship JW, Morgan JE, Matthews HR. Mol. Biol. Rep. 1987;12:21. doi: 10.1007/BF00580646. [DOI] [PubMed] [Google Scholar]
- 11.Morgan JE, Blankenship JW, Matthews HR. Biochemistry. 1987;26:3643. doi: 10.1021/bi00386a058. [DOI] [PubMed] [Google Scholar]
- 12.Della Ragione F, Pegg AE. Biochemistry. 1982;21:6152. doi: 10.1021/bi00267a020. [DOI] [PubMed] [Google Scholar]
- 13.Erwin BG, Persson L, Pegg AE. Biochemistry. 1984;23:4250. doi: 10.1021/bi00313a036. [DOI] [PubMed] [Google Scholar]
- 14.Bolkenius FN, Seiler N. Int. J. Biochem. 1981;13:287. doi: 10.1016/0020-711x(81)90080-x. [DOI] [PubMed] [Google Scholar]
- 15.Marchant P, Manneh VA, Blankenship J. Biochim. Biophys. Acta. 1986;881:297. doi: 10.1016/0304-4165(86)90017-6. [DOI] [PubMed] [Google Scholar]
- 16.Fujishiro K, Ando M, Uwajima T. Biochem. Biophys. Res. Commun. 1988;157:1169. doi: 10.1016/s0006-291x(88)80997-5. [DOI] [PubMed] [Google Scholar]
- 17.Sakurada K, Ohta T, Fujishiro K, Hasegawa M, Aisaka K. J. Bacteriol. 1996;178:5781. doi: 10.1128/jb.178.19.5781-5786.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lombardi PM, Angell HD, Whittington DA, Flynn EF, Rajashankar KR, Christianson DW. Biochemistry. 2011;50:1808. doi: 10.1021/bi101859k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abendroth J, Gardberg AS, Robinson JI, Christensen JS, Staker BL, Myler PJ, Stewart LJ, Edwards TE. J. Funct. Struct. Genomics. 2011;12:83. doi: 10.1007/s10969-011-9101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dowling DP, Di Constanzo L, Gennadios HA, Christianson DW. Cell Mol. Life Sci. 2008;65:2039. doi: 10.1007/s00018-008-7554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Nature. 1996;383:554. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 22.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structure. 2004;12:1325. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Schuetz A, Min J, Allali-Hassani A, Schapira M, Schuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. J. Biol. Chem. 2008;283:11355. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkühler C, Gallinari P, Carfi A. J. Biol. Chem. 2008;283:26694. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Curr. Opin. Struct. Biol. 2011;21:735. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dredar SA, Blankenship JW, Marchant PE, Manneh V, Fries DS. J. Med. Chem. 1989;32:984. doi: 10.1021/jm00125a010. [DOI] [PubMed] [Google Scholar]
- 27.Huang TL, Dredar SA, Manneh VA, Blankenship JW, Fries DS. J. Med. Chem. 1992;35:2414. doi: 10.1021/jm00091a009. [DOI] [PubMed] [Google Scholar]
- 28.Jung M, Brosch G, Kölle D, Scherf H, Gerhaüser C, Loidl P. J. Med. Chem. 1999;42:4669. doi: 10.1021/jm991091h. [DOI] [PubMed] [Google Scholar]
- 29.Ilies M, Dowling DP, Lombardi PM, Christianson DW. Bioorg. Med. Chem. Lett. 2011;21:5854. doi: 10.1016/j.bmcl.2011.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 31.Gantt SL, Gattis SG, Fierke CA. Biochemistry. 2006;45:6170. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 32.Gantt SL, Joseph CG, Fierke CA. J. Biol. Chem. 2010;285:6036. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki T, Kouketsu A, Matsuura A, Kohara A, Ninomiya S, Kohda K, Miyata N. Bioorg. Med. Chem. Lett. 2004;14:3313. doi: 10.1016/j.bmcl.2004.03.063. [DOI] [PubMed] [Google Scholar]
- 34.Jose B, Oniki Y, Kato T, Nishino N, Sumida Y, Yoshida M. Bioorg. Med. Chem. Lett. 2004;14:5343. doi: 10.1016/j.bmcl.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, Nakagawa H, Miyata N. J. Med. Chem. 2005;48:1019. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- 36.Bhuiyan MPI, Kato T, Okauchi T, Nishino N, Maeda S, Nishino TG, Yoshida M. Bioorg. Med. Chem. 2006;14:3438. doi: 10.1016/j.bmc.2005.12.063. [DOI] [PubMed] [Google Scholar]
- 37.Gu W, Nusinzon I, Smith RD, Jr, Horwath CM, Silverman RB. Bioorg. Med. Chem. 2006;14:3320. doi: 10.1016/j.bmc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki N, Suzuki T, Ota Y, Nakano T, Kurihara M, Okuda H, Yamori T, Tsumoto H, Nakagawa H, Miyata N. J. Med. Chem. 2009;52:2909. doi: 10.1021/jm900125m. [DOI] [PubMed] [Google Scholar]
- 39.Islam MS, Bhuiyan MPI, Islam MN, Nsiama TK, Oishi N, Kato T, Nishino N, Ito A, Yoshida M. Amino Acids. 2012;42:2103. doi: 10.1007/s00726-011-0947-6. [DOI] [PubMed] [Google Scholar]
- 40.Gelb MH, Svaren JP, Abeles RH. Biochemistry. 1985;24:1813. doi: 10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- 41.Huntington KM, Yi T, Wei Y, Pei D. Biochemistry. 2000;39:4543. doi: 10.1021/bi992452y. [DOI] [PubMed] [Google Scholar]
- 42.Boularot A, Giglione C, Petit S, Duroc Y, Alves de Sousa R, Larue V, Cresteil T, Dardel F, Artaud I, Meinnel T. J. Med. Chem. 2007;50:10. doi: 10.1021/jm060910c. [DOI] [PubMed] [Google Scholar]
- 43.Baggio R, Elbaum D, Kanyo ZF, Carroll PJ, Cavalli RC, Ash DE, Christianson DW. J. Am. Chem. Soc. 1997;119:8107. [Google Scholar]
- 44.Rossiter BE, Katsuki T, Sharpless KB. J. Am. Chem. Soc. 1981;103:464. [Google Scholar]
- 45.Carlsen PHJ, Katsuki T, Martin VS, Sharpless BK. J. Org. Chem. 1981;46:3936. [Google Scholar]
- 46.Zimmermann F, Meux E, Mieloszynski JL, Lecuire JM, Oget N. Tetrahedron Lett. 2005;46:3201. [Google Scholar]
- 47.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815. [Google Scholar]
- 48.Usachova N, Leitis G, Jirgensons A, Kalvinsh I. Synth. Commun. 2010;40:927. [Google Scholar]
- 49.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973. [Google Scholar]
- 50.Ruppert I, Schlich K, Volbach W. Tetrahedron Lett. 1984;25:2195. [Google Scholar]
- 51.Prakash GKS, Krishnamurti R, Olah GA. J. Am. Chem. Soc. 1989;111:393. [Google Scholar]
- 52.Singh RP, Shreeve JM. Tetrahedron. 2000;56:7613. [Google Scholar]
- 53.Dess DB, Martin JC. J. Org. Chem. 1983;48:4156. [Google Scholar]
- 54.Linderman RJ, Graves DM. J. Org. Chem. 1989;54:661. [Google Scholar]
- 55.Bajwa JS, Anderson RC. Tetrahedron Lett. 1991;32:3021. [Google Scholar]
- 56.Brown HC, Knights EF, Scouten CG. J. Am. Chem. Soc. 1974;96:7765. [Google Scholar]
- 57.Yamamoto Y, Fujikawa R, Umemoto T, Miyaura N. Tetrahedron. 2004;60:10695. [Google Scholar]
- 58.Suzuki T, Matsuura A, Kouketsu A, Nakagawa H, Miyata N. Bioorg. Med. Chem. Lett. 2005;15:331. doi: 10.1016/j.bmcl.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 59.Marchant P, Dredar S, Manneh V, Alshabanah O, Matthews H, Fries D, Blankenship J. Arch. Biochem. Biophys. 1989;273:12. doi: 10.1016/0003-9861(89)90170-7. [DOI] [PubMed] [Google Scholar]
- 60.Marks PA, Breslow R. Nat. Biotechnol. 2007;25:84. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 61.Christianson DW, Lipscomb WN. J. Am. Chem. Soc. 1986;108:4998. doi: 10.1021/ja00263a052. [DOI] [PubMed] [Google Scholar]
- 62.Christianson DW, Lipscomb WN. Proc. Natl. Acad. Sci. USA. 1985;82:6840. doi: 10.1073/pnas.82.20.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balwierczak JL, Kukkola PJ, Savage P, Jeng AY. Biochem. Pharmacol. 1995;49:291. doi: 10.1016/0006-2952(94)00508-j. [DOI] [PubMed] [Google Scholar]
- 64.Gurguis GNM, Antai-Otong D, Vo SP, Blakeley JE, Orsulak PJ, Petty F, Rush AJ. Neuropsychopharmacology. 1999;20:162. doi: 10.1016/S0893-133X(98)00062-1. [DOI] [PubMed] [Google Scholar]
- 65.Van Veen HW, Margolles A, Müller M, Higgins CF, Konings WN. EMBO J. 2000;19:2503. doi: 10.1093/emboj/19.11.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.