

Abstract
Gene-gene interactions may play an important role in the genetics of a complex disease. Detection and characterization of gene-gene interactions is a challenging issue that has stimulated the development of various statistical methods to address it. In this study, we introduce a method to measure gene interactions using entropy-based statistics from a contingency table of trait and genotype combinations. We also developed an exploration procedure by using graphs. We propose a standardized relative information gain (RIG) measure to evaluate the interactions between single nucleotide polymorphism (SNP) combinations. To identify the k th order interactions, contingency tables of trait and genotype combinations of k SNPs are constructed, with which RIGs are calculated. The RIGs are standardized using the mean and standard deviation from the permuted datasets. SNP combinations yielding high standardized RIG are chosen for gene-gene interactions. Detection of high-order interactions and comparison of interaction strengths between different orders are made possible by using standardized RIG. We have applied the proposed standardized entropy-based method to two types of data sets from a simulation study and a real genetic association study. We have compared our method and the multifactor dimensionality reduction (MDR) method through power analysis of eight different genetic models with varying penetrance rates, number of SNPs, and sample sizes. Our method shows successful identification of genetic associations and gene-gene interactions both in simulation and real genetic data. Simulation results suggest that the proposed entropy-based method is better able to detect high-order interactions and is superior to the MDR method in most cases. The proposed method is well suited for detecting interactions without main effects as well as for models including main effects.
Introduction
One of the major goals of human genetics is to identify the relationships between genotypes and disease status. Although single-locus approaches have successfully identified many genetic determinants of disease susceptibility, such approaches cannot adequately explain the genetic contribution to complex diseases such as hypertension, diabetes, and certain psychiatric disorders. This phenomenon may be a result of interactions between genetic factors and influences from environmental factors.
Various statistical methods have been proposed for the detection and characterization of gene-gene interactions in case-control studies [1]–[4]. Logistic regression is a traditional parametric approach to the modeling of relationships between genotypes and binary phenotypes. However, for high-order interactions, logistic regressions may produce large standard errors resulting in increased type I errors due to sparse and empty cells [5]. It is also known to have reduced power to detect high-order interactions [6]. Multifactor dimensionality reduction (MDR) is a popular non-parametric approach that characterizes the SNP combinations into “high risk” or “low risk” categories according to the ratio of the numbers of cases and controls. It converts a high-dimensional contingency table to a one-dimensional model without raising the issue of sparse cells [7]. However, this approach could be considered to be overly simplistic and is sensitive to small changes in cell frequencies. Several variants of MDR have been recently developed [8]–[10].
In this article, we focus on the entropy-based approach as an alternative method. Entropy is commonly used in information theory to measure the uncertainty of random variables [11]. There are several approaches that have adopted entropy-based measures to identify the relationships between genes and disease. Bush et al. [12] used the normalized mutual information (NMI) method as a measure to evaluate MDR model fitness. Kang et al. [13] proposed an entropy-based procedure to detect genetic associations for the case-only design method. Dong et al. [14] defined the gain ratio to combine a genetic model with two-locus gene-gene interactions. More recently, Chanda et al. [15] proposed an information-theoretic gene-gene and gene-environment interaction analysis of quantitative traits. In this study, we developed a more comprehensive and flexible framework for detecting and interpreting gene-gene interactions. Here we define the standardized relative information gain (RIG) by subtracting the mean values and dividing by the standard deviation of the relative information gain from permuted datasets and apply it to contingency tables of genotype combinations and disease status. It could account for the improper inflation of relative information gain commonly observed with higher order of interactions.
After a brief review of entropy in section 2.1, we have described a new entropy-based procedure for modeling genetic interactions in section 2.2. We have also illustrated the proposed method using two different genotype datasets in sections 3.1 and 3.2. In section 3.3, we have described the simulation study conducted to compare the powers of the proposed method and MDR. Discussions and final conclusions are included in section 4.
Methods
Definition of Entropy
The term entropy usually refers to the Shannon entropy, which plays a central role in information theory as a measure of information, choice, and uncertainty contained in a system consisting of a random variable [11]. It quantifies the amount of average information necessary to remove any uncertainty from the system.
If X and Y are discrete random variables, then the following four entropy values can be computed: 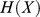 ,
, 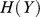 ,
, 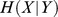 and
and 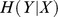 . The Shannon’s entropy of Y is defined by the following equation:
. The Shannon’s entropy of Y is defined by the following equation:
The conditional entropy 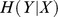 is defined as the average specific conditional entropy of Y:
is defined as the average specific conditional entropy of Y:
where 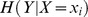 is the entropy of Y when
is the entropy of Y when 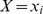 .
. 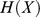 and
and 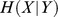 are defined similarly. The information gain (IG) and relative information gain (RIG) are given by
are defined similarly. The information gain (IG) and relative information gain (RIG) are given by
and
respectively [16]. The RIG value is often called the normalized mutual information (NMI). It quantifies the proportion of information contained in the X variable that is transferred to the Y variable. It is also interpreted as the amount by which the model reduces the uncertainty about the true state of affairs [12].
Entropy-based Procedure for Modeling Gene-gene Interactions
The procedure can be summarized in four stages as follows: (i) construction of a contingency table, (ii) calculation of initial relative information gain, (iii) standardization of relative information gain, and (iv) visualization.
[Step 1] Construction of a 2-way table
At the first stage, we constructed a 2-way contingency table of the genotypes and disease status. For two-locus interactions in the case-control study, we constructed a 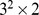 contingency table because there are
contingency table because there are 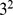 possible genotype combinations and dichotomous disease status. A
possible genotype combinations and dichotomous disease status. A 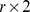 table was constructed for the k
th order interactions in the case-control study, where
table was constructed for the k
th order interactions in the case-control study, where 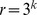 (Table 1).
(Table 1).
Table 1. Contingency table for kth order interaction.
| SNP | Disease status | total | |||||
| Combination | SNP1 | SNP2 | … | SNPk | case | control | |
| 1 | AA | BB | KK |
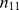
|
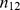
|
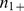
|
|
| 2 | Aa | BB | KK |
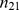
|
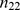
|
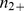
|
|
| 3 | aa | BB | KK |
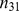
|
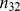
|
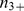
|
|
| : | : | : | : | : | : | : | : |
| : | : | : | : | : | : | : | : |
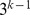
|
aa | bb | Kk | : | : | : | |
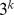
|
aa | bb | kk |
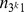
|
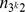
|
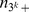
|
|
| Total |
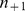
|
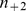
|
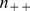
|
||||
nij means the number of samples with i th joint genotype for each SNP combination and j th disease status.
[Step 2] Calculation of Initial RIG: 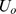
We calculated the initial relative information gain 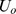 from the constructed contingency table. Let Y be the dichotomous disease status and X be the SNP combinations; then.
from the constructed contingency table. Let Y be the dichotomous disease status and X be the SNP combinations; then.
 |
and
where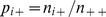 ,
, 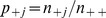 and
and 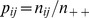 .
.
Note that 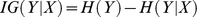 is equivalent to the log-likelihood ratio statistic divided by
is equivalent to the log-likelihood ratio statistic divided by 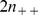 for the independence test of X and Y. Therefore,
for the independence test of X and Y. Therefore, 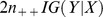 asymptotically follows a chi-square distribution with
asymptotically follows a chi-square distribution with 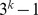 degree of freedom under the null hypothesis of independence. This approximation, however, is not expected to hold for sparse tables in higher order interaction analysis.
degree of freedom under the null hypothesis of independence. This approximation, however, is not expected to hold for sparse tables in higher order interaction analysis.
We calculated 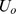 for all the possible combinations of SNPs. A larger
for all the possible combinations of SNPs. A larger 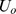 value indicates stronger association between a specific SNP combination and the disease status. It should be noted that empty cells do not cause any ambiguity in the definition. This feature proves advantageous when estimating high-order interactions, because empty cells are common in high-order SNP combinations.
value indicates stronger association between a specific SNP combination and the disease status. It should be noted that empty cells do not cause any ambiguity in the definition. This feature proves advantageous when estimating high-order interactions, because empty cells are common in high-order SNP combinations.
[Step 3] Standardization of RIG: Ur
An ensemble of datasets was generated from original data by repeated shuffling of the phenotypes while all genotypes remained fixed. Relative information gains (RIGs) were calculated for each permuted data set by following the same procedures given in Steps 1 and 2. To standardize the RIG, the empirical null distribution of the maximum value of RIG was obtained for each order of interaction. Let 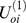 denote the maximum RIG of the i
th permuted data set. Then, the average and the standard deviation of
denote the maximum RIG of the i
th permuted data set. Then, the average and the standard deviation of 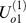 ,
,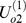 ,…,
,…, 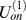 can be computed as follows:
can be computed as follows:
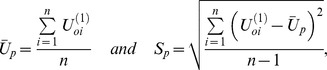 |
where n is the number of the permuted data sets in the ensemble. Standardized relative information gain, 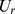 , corresponding to initial relative information gain of the original data,
, corresponding to initial relative information gain of the original data, 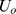 , is defined as follows:
, is defined as follows:
Note that 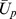 and
and 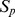 need to be computed for each order of interaction. The empirical null distribution of the maximum value of RIG was used for controlling the family-wise error rate of the multiple comparisons [17], [18]. The adjusted p-values could be obtained by counting the number of
need to be computed for each order of interaction. The empirical null distribution of the maximum value of RIG was used for controlling the family-wise error rate of the multiple comparisons [17], [18]. The adjusted p-values could be obtained by counting the number of 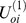 greater than
greater than 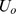 .
.
There is an additional advantage in using 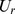 over
over 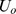 . The values of
. The values of 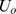 can be shown to increase with the order of interactions, when SNPs in a lower order interaction are a subset of SNPs in a higher order interaction. That is, the values of
can be shown to increase with the order of interactions, when SNPs in a lower order interaction are a subset of SNPs in a higher order interaction. That is, the values of 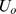 tend to increase regardless of the true additional contribution, as the number of SNPs increases. However, by using
tend to increase regardless of the true additional contribution, as the number of SNPs increases. However, by using 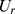 , a direct comparison of the association strengths between models with different orders was made possible. Therefore, for the main criteria of the association strength,
, a direct comparison of the association strengths between models with different orders was made possible. Therefore, for the main criteria of the association strength, 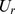 is a more appropriate candidate than
is a more appropriate candidate than 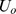 is.
is.
Figure 1 shows the properties of the proposed measures. As the order of interaction increased, the largest values of 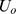 increased, which results from the way in which the mutual information is defined. Note that the empirical distributions for null hypothesis also shifted to the right and became wider as the order increased. Therefore, although the top ranked
increased, which results from the way in which the mutual information is defined. Note that the empirical distributions for null hypothesis also shifted to the right and became wider as the order increased. Therefore, although the top ranked 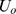 for 3rd order interactions was smaller than that for 4th order interactions, direct comparison of association strength by
for 3rd order interactions was smaller than that for 4th order interactions, direct comparison of association strength by 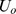 may be biased. In the next section, it will be shown that more reasonable comparison of the association strengths can be made by using the standardized measure
may be biased. In the next section, it will be shown that more reasonable comparison of the association strengths can be made by using the standardized measure  . Figure 1 is based on MDR open source data, where the association strengths have unusually large values.
. Figure 1 is based on MDR open source data, where the association strengths have unusually large values.
Figure 1. Visualization of the properties of the proposed measures using MDR open-source data.

The arrows on the upper side of the graph represent the largest observed 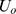 in each order of interactions. The distributions are the null distribution of
in each order of interactions. The distributions are the null distribution of 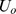 obtained by collecting the maximum
obtained by collecting the maximum 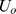 s from each permuted data. Order of interaction is denoted within the parentheses.
s from each permuted data. Order of interaction is denoted within the parentheses.
[Step 4] Visualization
To identify the SNP combinations with strong interactions at a glance, we use scree plot of 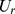 . After estimating
. After estimating 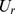 for all the possible k
th order SNP combinations, we ranked those values. The scree plot is drawn by plotting
for all the possible k
th order SNP combinations, we ranked those values. The scree plot is drawn by plotting 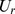 s against their rank; in that way the strength of the interactions may be visualized more clearly. The scree plot is known as an exploratory method to determine the optimal number of factors, and needs neither threshold nor fixed percentage. A typical “above the elbow” approach [19] could be adopted to choose the last substantial drop. The number of points before the last drop was taken as the number of SNP combinations with strong interactions. The line of cut-off value of
s against their rank; in that way the strength of the interactions may be visualized more clearly. The scree plot is known as an exploratory method to determine the optimal number of factors, and needs neither threshold nor fixed percentage. A typical “above the elbow” approach [19] could be adopted to choose the last substantial drop. The number of points before the last drop was taken as the number of SNP combinations with strong interactions. The line of cut-off value of 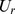 for the given significance level can be added to the plot to identify the significant SNP combinations. For example, the cut-off value of the 5% significance level can be calculated from the upper 5% point of the empirical null distribution of the maximum value of RIG.
for the given significance level can be added to the plot to identify the significant SNP combinations. For example, the cut-off value of the 5% significance level can be calculated from the upper 5% point of the empirical null distribution of the maximum value of RIG.
We have added one more step for the visualization of two-locus interactions. For p SNPs, a 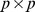 distance matrix was constructed, whose
distance matrix was constructed, whose 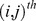 element is
element is 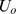 for two-locus interaction between i
th and j
th SNPs. Multi-dimensional scaling was applied to this matrix. Then, the distance between two SNPs in the graph approximated the strength of two-locus interactions measured by relative information gain. Multi-dimensional scaling (MDS) analysis displays the distance between two SNPs in the graph, which approximates the strength of two-locus interactions measured by the relative information gain. By keeping the point size proportional to the relative information gain from one locus model, 1st and 2nd order interactions could be presented simultaneously. MDS plots can be also constructed by
for two-locus interaction between i
th and j
th SNPs. Multi-dimensional scaling was applied to this matrix. Then, the distance between two SNPs in the graph approximated the strength of two-locus interactions measured by relative information gain. Multi-dimensional scaling (MDS) analysis displays the distance between two SNPs in the graph, which approximates the strength of two-locus interactions measured by the relative information gain. By keeping the point size proportional to the relative information gain from one locus model, 1st and 2nd order interactions could be presented simultaneously. MDS plots can be also constructed by 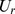 after adding a positive constant such that
after adding a positive constant such that 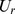 would be non-negative. The resulting MDS plot is equivalent to that produced by
would be non-negative. The resulting MDS plot is equivalent to that produced by 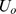 .
.
Results
In order to demonstrate the proposed entropy-based method, we applied it to two data sets. One is from the MDR open source site (http://www.multifactordimensionalityreduction.org/), and the other is from genetic association study of atopic dermatitis [20]. We generated an ensemble of 1000 permuted data sets with replacement.
Analysis of Data: Open Source MDR Data
The open-source MDR data consisted of 20 SNPs and 400 samples. By the MDR method, SNP combinations (1), (1, 8), (1, 6, 8), and (1, 2, 6, 8) were selected for the first to fourth order interactions, respectively. Figure 2 illustrates the initial relative information gain 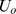 . The values of
. The values of 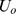 tended to increase with the interaction dimensions. For the 1st order interaction in Figure 1(a), SNP (1) shows the strongest association and followed by SNP (6). For the 2nd order interactions, there are three pairs that seem to be quite different from the others (Figure 2(b)). In the 3rd order interactions, a single SNP combination (1, 6, 8) shows eminent association strength (Figure 2(c)). For the 4th order interactions, all the upper group of SNP combinations included (1, 6, 8) as a subset. These combinations appeared to contain a carryover amount of association strength from that particular 3rd order interaction (Figure 2(d)). The adjusted p-value of
tended to increase with the interaction dimensions. For the 1st order interaction in Figure 1(a), SNP (1) shows the strongest association and followed by SNP (6). For the 2nd order interactions, there are three pairs that seem to be quite different from the others (Figure 2(b)). In the 3rd order interactions, a single SNP combination (1, 6, 8) shows eminent association strength (Figure 2(c)). For the 4th order interactions, all the upper group of SNP combinations included (1, 6, 8) as a subset. These combinations appeared to contain a carryover amount of association strength from that particular 3rd order interaction (Figure 2(d)). The adjusted p-value of 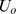 of the best combinations for the first order interaction is 0.292, while the corresponding adjusted p-values for the 2nd to 4th order interactions are less than 0.001.
of the best combinations for the first order interaction is 0.292, while the corresponding adjusted p-values for the 2nd to 4th order interactions are less than 0.001.
Figure 2. Scree plots of 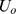 for MDR open-source data.
for MDR open-source data.

Main effects (A), 2nd order interactions (B), 3rd order interactions (C) and 4th order interactions (D) are shown. The observed relative information gain, 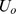 , is plotted against the rank determined by the magnitude of
, is plotted against the rank determined by the magnitude of 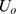 . Only the top 100 ranked
. Only the top 100 ranked 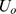 s are plotted for each order of interaction. Top ranked SNP names are denoted within the parentheses.
s are plotted for each order of interaction. Top ranked SNP names are denoted within the parentheses.
Figure 3 is the scree plot with the standardized measure, 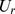 . The group of combinations including (1, 6, 8) in the 4th order interactions found to have lower values than (1,6,8) itself after the adjustment, while the (1, 6, 8) in 3rd order interaction maintained its prominence throughout the orders of interactions examined. It was understood that obtaining 4th order interactions by adding any single SNP into the combination of SNPs 1, 6, and 8 actually lowered the association strength from (1, 6, 8). We conclude that a 3-locus interaction involving SNPs identified as 1, 6, and 8 is the most appropriate model. The upper 5% cut-off values for
. The group of combinations including (1, 6, 8) in the 4th order interactions found to have lower values than (1,6,8) itself after the adjustment, while the (1, 6, 8) in 3rd order interaction maintained its prominence throughout the orders of interactions examined. It was understood that obtaining 4th order interactions by adding any single SNP into the combination of SNPs 1, 6, and 8 actually lowered the association strength from (1, 6, 8). We conclude that a 3-locus interaction involving SNPs identified as 1, 6, and 8 is the most appropriate model. The upper 5% cut-off values for 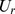 were 1.877, 1.933, 1.761 and 1.634 from the 1st to 4th order interactions, respectively, and were represented by the dotted lines. SNP combinations above the line may be interpreted as significant at the 5% significance level. According to this criterion, only the three combinations pointed by arrows in Figure 3(b) were found to be significant among the 2nd order interactions. In addition to the most promising combination of (1, 6, 8), a few more combinations were significant, as shown in Figure 3(c). All of them were found to share the SNP pairs of (1, 6) or (1, 8) as a subset.
were 1.877, 1.933, 1.761 and 1.634 from the 1st to 4th order interactions, respectively, and were represented by the dotted lines. SNP combinations above the line may be interpreted as significant at the 5% significance level. According to this criterion, only the three combinations pointed by arrows in Figure 3(b) were found to be significant among the 2nd order interactions. In addition to the most promising combination of (1, 6, 8), a few more combinations were significant, as shown in Figure 3(c). All of them were found to share the SNP pairs of (1, 6) or (1, 8) as a subset.
Figure 3. Scree plots of 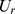 for MDR open-source data.
for MDR open-source data.

Main effects (A), 2nd order interactions (B), 3rd order interactions (C) and 4th order interactions (D) are shown. The standardized relative information gain, 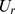 , is plotted against the rank determined by the magnitude of
, is plotted against the rank determined by the magnitude of 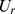 . Open-source sample set is used to show the plausibility of using
. Open-source sample set is used to show the plausibility of using 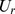 . Only the top 100 ranked
. Only the top 100 ranked 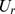 s are plotted for each order of interaction. Top ranked SNP names are denoted in parentheses. The dotted lines show the upper 5% cut-off values of
s are plotted for each order of interaction. Top ranked SNP names are denoted in parentheses. The dotted lines show the upper 5% cut-off values of 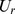 in the empirical null distribution. SNP combinations above the line may be interpreted as significant at 5% significance level.
in the empirical null distribution. SNP combinations above the line may be interpreted as significant at 5% significance level.
Figure 4 is a multi-dimensional scaling (MDS) plot for 2-locus gene-gene interactions. We used 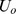 for the construction of the distance matrix. Sizes of the points represent the strength of the main effect of each SNP to the disease. The distances between the two points approximate the degree of gene-gene interactions, although there is loss of information due to dimension reduction via MDS. Point 1 shows the strongest main effect and also the points 6, 7, and 8 show large main effects. The distances between them are prominent among others and represent strong gene-gene interactions.
for the construction of the distance matrix. Sizes of the points represent the strength of the main effect of each SNP to the disease. The distances between the two points approximate the degree of gene-gene interactions, although there is loss of information due to dimension reduction via MDS. Point 1 shows the strongest main effect and also the points 6, 7, and 8 show large main effects. The distances between them are prominent among others and represent strong gene-gene interactions.
Figure 4. MDS plot for MDR open-source data.

Multi-dimensional scaling plot is produced using 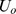 of the 2nd order interactions. The distance between two points approximates the interaction between the corresponding SNPs. The size of the points is proportional to the size of the main effects.
of the 2nd order interactions. The distance between two points approximates the interaction between the corresponding SNPs. The size of the points is proportional to the size of the main effects.
Analysis of Real Data: ATOPIC DERMATITIS DATA
This data set was collected from 433 atopic dermatitis patients with allergic type and 474 normal subjects [20]. A total of 17 SNPs were genotyped from 5 genes (IL5, IL8, IL5R, IL8RA, and IL8RB). In this study, 385 cases and 440 controls with complete genotype data were included, as done in the study by Namkung et al. [21]. The best combinations chosen by MDR evaluated by balanced accuracy in each dimension are (rs2522411), (rs2522411, rs340808), (rs2290610, rs17882210, rs340808), and (rs17026903, rs340808, rs334809, rs4073), respectively. The corresponding average cross validation consistencies (CVCs) for 10 replications are 7.0, 5.9, 3.5, and 4.5, respectively [21].
Figure 5 is a scree plot of 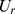 . For the 1st order interactions, rs2522411 shows the strongest association with the phenotype and the rs334809, rs340808, and rs17882210 are followed by. For the 2nd order interactions, (rs2522411, rs340808) pair shows the strongest interaction. (rs2522411, rs17026903, rs340808) and (rs2290610, rs17882210, rs340830, rs4073) combinations are the best SNP combinations in the 3rd and 4th order interactions, respectively. Adding rs340808 to the top ranked SNP of rs2522411 in the main effect to convert it into a 2nd order interaction resulted in comparable association strength. On the other hand, adding another SNP, rs17026903 to the pair (rs2522411, rs340808) to make a 3rd order interaction effectively lowered the association strength. This suggested that rs17026903 gave no additional information. For 1st and 2nd order interactions, the best SNP combinations obtained from the proposed method were the same as those obtained from MDR, while the SNP combinations in the best models for 3rd and 4th order interactions were different in the two methods. No significant SNP combinations were detected after adjustment for multiple comparisons. The adjusted p-values of the largest
. For the 1st order interactions, rs2522411 shows the strongest association with the phenotype and the rs334809, rs340808, and rs17882210 are followed by. For the 2nd order interactions, (rs2522411, rs340808) pair shows the strongest interaction. (rs2522411, rs17026903, rs340808) and (rs2290610, rs17882210, rs340830, rs4073) combinations are the best SNP combinations in the 3rd and 4th order interactions, respectively. Adding rs340808 to the top ranked SNP of rs2522411 in the main effect to convert it into a 2nd order interaction resulted in comparable association strength. On the other hand, adding another SNP, rs17026903 to the pair (rs2522411, rs340808) to make a 3rd order interaction effectively lowered the association strength. This suggested that rs17026903 gave no additional information. For 1st and 2nd order interactions, the best SNP combinations obtained from the proposed method were the same as those obtained from MDR, while the SNP combinations in the best models for 3rd and 4th order interactions were different in the two methods. No significant SNP combinations were detected after adjustment for multiple comparisons. The adjusted p-values of the largest 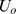 are 0.183, 0.201, 0.388 and 0.162, respectively for the 1st to 4th order interactions. All the points were located under the cut-off line (Figure 5).
are 0.183, 0.201, 0.388 and 0.162, respectively for the 1st to 4th order interactions. All the points were located under the cut-off line (Figure 5).
Figure 5. Scree plot of 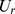 for atopic dermatitis data set.
for atopic dermatitis data set.

Same plot arrangement as in Figure 3.
Simulation Results
We evaluated the power of the proposed method in comparison with MDR on the basis of the balanced accuracy (BA) and cross-validation consistency (CVC) for two-locus interactions to assess the performance of the proposed method. We used the same simulation scheme as that of Namkung et al. [21]. We took into consideration three different sample sizes (400, 1000, 2000) and five different numbers of SNPs (10, 50, 100, 500, 1000). The numbers of case and control samples were balanced. One pair of SNPs was simulated as a causal factor among all possible combinations. The genotype data of the causal SNPs were generated based on 8 different genetic models (Tables 2, 3). For models 1, 2, and 3, the odds ratio (OR) varied with fixed interaction structure, minor allele frequency, and prevalence. Models 4, 5, and 6 were obtained from Ritchie et al. [22], and models 7 and 8 from Bush et al. [12]. All the models had little marginal effects. Fifteen different groups of input parameters were used for each model. Total of 100 replicated data sets for each combination of models and groups was used for the power comparison.
Table 2. Simulation Scheme based on eight genetic models.
| Model 1 | Model 2 | Model 3 | *Model 4 | |||||||||
| MAF/Prevalence | 0.1/0.050 | 0.1/0.050 | 0.1/0.050 | 0.1/0.046 | ||||||||
| AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | |
| BB | 1.21 | 0.2 | 0.2 | 1.23 | 0.33 | 0.33 | 1.22 | 0.4 | 0.4 | 0.55 | 1.75 | 1.33 |
| Bb | 0.2 | 5 | 5 | 0.33 | 3 | 3 | 0.4 | 2.5 | 2.5 | 1.54 | 0.18 | 0.74 |
| bb | 0.2 | 5 | 5 | 0.33 | 3 | 3 | 0.4 | 2.5 | 2.5 | 1.75 | 0.18 | 0 |
| * Model 5 | * Model 6 | ** Model 7 | ** Model 8 | |||||||||
| MAF/Prevalence | 0.1/0.026 | 0.1/0.017 | 0.2/0.052 | 0.4/0.048 | ||||||||
| AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | AA | Aa | aa | |
| BB | 1.16 | 0.38 | 0.76 | 1.15 | 0.40 | 0.17 | 0.84 | 1.35 | 0.80 | 0.52 | 1.07 | 1.89 |
| Bb | 0.38 | 3.70 | 1.97 | 0.28 | 4.23 | 4.89 | 1.30 | 0.39 | 1.45 | 1.30 | 0.92 | 0.59 |
| bb | 0.76 | 1.97 | 2.82 | 1.15 | 0.06 | 5.56 | 1.45 | 0.13 | 1.04 | 1.21 | 1.08 | 0.33 |
Table 3. Definition of data groups in simulation.
| Group | n_SNP | |||||
| 10 | 50 | 100 | 500 | 1000 | ||
| n_sample | 400 | 1 | 4 | 7 | 10 | 13 |
| 1000 | 2 | 5 | 8 | 11 | 14 | |
| 2000 | 3 | 6 | 9 | 12 | 15 | |
n_SNP: number of SNPs; n_sample: total number of samples (1∶1 for case:control).
Empirical power is defined as the proportion of replicated datasets with which the true causal SNP pair is detected as the best pair among all possible two-locus SNP pairs. Each model was run through fifteen groups, varying the number of SNPs and samples. Power of 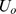 and
and 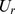 are equivalent because the rank is not changed by standardization. Figure 6 is the graph for empirical power. Two MDR results are clustered together in the plot, although MDR by CVC showed slightly better power than MDR by BA.
are equivalent because the rank is not changed by standardization. Figure 6 is the graph for empirical power. Two MDR results are clustered together in the plot, although MDR by CVC showed slightly better power than MDR by BA. 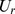 is consistently located well above the CVC, BA groups except for model 4. Groups 1, 2, and 3 have the same numbers of SNPs, and the number of samples increases with the group numbers. The same patterns of number of samples are repeated for the next three groups with an increased number of SNPs, and so on (Table 3). Therefore, there are five subgroups showing similar patterns in these plots. In general, using
is consistently located well above the CVC, BA groups except for model 4. Groups 1, 2, and 3 have the same numbers of SNPs, and the number of samples increases with the group numbers. The same patterns of number of samples are repeated for the next three groups with an increased number of SNPs, and so on (Table 3). Therefore, there are five subgroups showing similar patterns in these plots. In general, using 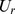 as a measure to find the causal pair seems to be superior to using MDR with CVC or BA. The superiority is clearer, especially for the groups 1, 4, 7, 10, and 13, in which the number of samples is insufficient when compared to the number of SNPs. As the number of SNPs increased, the power difference became larger, which would be a great advantage when dealing with a real data set in which the number of samples is usually far less than the number of SNPs.
as a measure to find the causal pair seems to be superior to using MDR with CVC or BA. The superiority is clearer, especially for the groups 1, 4, 7, 10, and 13, in which the number of samples is insufficient when compared to the number of SNPs. As the number of SNPs increased, the power difference became larger, which would be a great advantage when dealing with a real data set in which the number of samples is usually far less than the number of SNPs.
Figure 6. Power comparison between the methods based on entropy and MDR.

Hit ratio is used as the empirical power for the fifteen groups each for the eight models. Hit ratio is defined as the ratio at which the incorporated causal pair is identified to have the strongest association. Three different measures, 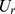 , CVC, and BA are compared. Groups 1, 2, and 3 have the same number of SNPs (10), and the numbers of samples increase with the group numbers (400, 1000, 2000), repeating the same for the next 3 groups with an increased number of SNPs (50), and so on. See Table 3 for details. The power of
, CVC, and BA are compared. Groups 1, 2, and 3 have the same number of SNPs (10), and the numbers of samples increase with the group numbers (400, 1000, 2000), repeating the same for the next 3 groups with an increased number of SNPs (50), and so on. See Table 3 for details. The power of 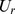 is shown to be higher than the powers of MDR with CVC or BA. The superiority is clearer, especially for the groups 1, 4, 7, 10, 13 in which the number of samples are insufficient when compared to the number of SNPs. As the number of SNPs increases, the difference in power becomes larger.
is shown to be higher than the powers of MDR with CVC or BA. The superiority is clearer, especially for the groups 1, 4, 7, 10, 13 in which the number of samples are insufficient when compared to the number of SNPs. As the number of SNPs increases, the difference in power becomes larger.
Discussion
In this study, we proposed an entropy-based method, which could identify the high-order gene-gene interactions efficiently. The proposed method utilizes the relative information gain and its standardized measure. Scree plots of the measures enabled us to identify the significant SNP combinations. Direct comparison of the association strengths was possible between different orders of locus interactions. An MDS plot represented 2nd order interactions while representing the degree of the main effects simultaneously. One could calculate the empirical p-values for 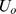 from the permuted datasets in Step 3 of the proposed method.
from the permuted datasets in Step 3 of the proposed method.
The proposed method and the MDR with different evaluation criteria were compared by simulation. In this simulation, we focused on the 2nd order interactions. The power obtained varied across the different measures as well as across the genetic models that describe the effect size and the patterns of interactions. The proposed method shows consistent superiority to MDR throughout the examined simulation models. This pattern became clearer in the groups with insufficient numbers of samples when compared to the given numbers of the SNPs.
Computing time of the proposed method would depend directly on the size of the ensemble of the permuted data sets. With 1000 permutation, the computational time from the first to fourth order interactions of the open source MDR data was 1.5 minute using Intel 2.33GHz Quad Core CPU. Atopic dermatitis data took only 1 minute.
The proposed method has been devised mainly for the candidate gene sets. Thus, applying it directly to the genome wide association studies (GWAS) would be infeasible in its current form. For the joint identification of SNPs for GWAS, Cho et al. [23] proposed using a pre-screening step and then applying a joint identification step. Our proposed method can be easily applied to the GWAS data after a pre-screening step. For example, when top 1000 SNPs are pre-screened with 2000 samples, our method would take about 1.3 hour for two locus interactions with 1000 permutation. Since the proposed method uses only the mean and variance from the permuted data sets, a large number of permutations is not required. We are investigating more efficient computational approaches for GWAS data such as an adaptive permutation approach [24] which repeats the permutations up until both mean and standard deviation of empirical null distribution are converged. An alternative approach using approximation to the known distribution is also under investigation. The entropy method for GWAS data will be reported separately.
In summary, there are several advantages of the proposed method. It is a non-parametric method and does not assume any prior distribution or any particular genetic model. It provides a list of ranked interactions on the basis of their information gain. It demonstrated better power in most simulation settings. It could perform well with the data that had sparse cells. The performance of our method in the case of sparse data would be considered as a future study.
However, there are some limitations of our proposed method, which can be summarized as follows. First, it is basically an exhaustive search technique as is MDR, and therefore is more suitable for candidate gene studies to find higher-order interactions. Second, it does not separate the main effects from pure interaction effects. Consequently, the SNPs with strong marginal effects but little interaction effects may not be discriminated, although the simulation results suggest that the proposed method is well suited for detecting interactions without main effects.
Funding Statement
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2010-0012133, 2012R1A3A2026438). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zhang H, Bonney G (2000) Use of classification trees for association studies. Genet. Epidemiol. 19: 323–332. [DOI] [PubMed] [Google Scholar]
- 2. Sheriff A, Ott J (2001) Applications of neural networks for gene finding. Adv. Genet. 42 287–297. [DOI] [PubMed] [Google Scholar]
- 3. Kooperberg C, Ruczinski I (2005) Identifying interacting SNPs using Monte Carlo logic regression. Genet. Epidemiol. 28: 157–170. [DOI] [PubMed] [Google Scholar]
- 4. Cordell HJ (2009) Detecting gene-gene interaction that underlies human diseases, Nature Reviews Genetics. 10: 392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosmer DW, Lemeshow D (2000) Applied logistic regression, 2nd edn. New York: John Wiley and Sons.
- 6. Moore JH, Williams SM (2002) New strategies for identifying gene-gene interactions in hypertension. Ann. Med. 34: 88–95. [DOI] [PubMed] [Google Scholar]
- 7. Ritchie MD, Hahn L, Roodi L, Bailey L, Dupont W, et al. (2001) Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer, Am. J. Hum. Genet. 69(1): 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung Y, Lee SY, Elston RC, Park T (2007) Odds ratio based multifactor-dimensionality reduction method for detecting gene-gene interactions. Bioinformatics 23: 71–76. [DOI] [PubMed] [Google Scholar]
- 9. Lee SY, Chung Y, Elston RC, Kim Y, Park T (2007) Log-linear model-based multifactor dimensionality reduction method to detect gene-gene interactions. Bioinformatics 23: 2589–2595. [DOI] [PubMed] [Google Scholar]
- 10. Calle ML, Urrea V, Vellalta G, Malats N, Steen KV (2008) Improving strategies for detecting genetic patterns of susceptibility in association studies. Stat. Med. 27: 6532–6546. [DOI] [PubMed] [Google Scholar]
- 11. Shannon CE (1948) A mathematical theory of communication. Bell Syst. Tech. J. 27: 379–423. [Google Scholar]
- 12. Bush WS, Edwards TL, Dudek SM, McKinney BA, Ritchie MD (2008) Alternative contingency table measures improve the power and detection of multifactor dimensionality reduction. BMC Bioinformatics 9: 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kang G, Yue W, Zhang J, Cui Y, Zuo Y, et al. (2008) An entropy-based approach for testing genetic epistasis underlying complex diseases, J. Theor. Biol. 250: 362–374. [DOI] [PubMed] [Google Scholar]
- 14. Dong C, Chu X, Wang Y, Wang Y, Jin L, et al. (2008) Exploration of gene–gene interaction effects using entropy-based methods. Eur. J. Hum. Genet. 16: 229–235. [DOI] [PubMed] [Google Scholar]
- 15. Chanda P, Sucheston L, Liu S, Zhang A, Ramanathan M (2009) Information-theoretic gene-gene and gene-environment interaction analysis of quantitative traits. BMC Genomics 10: 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray RM (2011) Entropy and information theory, 2nd edn. New York: Springer.
- 17. Jung SH, Bang H (2005) Sample size calculation for multiple testing in microarray data analysis. Biostatistics 6(1): 157–169. [DOI] [PubMed] [Google Scholar]
- 18. Jung SH, Jang W (2006) How accurately can we control the FDR in analyzing microarray data? Bioinformatics 22(14): 1730–1736. [DOI] [PubMed] [Google Scholar]
- 19. Cattell RB (1966) The scree test for the number of factors, Multivariate Behavioral Research. 1: 245–276. [DOI] [PubMed] [Google Scholar]
- 20. Namkung JH, Lee J, Kim E, Cho HJ, Kim S, et al. (2007) IL-5 and IL-5 receptor alpha polymorphisms are associated with atopic dermatitis in Koreans. Allergy 62: 934–942. [DOI] [PubMed] [Google Scholar]
- 21. Namkung J, Elston RC, Yang JM, Park T (2009) Identification of gene-gene interactions in the presence of missing data using the multifactor dimensionality reduction method. Genet. Epidemiol. 33: 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ritchie MD, Hahn LW, Mooren JH (2003) Power of multifactor dimensionality reduction for detecting gene-gene interactions in the presence of genotyping error, missing data, phenocopy, and genetic heterogeneity. Genet. Epidemiol. 24: 150–157. [DOI] [PubMed] [Google Scholar]
- 23. Cho S, Kim K, Kim YJ, Kim J-K, Cho YS, et al. (2010) Joint identification of multiple genetic variants via elastic-net variable selection in a genome-wide association analysis. Ann. of Human Genetics 74: 416–28. [DOI] [PubMed] [Google Scholar]
- 24. Knijnenburg TA, Wessels LFA, Reinders MJT, Shmulevich I (2009) Fewer permutations, more accurate p-values. Bioinformatics 25: i161–i168. [DOI] [PMC free article] [PubMed] [Google Scholar]