

Abstract
β subunits of mammalian sodium channels play important roles in modulating the expression and gating of mammalian sodium channels. However, there are no orthologs of β subunits in insects. Instead, an unrelated protein, TipE in Drosophila melanogaster and its orthologs in other insects, is thought to be a sodium channel auxiliary subunit. In addition, there are four TipE-homologous genes (TEH1-4) in D. melanogaster and three to four orthologs in other insect species. TipE and TEH1-3 have been shown to enhance the peak current of various insect sodium channels expressed in Xenopus oocytes. However, limited information is available on how these proteins modulate the gating of sodium channels, particularly sodium channel variants generated by alternative splicing and RNA editing. In this study, we compared the effects of TEH1 and TipE on the function of three Drosophila sodium channel splice variants, DmNav9-1, DmNav22, and DmNav26, in Xenopus oocytes. Both TipE and TEH1 enhanced the amplitude of sodium current and accelerated current decay of all three sodium channels tested. Strikingly, TEH1 caused hyperpolarizing shifts in the voltage-dependence of activation, fast inactivation and slow inactivation of all three variants. In contrast, TipE did not alter these gating properties except for a hyperpolarizing shift in the voltage-dependence of fast inactivation of DmNav26. Further analysis of the gating kinetics of DmNav9-1 revealed that TEH1 accelerated the entry of sodium channels into the fast inactivated state and slowed the recovery from both fast- and slow-inactivated states, thereby, enhancing both fast and slow inactivation. These results highlight the differential effects of TipE and TEH1 on the gating of insect sodium channels and suggest that TEH1 may play a broader role than TipE in regulating sodium channel function and neuronal excitability in vivo.
Introduction
Voltage-gated sodium channels are transmembrane proteins that are critical for the initiation and propagation of action potentials in neurons and other excitable cells [1]. Upon membrane depolarization, sodium channels open, resulting in sodium ion influx and further depolarization of the membrane potential. This process is called channel activation, which is responsible for the rapidly rising phase of action potentials. After channel opening, sodium channels inactivate rapidly, within a few milliseconds in a process known as fast inactivation. Fast inactivation plays an important role in the termination of action potentials. Furthermore, in response to prolonged depolarization (seconds to minutes), sodium channels progressively enter into more stable, slow-inactivated states. This process is known as slow inactivation, which is important for regulating membrane excitability, action potential patterns and spike frequency adaptation [2].
Mammalian sodium channels are composed of a pore-forming α subunit and one or more β subunits. Sodium channel α subunits have four homologous domains (I–IV), each containing six transmembrane segments (S1-S6). Mammals have nine α-subunit genes which encode sodium channel isoforms with different gating properties and different expression patterns in various cell types, tissues, and developmental stages, presumably to fulfill unique physiological functions in specific neuronal and non-neuronal cells [1], [3] [4]. Four homologous β subunits (β1-β4) have been identified and characterized [5]. They are small transmembrane proteins that possess an extracellular immunoglobulin (Ig) domain, a single transmembrane segment, and a short intracellular C-terminal domain [6]. β subunits are widely recognized as both channel modulators and cell adhesion molecules [6], [7]. They modulate sodium channel expression and channel gating; and also regulate cell adhesion and migration [6], [7]. A particular β subunit can have variable effects on different sodium channel isoforms. For instance, β2 causes a depolarizing shift in the steady-state inactivation of Nav1.2 channels, but has little effect on Nav1.3 channels [8], [9]. Different β subunits can also have different effects on a given sodium channel isoform. For instance, β1, β2 and β3 all accelerate fast inactivation kinetics of Nav1.8 channels. However, β1 enhances peak sodium current of Nav1.8 channels and causes hyperpolarizing shifts in the voltage-dependences of activation and inactivation, whereas β2 and β3 have no effect on peak sodium current and cause depolarizing shifts in the voltage-dependence of activation and inactivation of Nav1.8 channels [10].
In contrast to mammals, insects appear to have only a single sodium channel gene that encodes the α-subunit equivalent of mammalian sodium channels [11], [12]. Despite having only a single gene, insects employ alternative splicing and RNA editing to generate many sodium channel variants with different gating and pharmacological properties [12], [13]. Interestingly, there are no orthologs of mammalian β subunit in insects [14]. Instead, a transmembrane protein, TipE, is considered to be an auxiliary subunit of insect sodium channels because it increases the functional expression of insect sodium channels in Xenopus oocytes; and TipE - mutants exhibit a temperature-sensitive paralytic phenotype, similar to sodium channel mutants [12], [15] [16], [17].
Derst and associates identified four TipE-homologous genes (TEH1-4) in the genome of Drosophila melanogaster [18]. TEH1 is expressed in the central nervous system, whereas the transcripts of the other three were also detected in non-neuronal tissues, such as fat body and gut [18]. TEH1-3 proteins have been shown to increase the amplitude of sodium currents of a Drosophila sodium channel in Xenopus oocytes [18]. TEH1 has also been shown to shift the voltage-dependence of fast inactivation in the hyperpolarizing direction and slow the recovery from fast inactivation of a Drosophila sodium channel (different from sodium channel variants in this study) [18]. TipE accelerates the inactivation kinetics of the same Drosophila sodium channel [17]. However, the extent of TipE- or TEH1-mediated gating modification and whether their effects are variant-specific remains unclear.
Sodium channels are the primary target of pyrethroid insecticides [19], [20]. Because of the involvement of sodium channel mutations in pyrethroid resistance, intense research has been carried out in the past two decades to functionally express and characterize the effects of pyrethroids on the gating properties of insect sodium channels in Xenopus oocytes [12], [21]. Prior to this study, almost all functional and pharmacological analyses of insect sodium channels were conducted by co-expression of insect sodium channels with TipE, and it is not clear whether TipE or TEH1 modulate the action of pyrethroids.
In a previous study, we identified 33 functional Drosophila sodium channel (DmNav) splice variants with a wide range of voltage dependences of activation and inactivation [22]. In this study, we used three of these splice variants, DmNav9-1, DmNav22 and DmNav26, to compare the effects of TipE and TEH1 on DmNav channels. We chose these three variants because in the absence of TipE or TEH1, they generate sufficient currents for electrophysiological analysis, which made it possible to evaluate the gating-modifying effects of TipE or TEH1. In addition, these variants belong to three different splice types and exhibit different functional properties [22], which potentially allows us to determine variant-specific gating modulation by TipE and/or TEH1. Our results show that, like TipE, TEH1 enhanced the expression of sodium currents and accelerated current decay of all three variants. Furthermore, we found that TEH1 extensively modified sodium channel functional properties of all three variants, whereas TipE only modified the gating of one of the variants. TEH1, but not TipE, also reduced DmNav9-1 sensitivity to deltamethrin by reducing the duration of sodium channels in the open state. Our findings raise the possibility that TEH1 may play a broader role in regulating sodium channel gating and neuronal excitability in vivo.
Materials and Methods
Ethics statement
All animal protocols used in this study were approved by the Institutional Animal Care and Use Committee at Michigan State University.
Xenopus oocyte expression system
Oocytes were obtained surgically from female Xenopus laevis (Nasco, Ft. Atkinson. WI) and incubated with 1 mg/ml Type IA collagenase (Sigma Co., St. Louis, MO) in Ca2+-free ND-96 medium (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES, pH 7.5). Follicle still remaining on the oocytes following digestion was removed with forceps. Isolated oocytes were incubated in ND-96 medium containing 1.8 mM CaCl2 supplemented with 50 µg/ml gentamicin, 5 mM pyruvate, and 0.5 mM theophylline [23]. Healthy stage V-VІ oocytes were used for cRNA injection. TipE/TEH1 cRNA or H2O (as control) was injected together with DmNa v cRNA at a 1:1 ratio.
Electrophysiological recording and analysis
Methods for two-electrode recording and data analysis were similar to those described previously [24]. The borosilicate glass electrodes were filled with filtered 3 M KCl in 0.5% agarose and had a resistance of 0.5 to 1.0 MΩ. The recording solution was ND-96 recording solution (96 mM NaCl, 2.0 mM KCl, 1.0 mM MgCl2, 1.8 mM CaCl2, and 10 mM HEPES, pH adjusted to 7.5 with NaOH). Sodium currents were measured with a Warner OC725C oocyte clamp amplifier (Warner Instrument, Hamden, CT) and processed with a Digidata 1440 (Axon Instruments Inc., Foster City, CA). Data were sampled at 50 kHz and filtered at 2 kHz. Leak currents were corrected by p/4 subtraction. pClamp 10.2 software (Axon Instruments Inc., CA) was used for data acquisition and analysis. The maximal peak sodium current was about 2 µA to achieve optimal voltage control by adjusting the incubation time after injection.
The voltage dependence of sodium channel conductance (G) was calculated by measuring the peak current at test potentials ranging from −80 mV to +65 mV in 5-mV increments and divided by (V−V rev), where V is the test potential and V rev is the reversal potential for sodium ion. Peak conductance values were normalized to the maximal peak conductance (G max) and fitted with a two-state Boltzmann equation of the form G/Gmax = [1 + exp(V−V1/2)/k]−1, in which V is the potential of the voltage pulse, V 1/2 is the voltage for half-maximal activation, and k is the slope factor.
The voltage dependence of sodium channel fast inactivation was determined by using 100-ms inactivating pre-pulses ranging from -120 mV to 0 mV in 5 mV increments from a holding potential of −120 mV, followed by test pulses to -10 mV for 20 ms. The peak current amplitude during the test depolarization was normalized to the maximum current amplitude and plotted as a function of the pre-pulse potential. Data were fitted with a two-state Boltzmann equation of the form I/Imax = [1 + (exp(V−V1/2)/k)]−1, in which I is the peak sodium current, I max is the maximal current evoked, V is the potential of the voltage pre-pulse, V1/2 is the half-maximal voltage for inactivation, and k is the slope factor.
The voltage dependence of sodium channel slow inactivation was measured with 60 s conditioning pulses ranging from −100 mV to 0 mV in 10 mV increments, followed by repolarization to a holding potential of -120 mV for 100 ms to remove fast inactivation, and at last a -10 mV test pulse for 20 ms. The peak current amplitude during the test depolarization was normalized to the maximum current amplitude and plotted against the pre-pulse potential. Data were fitted with a two-state Boltzmann equation as above for fast inactivation.
Development of fast inactivation was measured by holding oocytes at -120 mV, followed by a depolarization to -45 mV for 0 to 80 ms, and then a -10 mV test pulse for 20 ms to measure the fraction of sodium current inactivated during the pre-pulse. The peak current during the test pulse was divided by the peak current which has a pre-pulse duration of 0 ms and plotted as a function of duration time of pre-pulse. Time constant (τ) was calculated by fitting the plot with a single exponential decay function.
Recovery from fast inactivation was tested with a conditioning depolarization of -10 mV for 100 ms, which will drive all sodium channels into the fast inactivated state, then repolarization to -70 mV for 0-20 ms followed by a 20-ms test pulse to -10 mV. The peak current during the test pulse was divided by the peak current during the inactivating pulse and plotted as a function of duration time between two pulses. Time constant (τ) of recovery from fast inactivation was calculated by fitting the plot with a single exponential function.
Development of slow inactivation was measured by holding oocytes at -120 mV, followed with a -10-mV pre-pulse depolarization for 0 to 25 s, then repolarization to -120 mV for 100 ms to remove fast inactivation, and a -10 mV test pulse for 20 ms to measure the fraction of sodium current inactivated during the pre-pulse. The peak current during the test pulse was divided by the peak current which has a pre-pulse duration of 0 ms and plotted as a function of duration of pre-pulse. Time constant (τ) was calculated by fitting the plot with an exponential decay function.
Recovery from slow-inactivation was measured by holding oocytes at -120 mV, followed by a pre-pulse to -10 mV for 60 s to drive sodium channels into the slow inactivated state, followed by repolarization to -120 mV for 0 to 30 s, and finally a test pulse to -10 mV for 20 ms. The peak current during the test pulse was divided by the peak current which has a repolarizing duration of 30 s and plotted as a function of duration between the pre and test pulses. Recovery from slow inactivation was well fitted by a double exponential function.
Measurement of sodium channel sensitivity to deltamethrin
The method for application of deltamethrin in the recording system was identical to that described by Tan et al. [25]. The effect of deltamethrin was measured 10 min after toxin application. Deltamethrin-induced tail currents were recorded with a 100-pulse train of 5 ms step depolarizations from -120 to 0 mV at 66.7 Hz [26]. Additionally, deltamethrin-induced tail currents were measured using a single pulse protocol with a 500-ms step depolarization from -120 mV to -10 mV. The percentage of channels modified by deltamethrin was calculated using the equation M = {[I tail/(E h −E Na)]/[I Na/(E t −E Na)]}×100 [27], where I tail is the maximal tail current amplitude, E h is the potential to which the membrane is repolarized, E Na is the reversal potential for sodium current determined from the current-voltage curve, I Na is the amplitude of the peak current during depolarization before pyrethroids exposure, and E t is the potential of step depolarization.
Chemicals
Deltamethrin was kindly provided by Bhupinder Khambay (Rothamsted Research, Harpenden, UK). Deltamethrin was dissolved in dimethyl sulfoxide (DMSO). The working concentration was prepared in ND-96 recording solution immediately prior to experiments. The concentration of DMSO in the final solution was <0.5%, which had no effect on the function of sodium channels.
Statistical analysis
Results are reported as mean ± SEM. Statistical significance was determined by using one-way analysis of variance (ANOVA) with Scheffe’s post hoc analysis, and significant values were set at p<0.05.
Results
TipE and TEH1 increase the peak sodium current and accelerate the current decay of all three DmNav variants
To compare the expression of sodium current in Xenopus oocytes, equal amounts of cRNA synthesized in vitro from the DmNav9-1, DmNav22, or DmNav26 plasmids were injected into oocytes with or without TipE or TEH1 cRNA. Sodium currents were recorded 48 hours after injection by step depolarizations to a series of voltages ranging from -80 mV to +25 mV in 5-mV increments. Both TipE and TEH1 increased the amplitude of peak sodium current of all three variants by 4 to 9 fold (Figure 1A and B).
Figure 1. Modulatory effects of TipE and TEH1 on peak sodium currents of DmNav9-1, DmNav22, or DmNav26 channels.

(A) Representative traces of peak sodium currents from oocytes expressing DmNav9-1, DmNav9-1+TipE, and DmNav9-1+TEH1 sodium channels. Note that the sodium current of the DmNav9-1 channel possesses a non-inactivating component, known as persistent current (10% of the maximal transient peak current). Both TipE and TEH1 enhanced the persistent current. However, the persistent current remained to be about 10% of the maximal transient peak current. (B) Both TipE and TEH1 significantly increased peak sodium currents of all three Para sodium variants tested: DmNav9-1, DmNav26, and DmNav22, but there was no significant difference between the effects of TipE and TEH1. Sodium currents were recorded 48 hours after cRNA injection. Sodium currents were recorded by a step depolarization to from -80 to 65 mV in 5mV increments with a holding potential of -120 mV. Data are presented as means ± SEM for 12-15 oocytes. * indicates a significant difference compared to peak of DmNav channel only using one-way ANOVA with Scheffe’s post hoc analysis (p<0.05).
We then measured the effect of TipE and TEH1 on the decay of sodium currents for all three variants. Current decay was well fitted by a single exponential with or without TipE or TEH1. TipE slightly but significantly increased the rate of current decay of all three variants between -40 mV and -5 mV (Figure 2A–C). Similarly, TEH1 also slightly accelerated current decay for all three variants, but over different voltage ranges (Figure 2D–F). TEH1 affected the current decay of DmNav22 channels over a broader voltage range (between -40 mV to 10 mV) than the other two variants which were affected between -40 mV and -30 mV for DmNav9-1, and -40 mV and -20 mV for DmNav26 (Figure 2D–F).
Figure 2. Enhancement of sodium current decay by co-expression of TipE or TEH1 with DmNav channel variants.

Sodium current decay in DmNav9-1, DmNav22, or DmNav26 co-expressed with TipE (A, B, C, respectively) or TEH1 (D, E, and F, respectively). The decay of sodium current was fitted by a single exponential to generate time constants of current decay (τdecay). Each data point represents mean ± SEM for 12-20 oocytes. * indicates a significant difference compared to that of DmNav channel only (p<0.05).
Differential effects of TEH1 and TipE on the voltage-dependence of activation and fast inactivation
Co-expression of DmNav9-1 with TEH1 induced a 12-mV hyperpolarizing shift in the voltage dependence of activation (Figure 3A and Table 1). Similarly, significant hyperpolarizing shifts were also detected with co-expression of TEH1 with DmNav26 or DmNav22 (Table 1). However, co-expression of TipE did not modify the voltage dependence of activation of any variants (Figure 3A and Table 1).
Figure 3. Effects of co-expression of TipE or TEH1 on the voltage-dependence of activation and fast inactivation of DmNa v9-1 channels.

(A) Voltage-dependences of activation. (B) Voltage-dependence of fast inactivation. Data were fitted with a two-state Boltzmann equation and fitting parameters are shown in Table 1. Data points are shown as mean ± SEM. Recording protocols are indicated and the details of the protocols and data analysis are described in the Materials and Methods.
Table 1. Gating properties of DmNav variants with or without TipE or TEH1.
| Activation |
Fast Inactivation |
Slow inactivation |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| V1/2 (mV) |
k (mV) |
V1/2 (mV) |
k (mV) |
V1/2 (mV) |
k (mV) |
||||
| DmNav9-1 | -29.0 ± 1.1 | 5.5 ± 0.7 | -41.4 ± 0.8 | 4.8 ± 0.2 | -44.5 ± 1.8 | 5.6 ± 0.8 | |||
| DmNav9-1 + TipE | -26.2 ± 0.5 | 5.2 ± 0.2 | -42.1 ± 0.7 | 5.1 ± 0.1 | -44.9 ± 1.4 | 5.5 ± 0.2 | |||
| DmNav9-1 + TEH1 | -41.1 ± 1.1* | 4.6 ± 0.2 | -51.7 ± 0.6* | 5.0 ± 0.1 | -60.2 ± 0.5* | 5.0 ± 0.3 | |||
| DmNav26 | -25.7 ± 0.2 | 3.4 ± 0.3 | -34.1 ± 0.1 | 4.1 ± 0.2 | -45.6 ± 0.2 | 5.0 ± 0.3 | |||
| DmNav26 + TipE | -24.7 ± 0.6 | 3.4 ± 0.4 | -40.8 ± 0.4* | 4.2 ± 0.2 | -44.1 ± 0.7 | 4.5 ± 0.4 | |||
| DmNav26 + TEH1 | -33.6 ± 0.6* | 3.1 ± 0.4 | -42.6 ± 0.6* | 4.6 ± 0.1 | -50.6 ± 0.3* | 4.5 ± 0.5 | |||
| DmNav22 | -26.7 ± 1.1 | 5.8 ± 0.5 | -40.7 ± 0.6 | 4.8 ± 0.1 | -49.2 ± 0.2 | 3.7 ± 0.2 | |||
| DmNav22 + TipE | -25.1 ± 1.2 | 7.2 ± 0.5 | -38.9 ± 0.5 | 4.8 ± 0.2 | -45.5 ± 0.8 | 5.3 ± 0.2 | |||
| DmNav22 + TEH1 | -32.1 ± 0.7* | 6.2 ± 0.3 | -48.3 ± 0.6* | 5.2 ± 0.1 | -57.9 ± 0.7* | 4.8 ± 0.1 | |||
Data represent mean ± SEM for 12-20 oocytes. DmNav 9 1, DmNa v 26, and DmNav 22 are treated as control of each group.
Significantly different from that of DmNav channel only using one-way ANOVA with Scheffe’s post hoc analysis (p<0.05).
Co-expression of DmNav9-1 with TEH1 induced a 9-mV hyperpolarizing shift in the voltage-dependence of fast inactivation compared with DmNa v9-1 alone (Figure 3B and Table 1). Similarly, co-expressing TEH1 with DmNav26 or DmNav22 also significantly shifted the voltage dependence of fast inactivation in the hyperpolarizing direction (Table 1). TipE did not alter the voltage dependence of fast inactivation of DmNav9-1 or DmNav22 channels, but caused a significant 6-mV hyperpolarizing shift in the voltage dependence of fast inactivation of DmNav26 channels (Table 1), indicating that TipE has a variant specific effect on the voltage dependence of fast inactivation.
Effect of TipE and TEH1 on the sensitivity of DmNav9-1 channels to deltamethrin
Deltamethrin, a pyrethroid insecticide, induces a slowly decaying tail current associated with repolarization in voltage clamp experiments [26]. To determine whether TipE and TEH1 differentially modulate the activity of deltamethrin, we used a train of depolarizing pulses to elicit deltamethrin-induced tail current in oocytes expressing DmNav9-1 with or without TipE or TEH1. At 1 µM, deltamethrin induced a large tail current (Figure 4A), which can be quantified as the percentage of channel modification by deltamethrin using the method developed by Tatebayashi and Narahashi [27]. Co-expression of TEH1 with DmNav9-1 channels significantly reduced the percentage of channels modified by deltamethrin (Figure 4B), whereas TipE had no effect on channel sensitivity to deltamethrin (Figure 4B).
Figure 4. Sensitivity of DmNa v9-1 to deltamethrin is modulated by co-expression of TEH1.

(A) A representative tail current induced by 1 µM deltamethrin. (B) Percentage of channel modification by deltamethrin (multiple-pulse test). Tail currents were elicited by a 66.7-Hz train of 100 5-ms depolarization from -120 to 0 mV. (C) Co-expression of TEH1 significantly reduced the stability of DmNav9-1 peak sodium current under repeated conditioning depolarizations. Sodium currents were recorded during 20-ms step depolarizations from -120 mV to -10 mV after 0-100 conditioning pulses (5-ms pulses from -120 mV to 0 mV at 66.7 Hz). (D) Percentage of channel modification by deltamethrin (single-pulse test). Tail currents were elicited by a 500 ms depolarization from -120 mV to 0 mV. All data are shown as mean ± SEM for 9-15 oocytes. * indicates significant difference compared to the DmNav9-1 channel using one-way ANOVA with Scheffe’s post hoc analysis (p<0.05).
Derst et al. [18] reported that the recovery from inactivation of a Drosophila sodium channel variant was slowed by TEH1. We hypothesized that, in the presence of TEH1, the trains of depolarizing pulses used in our study to evaluate the effect of deltamethrin may reduce the availability of open channels, thereby, reducing the gating modification by deltamethrin. To test whether changes in gating caused by TEH1 were responsible for the reduced channel sensitivity to deltamethrin, we examined the effect of the multiple depolarizing-prepulses on the stability of peak sodium current in channels co-expressed with TipE or TEH1. The peak current remained unchanged in oocytes expressing DmNav9-1 channels alone or with TipE, whereas the peak sodium current was gradually reduced after each conditioning pulse in oocytes coexpressing DmNav9-1channels with TEH1 (Figure 4C). These results support the hypothesis that the reduced deltamethrin sensitivity of DmNav9-1 channels in the presence of TEH1 is likely caused by reduced availability of open channels. We then examined the effect of deltamethrin without the conditioning pulsesand found that the percentage of channel modification by deltamethrin was not altered by either TEH1 or TipE (Figure 4D).
Co-expression of TEH1 with DmNav9-1 significantly enhanced entry into and stability of the fast-inactivated state
The results above prompted us to further characterize the effects of TEH1 and TipE on inactivation gating kinetics, particularly the development of and recovery from fast-inactivation. Figure 5A shows the time course of the development of fast inactivation at the pre-pulse voltage of -45 mV for DmNav9-1 alone or co-expressed with TipE or TEH1. Co-expression of TEH1 with DmNav9-1 greatly enhanced entry of DmNav9-1 channels into the fast inactivated state compared with that of DmNav9-1 alone or the combination of DmNav9-1 and TipE (Figure 5A). In addition, the accelerated entry into fast inactivation by TEH1 was observed at all three pre-pulse voltages tested (Figure 5B).
Figure 5. Effects of co-expression of TipE or TEH1 on entry into or recovery from fast inactivation of DmNav9-1 channels.

(A) Time course of development of fast inactivation with a pre-pulse of -45 mV. (B) τ values of development of fast inactivation under different pre-pulse voltages. τ values were determined by fitting time course of the development of fast inactivation with a single exponential decay (n ≥ 12). (C) Recovery from fast inactivation with a repolarizing voltage of -70 mV. (D) τ values of recovery from fast inactivation at different repolarizing voltages. τ values were calculated by fitting recovery from fast inactivation data from different repolarizing voltages by a single exponential function (n≥ 10). Recording protocols are indicated and the details of the protocols and data analysis are described in the Materials and Methods.
Co-expressing DmNav9-1 with TEH1 greatly inhibited the recovery of DmNav9-1 channels from fast inactivation all repolarization voltages tested (Figure 5C and D). In contrast, co-expression of DmNav9-1 with TipE had no effect on the recovery from fast inactivation (Figure 5C, and D).
Co-expression of TEH1 with DmNav9-1 inhibited recovery from slow inactivation
In addition to fast inactivation, sodium channels undergo slow inactivation which plays important roles in regulating firing frequency and pattern in response to sustained stimuli [2]. Therefore, we examined the effects of TEH1 and TipE on the voltage-dependence of slow inactivation, the rate of entry into the slow-inactivated state, and recovery from slow inactivation of DmNav9-1channels. Co-expression of DmNav9-1 with TEH1 induced a significant 16-mV hyperpolarizing shift compared with DmNav9-1 alone (Figure 6A and Table 1). TEH1 also induced significant hyperpolarizing shifts in voltage-dependence of slow inactivation in DmNav26 and DmNav22 channels (Table 1). In contrast, TipE had no effect on the voltage-dependence of slow inactivation in any of three variants tested (Table 1).
Figure 6. Co-expression of TEH1 inhibits recovery from slow inactivation of DmNav9-1 channels.
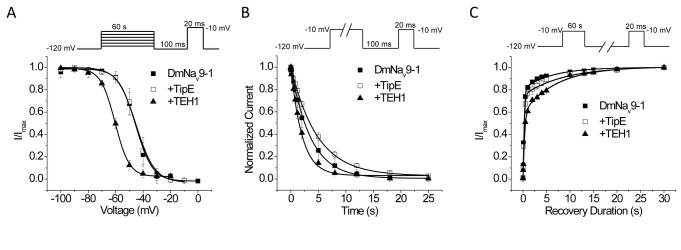
(A) Voltage-dependence of slow inactivation. (B) Time course of development of slow-inactivation. Development of slow inactivation was fitted by an exponential decay and the parameters are summarized in Table S1. (C) Time course of recovery from slow-inactivation. Recovery from slow inactivation was fitted by a double exponential function and the parameters are summarized in Table 2. Recording protocols are indicated and the details of the protocols and data analysis are described in the Materials and Methods.
The development of slow inactivation for DmNav9-1 with or without TipE or TEH1 all exhibited a monophasic time course and was well fitted by an exponential decay (Figure 6B and Table S1). We found that neither TipE nor TEH1 significantly alter the development of slow inactivation of DmNav9-1 channels (Figure 6B and Table S1). The rate of recovery from the slow inactivated state for DmNav9-1with or without TipE or TEH1 all followed a biphasic time course (Figure 6C and Table 2). DmNav9-1+TipE recovered from slow inactivation in a manner that was very similar to that of DmNav9-1 alone. However, co-expression with TEH1 significantly slowed the slow component (τ2) of recovery and also increased the fraction of the slow component, but did not alter the fast component (τ1) of recovery (Figure 6C and Table 2).
Table 2. Recovery from slow inactivation of DmNa v9-1 with or without TipE or TEH1.
| Na+ channel | τ1 | f1 | τ2 | f2 | n |
|---|---|---|---|---|---|
| DmNav9-1 | 0.23 ± 0.02 | 0.81 ± 0.01 | 5.10 ± 0.45 | 0.19 ± 0.01 | 9 |
| + TipE | 0.24 ± 0.02 | 0.79 ± 0.01 | 4.8 ± 0.42 | 0.21 ± 0.02 | 8 |
| + TEH1 | 0.24 ± 0.03 | 0.59 ± 0.02* | 7.1 ± 0.38* | 0.41 ± 0.01* | 10 |
Recovery from slow inactivation was fitted by double exponential function. Data represents mean ± SEM. τ: time constant, f relative fraction, n number of oocytes.
* Significant difference compared with DmNav9-1 channel using one-way ANOVA with Scheffe’s post hoc analysis (p<0.05).
Discussion
While the roles of mammalian sodium channel β subunits in modulating sodium channel activities have been extensively studied, research on auxiliary subunits of insect sodium channels is limited. This is particularly true with respect to how these auxiliary subunits modulate sodium channel gating and toxin pharmacology. In this study, we showed that while both TipE and TEH1 enhanced peak sodium currents and increased current decay, they modulated the gating of DmNav channels differently. First, TEH1 induced hyperpolarizing shifts in the voltage-dependences of activation, fast inactivation, and slow inactivation of all three DmNav sodium channel variants examined. In contrast, TipE did not alter these properties of the three variants, with one exception: TipE shifted the voltage-dependence of fast inactivation of DmNav26 channels in the hyperpolarizing direction. Second, TEH1, but not TipE, facilitated entry of sodium channels into fast inactivation and delayed their recovery from both fast and slow inactivation. Our findings therefore suggest distinct roles of TipE and TEH1 in regulating the function of sodium channels and neuronal excitability in vivo.
TipE is the first auxiliary subunit of insect sodium channels identified in D. melanogaster. tipE - mutants exhibit temperature-sensitive paralytic phenotypes [16], [28], suggesting an important role of TipE in regulating neuronal excitability. An earlier electrophysiological study on the activity of embryonic neurons from a tipE - mutant has shown that TipE modulates the activity of only certain neurons [29]. The percentage of embryonic neurons from a tipE - mutant capable of firing repetitively during a sustained depolarization was significantly reduced [29]. However, only a portion of the tipE - neurons was affected [29]. Additionally, a [H3] saxitoxin binding study showed that sodium channel density was reduced by about 30% to 40% in head membrane extracts from the tipE - mutants compared with wild type flies [30]. Furthermore, whole-cell patch clamp recordings indicated that sodium current density was decreased by about 40% to 60% in dissociated embryonic neurons of tipE - mutants [31]. Consistent with these findings, TipE was shown to enhance the peak current of insect sodium channels in heterologous expression ( Xenopus oocytes) studies [12], [16] [17],. TipE and TEH1 drastically increased the amplitude of peak current of all three sodium channel variants. These results suggest they increase sodium current density. Such effects may be exerted at the level of channel protein expression and/or channel conductance.
Although TipE accelerated the current decay of all three variants (Figure 2A-C) in our study, the effects on these three variants were not as drastic as that on the variant in Warmke et al. [17],, suggesting that the effect of TipE on inactivation kinetics may be variant-specific. Furthermore, the effects of TipE on sodium channel gating seem to be limited, compared to TEH1, and may also be variant-specific. The voltage dependence of inactivation of only one variant, DmNav26, that we examined was altered in the presence of TipE (Table 1). DmNav26 differs from the other two variants in the exclusion of one optional exon j, and inclusion of one optional exon f and one mutually exclusive exon k, and also contains five scattered amino acid changes which are possibly due to RNA editing. Which unique sequence(s) contributes to this variant-specific effect remains to be determined. It is known that DmNav and other insect sodium channel transcripts undergo extensively alternative splicing and RNA editing, generating a large collection of sodium channel variants [12], [13]. These variants exhibit unique gating and pharmacological properties [22], [25] [32], [33], [34]. They may be expressed in different tissues and cells to fulfill their unique roles in insect neurophysiology [32]. It is therefore possible that modulation of gating properties of selective DmNav variants by TipE provides a unique control of neuronal activities in specific neural circuits. On the other hand, extensive modification of sodium channel gating properties by TEH1 raises the possibility that TEH1 plays a broader role than TipE in modulating the gating of potentially diverse sodium channel variants in Drosophila . Enhanced fast and slow inactivation by TEH1 could lead to reduced availability of open sodium channels particularly in response to sustained stimulations of various durations, which could decrease firing frequency and alter firing patterns. As we showed, reduced availability of open channels by TEH1 decreased the potency of pyrethroids because pyrethroids preferably act on open sodium channels. It is possible that TEH1, but not TipE, may regulate sodium channel sensitivity to pyrethroids in neurons that encounter repetitive stimulations. It is also intriguing that TEH1 could modulate three different gating properties, activation, fast inactivation, and slow inactivation, which are thought to be controlled by distinct regions of the sodium channel protein [1], [35], [36]. Further characterization of how TEH1 modulates these three gating properties at the molecular level may uncover interconnecting molecular features that are critical for activation, fast inactivation, and slow inactivation of sodium channels.
Our data suggests that TEH1 has similar modulatory effects on DmNav variants as the β subunits of mammalian sodium channels. Aside from regulating the expression of sodium channels, mammalian β subunits modify the gating properties of sodium channels and modulate the electrical excitability of nerves and muscles [37]. For example, β1 subunits induce depolarizing shifts in the voltage-dependence of activation, fast inactivation, and slow inactivation of Nav1.2 channels [38]. Additionally, co-expression of β1 subunits with Nav1.7 and Nav1.8 sodium channels in Xenopus oocytes accelerates current kinetics and produces a hyperpolarizing shift in steady-state inactivation [39], and modulates activation, slow inactivation, and recovery from slow inactivation of Nav1.4 channels [40], [41] [42].
Modulation of the function of sodium channel variants by TEH1 indicates a potential functional coupling of sodium channel variants with TEH1. An earlier study has shown that the TEH1 transcript is detected exclusively in the central nervous system (CNS) [18], where DmNav transcripts are abundantly expressed [43], suggesting potential co-expression of TEH1 and DmNav in the CNS. However, further biochemical analysis is needed to confirm co-localization and/or direct physical interaction between TEH1 and DmNav channels in vivo. Structurally, TipE or TEH1 are different from mammalian sodium channel β subunits. Both TipE and TEH1 have intracellular N- and C-termini and two membrane segments connected by a large extracellular loop; whereas β subunits are composed of a single transmembrane segment with an extracellular N-terminus and a small intracellular C-terminus. Both extracellular and intracellular domains of mammalian β1 subunits have been shown to be essential for functional modulation of sodium channels [44], [45], [46]. Future molecular analyses are needed to address the molecular mechanism by which TipE and TEH1 modulate the function of insect sodium channels.
In conclusion, we showed that TipE and TEH1 differentially modulate key gating properties of DmNav, even though both TipE and TEH1 enhance the sodium current and accelerate current decay in all DmNav variants tested. Furthermore, although TipE and TEH1 are structurally different from mammalian sodium channel β subunits, our results show that these proteins appear to be functionally similar. Thus, not only TipE, but also TEH1, may play an important role in regulating neuronal activities in insects. Further understanding the role of TEH1 in vivo, including generation and characterization of TEH1 mutants, is expected to further advance our general knowledge of sodium channel function and neuronal excitability.
Supporting Information
(DOCX)
Acknowledgments
We thank Drs. Kris Silver and Frank D. Rinkevich for critical review of the manuscript.
Funding Statement
This study was supported by a grant to KD from National Institutes of Health (GM057440). LW was partially supported by a China Scholarship Council fellowship, #2008630022, via Northwest A & F University, China. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13-25. doi:10.1016/S0896-6273(00)81133-2. PubMed: 10798388. [DOI] [PubMed] [Google Scholar]
- 2. Vilin YY, Ruben PC (2001) Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys 35: 171-190. doi:10.1385/CBB:35:2:171. PubMed: 11892790. [DOI] [PubMed] [Google Scholar]
- 3. Frank HY, Catterall WA (2003) Overview of the voltage-gated sodium channel family. Genome Biol 4: 207. doi:10.1186/gb-2003-4-3-207. PubMed: 12620097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR et al. (2000) Nomenclature of voltage-gated sodium channels. Neuron 28: 365–368. doi:10.1016/S0896-6273(00)00116-1. PubMed: 11144347. [DOI] [PubMed] [Google Scholar]
- 5. Chahine M, Ziane R, Vijayaragavan K, Okamura Y (2005) Regulation of Nav channels in sensory neurons. Trends Pharmacol Sci 26: 496-502. doi:10.1016/j.tips.2005.08.002. PubMed: 16125256. [DOI] [PubMed] [Google Scholar]
- 6. Isom LL (2002) The role of sodium channels in cell adhesion. Front Biosci 7: 12-23. doi:10.2741/isom. PubMed: 11779698. [DOI] [PubMed] [Google Scholar]
- 7. Brackenbury WJ, Yuan Y, O’Malley HA, Parent JM, Isom LL (2011) Abnormal neuronal patterning occurs during early postnatal brain development of Scn1b-null mice and precedes hyperexcitability. Proc Natl Acad Sci U S A 110: 1089-1094. PubMed: 23277545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qu Y, Curtis R, Lawson D, Gilbride K, Ge P et al. (2001) Differential modulation of sodium channel gating and persistent sodium currents by the beta1, beta2, and beta3 subunits. Mol Cell Neurosci 18: 570-580. doi:10.1006/mcne.2001.1039. PubMed: 11922146. [DOI] [PubMed] [Google Scholar]
- 9. Meadows LS, Chen YH, Powell AJ, Clare JJ, Ragsdale DS (2002) Functional modulation of human brain Nav 1.3 sodium channels, expressed in mammalian cells, by auxiliary beta1, beta2 and beta3 subunits. Neuroscience 114: 745-753. doi:10.1016/S0306-4522(02)00242-7. PubMed: 12220575. [DOI] [PubMed] [Google Scholar]
- 10. Vijayaragavan K, Powell AJ, Kinghorn IJ, Chahine M (2004) Role of auxiliary beta1-, beta2-, and beta3-subunits and their interaction with Na(v)1.8 voltage-gated sodium channel. Biochem Biophys Res Commun 319: 531-540. doi:10.1016/j.bbrc.2004.05.026. PubMed: 15178439. [DOI] [PubMed] [Google Scholar]
- 11. Loughney K, Kreber R, Ganetzky B (1989) Molecular analysis of the para locus, a sodium channel gene in Drosophila . Cell 58: 1143-1154. doi:10.1016/0092-8674(89)90512-6. PubMed: 2550145. [DOI] [PubMed] [Google Scholar]
- 12. Dong K (2010) Recent progress in insect sodium channel research. In: Gilbert LI, [!(surname)!] Insect Pharmacology. New York: Elsevier; pp. 25-27. [Google Scholar]
- 13. Dong K (2007) Insect sodium channels and insecticide resistance. Invert Neurosci 7: 17-30. doi:10.1007/s10158-006-0036-9. PubMed: 17206406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Littleton JT, Ganetzky B (2000) Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron 26: 35-43. doi:10.1016/S0896-6273(00)81135-6. PubMed: 10798390. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki DT, Grigliatti T, Williamson R (1971) Temperature-sensitive mutations in Drosophila melanogaster, VII. A mutation (parats) causing reversible adult paralysis. Proc Natl Acad Sci USA 68: 890-893. doi:10.1073/pnas.68.5.890. PubMed: 5280526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng G, Deák P, Chopra M, Hall LM (1995) Cloning and functional analysis of tipE, a novel membrane protein that enhances drosophila para sodium channel function. Cell 82: 1001-1011. doi:10.1016/0092-8674(95)90279-1. PubMed: 7553842. [DOI] [PubMed] [Google Scholar]
- 17. Warmke JW, Reenan RAG, Wang P, Qian S, Arena JP et al. (1997) Functional expression of Drosophila para sodium channels. J Gen Physiol 110: 119-133. doi:10.1085/jgp.110.2.119. PubMed: 9236205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Derst C, Walther C, Veh RW, Wicher D, Heinemann SH (2006) Four novel sequences in Drosophila melanogaster homologous to the auxiliary Para sodium channel subunit TipE. Biochem Biophys Res Commun 339: 939-948. doi:10.1016/j.bbrc.2005.11.096. PubMed: 16325765. [DOI] [PubMed] [Google Scholar]
- 19. Soderlund DM, Bloomquist JR (1989) Neurotoxic actions of pyrethroid insecticides. Annu Rev Entomol 34: 77-96. doi:10.1146/annurev.en.34.010189.000453. PubMed: 2539040. [DOI] [PubMed] [Google Scholar]
- 20. Narahashi T (2000) Neuroreceptors and ion channels as the basis for drug action: past, present, and future. J Pharmacol Exp Ther 294: 1-26. PubMed: 10871290. [PubMed] [Google Scholar]
- 21. Soderlund DM (2005) Sodium channels. In: Gilbert LI, Iatrou K, Gill SS. Comprehensive molecular insect science. Amsterdam: Elsevier; pp. 1-24. [Google Scholar]
- 22. O’Donnell-Olson R, Liu Z, Nomura Y, Song W, Dong K (2008) Molecular and functional characterization of voltage-gated sodium channel variants from Drosophila melanogaster . Insect Biochem Mol Biol 38: 604-610. doi:10.1016/j.ibmb.2008.01.003. PubMed: 18405837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goldin AL (1992) Maintenance of Xenopus laevis and oocyte injection. Methods Enzymol 207: 266-279. doi:10.1016/0076-6879(92)07017-I. PubMed: 1528120. [DOI] [PubMed] [Google Scholar]
- 24. Tan J, Liu Z, Wang R, Huang ZY, Chen AC et al. (2005) Identification of amino acid residues in the insect sodium channel critical for pyrethroid binding. Mol Pharmacol 67: 513-522. PubMed: 15525757. [DOI] [PubMed] [Google Scholar]
- 25. Tan J, Liu Z, Tsai TD, Valles SM, Goldin AL et al. (2002) Novel sodium channel gene mutations in Blattella germanica reduce the sensitivity of expressed channels to deltamethrin. Insect Biochem Mol Biol 32: 445-454. doi:10.1016/S0965-1748(01)00122-9. PubMed: 11886779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vais H, Williamson MS, Goodson SJ, Devonshire AL, Warmke JW et al. (2000) Activation of Drosophila sodium channels promotes modification by deltamethrin: Reductions in affinity caused by knock-down resistance mutations. J Gen Physiol 115: 305-318. doi:10.1085/jgp.115.3.305. PubMed: 10694259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tatebayashi H, Narahashi T (1994) Differential mechanism of action of the pyrethroid tetramethrin on tetradotoxin-sensitive and tetradotoxin-resistant sodium channels. J Pharmacol Exp Ther 270: 595-603. PubMed: 8071852. [PubMed] [Google Scholar]
- 28. Kulkarni SJ, Padhye A (1982) Temperature-sensitive paralytic mutations on the second and third chromosomes of Drosophila melanogaster. Genet Res 40: 191-199. doi:10.1017/S0016672300019054. [Google Scholar]
- 29. Hodges DD, Lee D, Preston CF, Boswell K, Hall LM et al. (2002) tipE regulates Na+-dependent repetitive firing in Drosophila neurons. Mol Cell Neurosci 19: 402-416. doi:10.1006/mcne.2001.1088. PubMed: 11906212. [DOI] [PubMed] [Google Scholar]
- 30. Jackson FR, Wilson SD, Hall LM (1986) The tip-E mutation of Drosophila decreases saxitoxin binding and interacts with other mutations affecting nerve membrane excitability. J Neurogenet 3: 1-17. doi:10.3109/01677068609106891. PubMed: 2420952. [DOI] [PubMed] [Google Scholar]
- 31. O’Dowd DK, Aldrich RW (1988) Voltage-clamp analysis of sodium channels in wild-type and mutant Drosophila neurons. J Neurosci 8: 3633-3643. PubMed: 2848103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song W, Liu Z, Tan J, Nomura Y, Dong K (2004) RNA editing generates tissue-specific sodium channels with distinct gating properties. J Biol Chem 279: 32554-32561. doi:10.1074/jbc.M402392200. PubMed: 15136570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song W, Liu Z, Dong K (2006) Molecular basis of differential sensitivity of insect sodium channels to DCJW, a bioactive metabolite of the oxadiazine insecticide indoxacarb. Neurotoxicology 27: 237-244. doi:10.1016/j.neuro.2005.10.004. PubMed: 16325912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin WH, Wright DE, Muraro NI, Baines RA (2009) Alternative splicing in the voltage-gated sodium channel DmNav regulates activation, inactivation, and persistent current. J Neurophysiol 102: 1994-2006. doi:10.1152/jn.00613.2009. PubMed: 19625535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goldin AL (2003) Mechanisms of sodium channel inactivation. Curr Opin Neurobiol 13: 284-290. doi:10.1016/S0959-4388(03)00065-5. PubMed: 12850212. [DOI] [PubMed] [Google Scholar]
- 36. Catterall WA (2002) Molecular mechanisms of gating and drug block of sodium channels. Novartis Found Symp 241: 206-218. PubMed: 11771647. [PubMed] [Google Scholar]
- 37. Chahine M, Chatelier A, Babich O, Krupp JJ (2008) Voltage-gated sodium channels in neurological disorders. CNS Neurol Disord Drug Targets 7: 144-158. doi:10.2174/187152708784083830. PubMed: 18537643. [DOI] [PubMed] [Google Scholar]
- 38. Xu R, Thomas EA, Gazina EV, Richards KL, Quick M et al. (2007) Generalized epilepsy with febrile seizures plus-associated sodium channel beta1 subunit mutations severely reduce beta subunit-mediated modulation of sodium channel function. Neuroscience 148: 164-174. doi:10.1016/j.neuroscience.2007.05.038. PubMed: 17629415. [DOI] [PubMed] [Google Scholar]
- 39. Vijayaragavan K, O’Leary ME, Chahine M (2001) Gating properties of Na(v)1.7 and Na(v)1.8 peripheral nerve sodium channels. J Neurosci 21: 7909-7918. PubMed: 11588164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Balser JR, Nuss HB, Chiamvimonvat N, Pérez-García MT, Marban E et al. (1996) External pore residue mediates slow inactivation in mu 1 rat skeletal muscle sodium channels. J Physiol 494: 431-442. PubMed: 8842002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Webb J, Wu FF, Cannon SC (2009) Slow inactivation of the NaV1.4 sodium channel in mammalian cells is impeded by co-expression of the beta1 subunit. Pflugers Arch 457: 1253-1263. doi:10.1007/s00424-008-0600-8. PubMed: 18941776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vilin YY, Makita N, George AL Jr, Ruben PC (1999) Structural determinants of slow inactivation in human cardiac and skeletal muscle sodium channels. Biophys J 77: 1384-1393. doi:10.1016/S0006-3495(99)76987-0. PubMed: 10465750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hong CS, Ganetzky B (1994) Spatial and temporal expression patterns of two sodium channel genes in Drosophila. J Neurosci 14: 5160-5169. PubMed: 8083728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen C, Cannon SC (1995) Modulation of Na+ channel inactivation by the beta 1 subunit: a deletion analysis. Pflugers Arch 431: 186-195. doi:10.1007/BF00410190. PubMed: 9026778. [DOI] [PubMed] [Google Scholar]
- 45. McCormick KA, Srinivasan J, White K, Scheuer T, Catterall WA (1999) The extracellular domain of the beta1 subunit is both necessary and sufficient for beta1-like modulation of sodium channel gating. J Biol Chem 274: 32638-32646. doi:10.1074/jbc.274.46.32638. PubMed: 10551818. [DOI] [PubMed] [Google Scholar]
- 46. Zhao J, O’Leary ME, Chahine M (2011) Regulation of Nav1.6 and Nav1.8 peripheral nerve Na+ channels by auxiliary beta-subunits. J Neurophysiol 106: 608-619. doi:10.1152/jn.00107.2011. PubMed: 21562192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)