

Abstract
Background
Bacterial products add to mechanical ventilation in enhancing lung injury. The role of endogenous triggers of innate immunity herein is less well understood. S100A8/A9 proteins are released by phagocytes during inflammation. The present study investigates the role of S100A8/A9 proteins in ventilator-induced lung injury.
Methods
Pulmonary S100A8/A9 levels were measured in samples obtained from patients with and without lung injury. Furthermore, wild-type and S100A9 knock-out mice, naive and with lipopolysaccharide-induced injured lungs, were randomized to 5 hours of spontaneously breathing or mechanical ventilation with low or high tidal volume (VT). In addition, healthy spontaneously breathing and high VT ventilated mice received S100A8/A9, S100A8 or vehicle intratracheal. Furthermore, the role of Toll-like receptor 4 herein was investigated.
Results
S100A8/A9 protein levels were elevated in patients and mice with lung injury. S100A8/A9 levels synergistically increased upon the lipopolysaccharide/high VT MV double hit. Markers of alveolar barrier dysfunction, cytokine and chemokine levels, and histology scores were attenuated in S100A9 knockout mice undergoing the double-hit. Exogenous S100A8/A9 and S100A8 induced neutrophil influx in spontaneously breathing mice. In ventilated mice, these proteins clearly amplified inflammation: neutrophil influx, cytokine, and chemokine levels were increased compared to ventilated vehicle-treated mice. In contrast, administration of S100A8/A9 to ventilated Toll-like receptor 4 mutant mice did not augment inflammation.
Conclusion
S100A8/A9 proteins increase during lung injury and contribute to inflammation induced by HVT MV combined with lipopolysaccharide. In the absence of lipopolysaccharide, high levels of extracellular S100A8/A9 still amplify ventilator-induced lung injury via Toll-like receptor 4.
Introduction
Acute lung injury (ALI) and its most severe form the acute respiratory distress syndrome (ARDS) is a devastating pulmonary condition with a high mortality rate, characterized by acute lung inflammation and edema [1]. Although mechanical ventilation (MV) is a lifesaving intervention in the management of these patients, it is well known that MV can contribute to the pathogenesis of ARDS [2]. Studies demonstrated that conventional MV could enhance but also initiate lung inflammation referred to as ventilator-induced lung injury (VILI) [2]–[4]. To date, low tidal volume (VT) MV is recommended. However, it has been reported that also MV with lung protective ventilator settings can cause (regional) hyperinflation and VILI [5]. The need for additional pharmacological interventions demands further research investigating potential new therapeutic targets.
Although the exact underlying mechanisms are incompletely elucidated, accumulating evidence indicates that mechanical stress and innate immunity pathways interact and compound lung injury [6]. One of the key findings in understanding the development of lung injury in the presence of MV was the discovery that microbial molecules, like lipopolysaccharide (LPS), have synergistic effects with MV in initiating or enhancing lung injury [7]–[9]. Innate immunity plays a central role in orchestrating pulmonary inflammation. Pathogen-associated molecular patterns are recognized by pattern recognition receptors and activation triggers an intense inflammatory response necessary to combat infection [10]. In the last decade it became clear that endogenous molecules released at sites of tissue injury, termed “alarmins” or “damage-associated molecular patterns” (DAMPs), also initiate and further activate innate immunity via the same receptors [10]. Of particular interest are S100A8 (also referred to as myeloid related protein 8) and S100A9 (also referred to as myeloid related protein 14) proteins, released into the extracellular space by activated phagocytes [11]. Indeed, neutrophils are among the first cells to infiltrate inflammatory regions and play a central role in the pathogenesis of ARDS and VILI [4]. The physiologically relevant form of S100A8 and S100A9 proteins is the S100A8/A9 complex in which S100A8 is thought to be the most active component [12]–[14]. S100A8/A9 levels correlate with disease activity in several inflammatory disorders, especially in rheumatoid arthritis, juvenile idiopathic arthritis, and inflammatory bowel disease [11]. Recently it became clear that these proteins are not only excellent biomarkers of inflammation, they also amplify the pro-inflammatory cascade via activation of innate immunity [12]; [15]. In lung tissue, up-regulation of S100A9 mRNA due to injurious MV was demonstrated [16]. Also, S100A8 and S100A9 proteins were detected in bronchoalveolar lavage fluid (BALF) of ARDS patients [17]–[19]. However, knowledge about the impact of S100A8/A9 proteins in inflamed ventilated lungs is limited.
We hypothesized that S100A8/A9 proteins are released during human and murine lung injury and contribute to the inflammatory response in a 2-hit model of LPS-induced lung injury combined with MV. In addition, we analyzed the effects of exogenous administered S100A8/A9 and S100A8 proteins in naïve mice, the impact of these proteins when combined with HVT MV, and the role of TLR4 herein.
Materials and Methods
Patients
The study represents a secondary analysis of a previous prospective nested case control study where the relation between transfusion and the onset of ALI was investigated [20]. The Medical Ethics Committee from the University of Amsterdam approved the study protocol and written informed consent was obtained from all patients. Cardiac surgery patients were observed for the onset of ALI up to 30 hours after the surgical procedure. At onset of ALI a non-directed lung lavage was performed. Patients without ALI who were lavaged within 30 hours of ICU admission served as controls. In total 16 cases of ALI and 62 controls were identified. Study design and methods were described in detail previously [20].
In addition, to illustrate S100A8/A9 presence we stained paraffin-embedded lung biopsies for S100A9. Sections were obtained from 12 ICU patients during autopsy: 5 patients died without ALI and 7 patients died with ALI. Lung sections shown were from 2 patients: both were admitted to the ICU with an intracerebral bleeding, 1 patient developed ALI, the other patient succumbed without ALI.
Mice
The Animal Care and Use Committee of the University of Amsterdam approved all experiments. Deficiency of the S100A8 gene in mice results in a lethal phenotype during embryogenesis [21]. Therefore S100A9 knockout (KO) mice, which lack both S100A9 and S100A8 proteins, despite normal S100A8 mRNA levels, were used [22]. It is thought that the turnover of isolated S100A8 is higher in the absence of its binding partner S100A9. Eight- to eleven week old S100A9 KO mice, generated as described previously [22] and backcrossed 10 times to a C57Bl/6 background, were bred in the animal facility of the Academic Medical Center of Amsterdam, The Netherlands. Age and sex matched wild-type (WT) mice were purchased from Harlan Sprague Dawley (Horst, The Netherlands) one week prior to the experiments and were also housed in the animal facility of the Academic Medical Center of Amsterdam.
Experimental Groups
1) WT versus S100A9 KO mice in (ventilator-induced) lung injury
Lungs of critically ill patients are exposed to diverse insults such as MV, infections, and systemic inflammation. These insults may interact and culminate in overwhelming lung inflammation often seen in ARDS patients. To study the role of S100A8/A9 proteins in these settings we used WT and S100A9 KO mice in a 1-hit and 2-hit lung injury model. Healthy animals and mice with pre-injured lungs, induced by 0.25 mg/kg lipopolysaccharide (LPS) (E. coli L4130, Sigma Aldrich) intranasally 1 hour before randomization, were assigned to a spontaneously breathing group (n = 6–8/group) or to a mechanically ventilated group (n = 6–8/group). Mice were ventilated with high (H)VT or with a relatively low (L)VT (see below MV strategy). After 5 hours all mice were sacrificed by exsanguination under general anesthesia. Lungs were used for histopathology, BALF, and lung tissue homogenates.
2) Exogenous S100A8/A9 or S100A8 in vivo
To study the impact of extracellular S100A8/A9 or S100A8 proteins in the lung, healthy WT mice received recombinant mouse S100A8/A9 (30 µg/mouse) or S100A8 (30 µg/mouse) or vehicle (PBS) intratracheal. Mice were subsequently randomized to a spontaneously breathing group (n = 6) or to HVT MV (n = 7). After 5 hours all mice were sacrificed by exsanguination under general anesthesia and lungs were used for BALF.
3) Exogenous S100A8/A9 in TLR4 mutant mice
Next, we analyzed the role of TLR4 in pulmonary S100A8/A9 signaling. For this we used C3H/HeN mice (WT mice) and C3H/HeJ mice, that have homozygous mutated TLR4 genes leading to a TLR4 null phenotype. At start of 5 hours of HVT MV, we administered S100A8/A9 proteins (30 µg/mouse) or vehicle (PBS) intratracheal to C3H/HeN mice (Charles River, Someren, the Netherlands) and C3H/HeJ mice (Jackson Laboratory, Bar Harbor, Maine). After 5 hours all mice were sacrificed by exsanguination under general anesthesia and lungs were used for BALF (n = 7–8/group).
Instrumentation and anesthesia
LPS, S100A8/A9, S100A8, or PBS were administered under 2–3% isoflurane anesthesia. In ventilated animals, a tracheotomy was performed and an Y–tube connector (1.0 mm outer diameter and 0.6 mm inner diameter, VBM Medizintechnik GmbH, Sulz am Neckar, Germany) was inserted into the trachea under general anesthesia with an intraperitoneal injection of KMA “induction”–mix: 7.5 µl per 10 gram of body weight of 1.26 ml 100 mg/ml ketamine, 0.2 ml 1 mg/ml medetomidine, and 1 ml 0.5 mg/ml atropine in 5 ml normal saline. Maintenance anesthesia consisted of 10 µl per 10 gram body weight of KMA “maintenance”-mix of 0.72 ml 100 mg/ml ketamine, 0.08 ml 1 mg/ml medetomidine and 0.3 ml 0.5 mg/ml atropine in 20 ml normal saline. Maintenance mix was administered via an intraperitoneal catheter (PE 10 tubing, BD, Breda, the Netherlands) hourly, every 30 minutes 0.2 ml sodium carbonate (200 mmol/l NaHCO3) was administered via the same intraperitoneal catheter throughout the experiment. Rectal temperature was maintained between 36.5–37.5°C using a warming pad. In a subset of the experiment heart rate and systolic blood pressure were non-invasively monitored using a murine tail-cuff system (ADInstruments, Spenbach, Germany). Directly after start of MV, after 2.5, and 5 hours of MV. Both remained stable throughout the experiment (See Data S1).
MV strategy
Methods of the murine MV-model used were published in detail previously [23]. Animals were placed in supine position and connected to a ventilator (Servo 900 C, Siemens, Sweden). During 5 hours mice were pressure controlled ventilated with an inspiratory pressure of 18 cm H2O (resulting in VT ∼15 ml/kg (HVT)) or with an inspiratory pressure of 10 cm H2O (resulting in VT∼7.5 ml/kg (LVT)) under general anesthesia. Respiratory rate was set at 70 or 110 breaths per minute respectively, PEEP was set at 2 cm H2O, the fraction of inspired oxygen was kept at 0.5, and inspiration to expiration ratio was set at 1∶1. A sigh (sustained inflation with 30 cm H2O) for 5 breaths was performed hourly. After 5 hours of MV mice were sacrificed under general anesthesia by withdrawing blood from the carotid artery. This was used for blood gas analysis in a subset of the experiment, demonstrating adequate gas exchange in ventilated animals with no differences between WT and KO mice (See Data S2).
Purification of S100A8 and S100A8/A9
Murine S100A8 and S100A9 proteins were purified as described earlier for the human S100 proteins [24]. To obtain heterodimer complexes, purified homodimers were denatured in 8 M urea and mixed in equal amounts. Renaturation was allowed during extensive dialysis from acid pH to neutral pH in different steps. Protein identification was performed by electrospray ionization mass spectrometry. Possible endotoxin contaminations were eliminated by Endotrap column and quantified by limulus amebocyte lysate assay (BioWhittaker) and in blocking experiments using polymyxin B (Sigma). Limulus amebocyte assay did not detect LPS in the protein preparations (sensitivity ∼5 pg/µg protein).
Sampling
BALF was performed by instilling three times 0.5 ml of saline into the trachea. Cell counts were determined using a Coulter cell counter (Beckman Coulter, Fullerton, CA), differential cell counts were performed on cytospin preparations stained with Giemsa stain. Supernatant was stored at −20°C for further measurements. For histology, lungs were fixed in 4% formalin, embedded in paraffin, 4 µm sections were stained with hematoxylin–eosin and analyzed by a pathologist who was blinded for group identities. To score lung injury, 4 pathologic parameters were scored on a scale of 0–4: (a) oedema, (b) haemorrhage, (c) interstitial infiltration and (d) hyaline membranes [23]. Total histology score was expressed as the sum of the score for all parameters. In addition, S100A8 and S100A9 stainings were performed on lung sections as described previously [22]. Lung tissue homogenates were prepared by homogenizing lungs in 4 volumes of sterile 0.9% NaCl and these samples were subsequently lysed in 1∶2 lysis buffer containing 300 mM NaCl, 30 mM Tris, 2 mM MgCl2, 2 mM CaCl2, 1% Triton x-100 and Pepstatin A, Leupeptin and Aprotinin (all 20 ng/ml; pH 7.4). Homogenates were centrifuged and supernatants were stored at −20°C until further analysis.
Assays
Total protein levels were determined in BALF using a Bradford Protein Assay Kit (OZ Biosciences, Marseille, France). Interleukin (IL)–6, IL–1β, tumor necrosis–factor (TNF)–α, keratinocyte–derived chemokine (KC) and macrophage inflammatory protein (MIP)-2 levels were measured by enzyme–linked immunosorbent assay (R&D systems, Mineapolis, MN). Detection limits were 51 pg/ml for KC, IL-6, TNF-α and IL-1β. MIP-2 had a detection limit of 153 pg/ml. Immunoglobulin M (IgM) levels were analyzed as previously described [25]. S100A8/A9 concentrations were measured by sandwich enzyme–linked immunosorbent assay as previously described: human [26], mouse [14].
Statistical analysis
All data are presented as mean ± SEM. Two group comparisons were analyzed with a student t-test or Mann Whitney U-test depending on data distribution (ARDS versus no ARDS and WT versus KO). A secondary analysis compared WT mice of control, HVT MV-only, LPS-only and HVT MV/LPS groups. For this we used analysis of variance in conjunction with Bonferroni post hoc testing or a Kruskal-Wallis test with Mann-Whitney U-test, depending on data distribution. For the experiments were intratracheal vehicle was compared with S100A8/A9 or S100A8 protein exposure in naïve and ventilated mice we also used analysis of variance with Bonferroni post hoc analysis or a Kruskal-Wallis in conjunction with a Mann-Whitney U-test, depending on data distribution. All statistical analyses were carried out using GraphPad Prism version 5 (Graphpad Software; San Diego, CA). P values<0.05 were considered significant.
Results
S100A8/A9 Levels Increase in Clinical and Experimental Lung Injury
In the prospective nested case control study, baseline characteristics of the cases (n = 16), and their controls (n = 62), were described in detail previously [20]. There were no differences in cardiac or pulmonary function between the groups pre-operatively. Patients who developed ALI were older, received more transfusions, and total operation time, clamptime, and pumptime were longer when compared to controls [20]. Also, PaO2/FiO2 ratios were reduced in ALI patients and several outcome parameters were also different: patients with ALI were mechanically ventilated longer and had a longer stay on the ICU and hospital (See Data S3) [20]. To determine whether levels of S100A8/A9 proteins increase during lung injury in these patients we analyzed S100A8/A9 concentrations in lavage samples. ALI patients had increased levels of S100A8/A9 proteins in BALF when compared to patients without ALI (fig. 1). In addition, we illustrated increased S100A9 presence in lung tissue of a patient who succumbed with ALI by immuno-histochemical staining, which was clearly more intense compared to an ICU patient who died without ALI (fig. 1b,c).
Figure 1. S100A8/A9 proteins increase in patients with mild ARDS.

Presence of S100A8/A9 proteins in lung lavage fluid of patients with acute lung injury (ALI) (n = 16) and patients without ALI (n = 62) (A). Representative immunohistochemical stainings of S100A9 (staining in red, background in blue) of human lung tissue obtained from an ICU patient without ALI (B) and with ALI (C). Data are shown as mean ± SEM. *P<0.05.
In our mouse model, we found that both HVT MV and LPS-induced injury resulted in significantly increased S100A8/A9 levels compared to non-ventilated control mice (fig. 2). The combination of both LPS and HVT MV resulted in synergistically increased concentrations of S100A8/A9 in BALF; levels were higher when compared to non-ventilated controls, HVT MV-only and LPS-only groups. To visualize S100A8 and S100A9 in the pulmonary compartment immuno-histochemical staining of mouse lung slides was performed. The expression of S100A8 and S100A9 increased with LPS administration or HVT MV separately (fig. 3). In line with the S100A8/A9 protein levels in BALF, the most intense staining was seen in lung tissue of mice that received both HVT MV and LPS. Higher magnification revealed that infiltrating neutrophils were the main S100A8 and S100A9-expressing cells (fig. 3). Healthy S100A9 KO mice lack both S100A9 and S100A8 proteins and thus biologically active S100A8/A9 heterodimers in myeloid cells [19]. For additional control purposes we also measured BALF S100A8 levels and stained lung tissue for S100A8 in S100A9 KO mice undergoing the 1 or 2-hit injury. S100A8 could not be detected in BALF. Moreover, in contrast to WT mice, no increased S100A8 staining was seen in KO mice undergoing the 1 or 2-hit injury (see Data S4).
Figure 2. S100A8/A9 protein levels increase in mice with lung injury.

S100A8/A9 levels in lung lavage fluid in a murine 2-hit lung injury model. Mice were spontaneously breathing (C), mechanically ventilated for 5 hours with high tidal volume (HVT MV), received intranasal lipopolysaccharide (LPS; 0.25 mg/kg) followed by 5 hours spontaneously breathing (LPS), or received intranasal LPS followed by 5 hours of HVT ventilation (HVT MV+LPS). Data represent mean ± SEM of 8 mice per group. ###p<0.001 versus control, ++p<0.01 versus MV, §§p<0.01 versus LPS.
Figure 3. S100A8/A9 presence in lung tissue increases in mice with lung injury.

Representative images of immunohistochemical stainings of S100A8 and S100A9 (specific staining in red, background staining in blue) of murine lung sections. Wild-type mice were spontaneously breathing (C), mechanically ventilated for 5 hours with high tidal volume (HVT MV), received intranasal lipopolysaccharide (LPS; 0.25 mg/kg) followed by spontaneously breathing for 5 hours (LPS), or received intranasal LPS followed by 5 hours of HVT mechanical ventilation (HVT MV+LPS). Magnification 10×, detailed view of the HVT MV+LPS group: magnification 100×.
S100A9 Deficient Mice are Partially Protected in a HVT MV/LPS 2-hit Lung Injury Model
To determine if the increased presence of S100A8/A9 proteins influenced lung injury and inflammation, we compared WT with S100A9 KO mice. HVT MV-only and LPS-only both induced lung injury and the HVT MV/LPS double-hit boosted lung injury: total protein levels, IgM concentrations, and neutrophil influx into the alveolar compartment were significantly increased when compared to control, HVT MV-only, and LPS-only groups (fig. 4). No differences between S100A9 KO and WT mice were found in the control, HVT MV-only and LPS-only groups. However, S100A9 KO mice undergoing the double hit demonstrated attenuated alveolar-epithelial permeability when compared to WT mice. This was demonstrated by a lower total protein content and IgM concentration in BALF. The same trend was seen for neutrophil influx although this did not reach statistical significance. To further analyze the lung inflammatory response in S100A9 KO mice, concentrations of cytokines and chemokines were measured in BALF. In line with the lung permeability measurements, inflammation was most severe in mice that were exposed to both LPS and overinflation. HVT MV/LPS double hit increased the concentrations of BALF IL-6, MIP-2, Il-1β, TNF-α, and KC compared to the control, HVT MV-only and LPS-only groups (fig. 5). As compared to WT mice, S100A9 KO mice had reduced levels of IL-6, MIP-2, IL-1β and TNF-α after both LPS and HVT MV (fig. 5). Furthermore, inflammation was also attenuated in the LPS-only group demonstrated by lower IL-6, KC, MIP-2, and TNF-α concentrations compared to WT mice. Again, no significant differences were found in cytokine concentrations of WT and KO mice in the control group, and the HVT MV-only group. In addition, we analyzed inflammation in lung parenchyma (See Data S5). In line with levels in BALF, IL-6, MIP-2 and TNF-α levels were significantly lower in lung tissue homogenates of S100A9 KO mice compared to WT mice of the HVT MV/LPS group. The difference between WT and KO mice of the LPS group was less prominent in lung parenchyma, only TNF-α concentrations were reduced in S100A9 KO mice. Next, we analyzed lung histopathology slides. Whereas no significant differences in histopathology scores were found between WT and KO mice of control, HVT MV-only, and LPS-only groups (table 1), histopathology scores of S100A9 KO mice undergoing the double hit were attenuated when compared to WT mice (table 1, fig. 6).
Figure 4. Barrier dysfunction is attenuated in S100A9 knockout mice undergoing a 2-hit lung injury model.
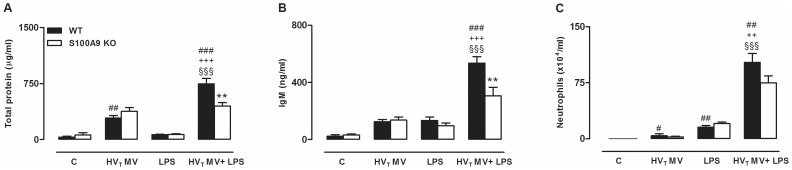
Total protein levels (A), immunoglobulin M (IgM) content (B) and neutrophil counts (C) in bronchalveolar lavage fluid of wild-type (WT) and S100A9 knockout (KO) mice. Animals were spontaneously breathing (C), high tidal mechanically ventilated (HVT MV), exposed to LPS followed by spontaneously breathing (LPS) or exposed to LPS followed by HVT mechanical ventilation (HVT MV+LPS). Data represent means (SEM) of 6–8 mice per group. **p<0.01 WT versus KO, ###p<0.001, ##p<0.01, #p<0.01 versus WT C. §§§p<0.001 versus LPS-only. +++p<0.001, ++p<0.01 versus HVT MV-only.
Figure 5. Inflammation is attenuated in S100A9 knockout mice undergoing a 2-hit lung injury model.

Cytokine and chemokine concentrations in lung lavage fluid of wild-type (WT) and S100A9 knockout (KO) mice. Animals were spontaneously breathing (C), high tidal mechanically ventilated (HVT MV), exposed to LPS followed by spontaneously breathing (LPS), or exposed to LPS followed by HVT mechanical ventilation (HVT MV+LPS). Levels of interleukin (IL)–6 (A), macrophage inflammatory protein (MIP)-2 (B), tumor necrosis factor-α (TNF-α) (C), IL-1β (D), and keratinocyte–derived chemokine (KC) (E) were determined. Data represent means (SEM) of 6–8 mice per group. *p<0.05, **p<0.01, ***p<0.001 KO versus WT mice, ###p<0.001, ##p<0.01, and #p<0.05 versus WT C, §§§p<0.001 and §§p<0.01 versus LPS-only, +++p<0.001, and ++p<0.01 versus HVT MV-only.
Table 1. Histopathology scores.
| Group | WT | S100A9 KO | |||
| C | 0.50 [0.3] | 1.50 [0.4] | |||
| HVT MV | 2.33 [0.3] | 2.67 [0.4] | |||
| LPS | 2.50 [0.6] | 2.14 [0.4] | |||
| HVT MV+LPS | 6.33 [0.61] | 4.43 [0.5]* | |||
Lung injury scores of wild-type (WT) and S100A9 knockout (KO) mice of the non-ventilated control group (C), high tidal mechanically ventilated group (HVT MV), LPS-exposed non-ventilated group (LPS), and LPS-exposed followed by mechanical ventilation group (HVT MV+LPS). Data represent mean ± [SEM] of n = 6–7 mice/group.
p<0.05 compared to WT mice of the HVT MV+LPS group.
Figure 6. Histopathological changes were reduced in S100A9 knockout mice undergoing 2-hit lung injury.
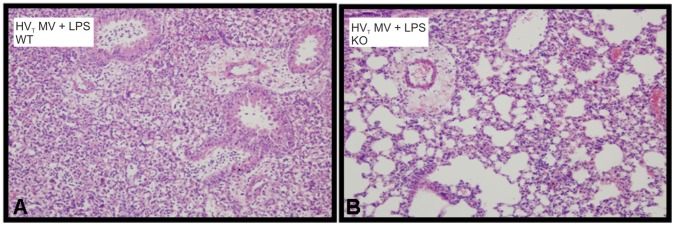
Representative histological sections of hematoxylin and eosin stained lungs of a wild-type (WT) mouse (A) and a S100A9 knockout (KO) (B) mouse exposed to lipopolysaccharide (LPS) and high tidal volume mechanical ventilation (HVT MV). Original magnification x20 (in set x40).
S100A8/A9 Proteins in LVT MV
After we have demonstrated that deficiency of S100A8/A9 reduces lung injury in overinflated inflamed lungs we analyzed the role of these proteins during LVT MV. Five hours of LVT MV induced S100A8/A9 proteins in BALF (See Data S6). Levels were higher in LPS-exposed LVT MV ventilated mice. Again, we analyzed total protein, IgM, neutrophil influx, and cytokines and chemokines in BALF. In mice with healthy lungs and also in animals with inflamed lungs, induced by LPS-inhalation, we observed no differences between WT and KO mice in these measures of injury and inflammation after 5 hours of LVT MV (table 2). These data indicate that S100A8/A9 proteins are less important in non-overstretched lung areas.
Table 2. S100A8/A9 in non-overstretched lung areas.
| Healthy | LPS-exposed | |||||||
| LVT WT | LVT KO | LVT WT | LVT KO | |||||
| Total protein (µg/ml) | 85 [17] | 64 [17] | 131 [15] | 164 [12] | ||||
| IgM (ng/ml) | 58 [13] | 30 [5] | 107 [11] | 126 [18] | ||||
| Neutrophils (×104/ml) | 4.1 [1.8] | 2.7 [0.8] | 25 [2.9] | 25 [2.9] | ||||
| IL-6 (pg/ml) | 206 [30] | 204 [38] | 532 [56] | 620 [63] | ||||
| KC (pg/ml) | 135 [26] | 189 [35] | 1382 [342] | 1767 [518] | ||||
| MIP-2 (pg/ml) | 81 [17] | 57 [19] | 1436 [439] | 1093 [266] | ||||
| IL-1β (pg/ml) | 62 [8] | 57 [18] | 152 [9] | 201 [35] | ||||
| TNF-α (pg/ml) | B.D. | B.D. | 614 [64] | 386 [77] | ||||
Total protein, immunoglobin M (IgM), neutrophil influx, cytokines and chemokines in bronchoalveolar lavage fluid of wild-type (WT) and S100A9 knockout (KO) mice. Animals with healthy lungs or with pre-existing lung injury induced by lipopolysaccharide exposure were ventilated for 5 hours with low tidal volumes (LVT) (∼7.5 ml/kg). Data represent mean (SEM) of n = 6–8 mice/group. Below detection limit: B.D.
Exogenous S100A8/A9 Proteins Induce Mild Lung Inflammation in Healthy Mice
In a second set of experiments we analyzed if S100A8/A9 proteins could elicit inflammation in otherwise healthy lungs. Within 5 hours, S100A8/A9 and S100A8 proteins induced neutrophil recruitment into the alveolar compartment (fig. 7). Total protein levels were not affected by S100A8/A9 or S100A8 protein instillation but BALF IgM levels were elevated. Cytokines and chemokines were not significantly affected. Additionally, no differences were detected between S100A8/A9 and S100A8 instillation.
Figure 7. Exogenous S100A8/A9 proteins amplify MV-induced lung inflammation.

Neutrophil influx (A), Total protein (B), immunoglobulin M (IgM) (C), interleukin (IL)-6 (D), keratinocyte-derived chemokine (KC) (E), macrophage inflammatory protein (MIP)-2 (F), IL-1β (G) and tumor necrosis factor (TNF)-α concentrations (H). Mice received S100A8/A9 (30 µg/mouse) or S100A8 (30 µg/mouse) or vehicle intra tracheal at start of five hours of spontaneously breathing (C) or high tidal volume mechanical ventilation (MV). Data represent mean (SEM) of 5–7 mice per group. *p<0.05, **p<0.01, ***p<0.001 versus vehicle treated mice. +p<0.05 S100A8 versus S100A8/A9 treated mice. #p<0.05, ##p<0.01 ventilated vehicle treated versus control vehicle treated mice.
S100A8/A9 Proteins have Synergistic Effects with Mechanical Stress
To determine if S100A8/A9 have additive effects during VILI in the absence of LPS, we administered these proteins also to naive mice and ventilated them with the HVT MV strategy. S100A8/A9 and S100A8 exposure both resulted in significantly more neutrophil influx into the alveolar compartment compared to vehicle-treated mice (fig. 7). Total protein levels were not influenced by administration of S100 proteins. IgM levels however, tended to be higher reaching significance for S100A8/A9 exposed mice. Concentrations of the inflammatory mediators IL-6, IL-1β, MIP-2, and TNF-α were significantly increased in BALF due to exogenous S100A8/A9 and S100A8 compared to vehicle treated ventilated mice. S100A8-exposed ventilated mice tended towards more inflammation which was significant for the IL-1β levels. These experiments suggest that when pulmonary S100A8/A9 or S100A8 levels are highly increased, these proteins have additive effects in overinflated lung areas in enhancing pulmonary inflammation.
S100A8/A9 Proteins Aggravate VILI via TLR4 Signaling
To evaluate if TLR4 signaling was required for S100A8/A9-induced aggravation of VILI we analyzed pulmonary inflammation in TLR4 mutant mice. First, we tested if presence of S100A8/A9 also aggravates HVT MV-induced inflammation in the C3H mouse strain. We observed, in line with the results above, significantly more neutrophils in the alveolar compartment in ventilated S100A8/A9 exposed C3H/HeN mice compared to vehicle exposed animals (fig. 8). Also, total protein, IgM, IL-6, IL-1β, MIP-2, KC, and TNF-α levels were increased in BALF by S100A8/A9 exposure. Strikingly, S100A8/A9 administration to ventilated C3H/HeJ mice did not affect any of the inflammatory parameters compared to vehicle exposed C3H/HeJ mice (fig. 8). These data clearly reveal that the TLR4 pathway is required for S100A8/A9 proteins to aggravate VILI. Noteworthy, we also found differences between neutrophil influx and cytokine and chemokine levels when comparing ventilated vehicle exposed C3H/HeN with C3H/HeJ mice which again underscores the importance of TLR4 signaling in VILI.
Figure 8. Exogenous S100A8/A9 proteins amplify VILI via Toll-like receptor 4.

Total protein (A), immunoglobulin M (IgM) (B) Neutrophil influx (C), interleukin (IL)-6 (D), keratinocyt-derived chemokine (KC) (E), macrophage inflammatory protein (MIP)-2 (F), IL-1β (G) and tumor necrosis factor (TNF)-α concentrations (H) in C3H-HeN and C3H-HeJ mice exposed to S100A8/A9 (30 µg/mouse) or vehicle intratracheally at start of high tidal volume mechanical ventilation. Mice were ventilated for 5 hours. Data represent mean (SEM) of 7–8 mice per group. *p<0.05, **p<0.01, ***p<0.001 versus vehicle treated mice. #p<0.05 S100A8 versus S100A8/A9 treated mice.
Discussion
The excessive pulmonary inflammatory response in ALI/ARDS is complex and knowledge about key modulators is limited. Here we reported that S100A8/A9 is an important DAMP able to amplify pulmonary inflammation during HVT MV: (1) S100A8/A9 proteins were increased in lungs of ALI patients and in mice with LPS- or MV-induced lung injury, (2) S100A8/A9 levels were synergistically increased upon HVT MV/LPS double hit and targeted deletion of S100A9 in this group attenuated pulmonary permeability and inflammation, (3) in gain of function experiments extracellular S100A8/A9 induced mild neutrophil influx in healthy recipients, and (4) in the absence of LPS S100A8/A9 clearly aggravated VILI via TLR4 dependent mechanisms.
The tidal volumes used in our mouse model are higher then those normally used in clinical practice. However, although the use of low tidal volume ventilation (6 ml/kg body weight) in ARDS is widely accepted, a recent multicenter multinational study indicated that higher tidal volumes (>8 ml/kg body weight) in patients with evidence for ARDS are still used [27]. More importantly, computed tomography studies demonstrated that during ARDS lungs are non-uniformly expanded [28]. Extensive areas are fluid filled and collapsed resulting in a reduced effective alveolar volume. Even the use of low tidal volume ventilation can lead to hyperinflation in less-affected lung areas, indicating that VILI is a regional phenomenon [5]. Studies on the effect of hyperinflation are therefore still relevant to reveal information on lung injury caused by MV, as hyperinflation is an important factor contributing to the development of VILI.
S100A8/A9 proteins are highly expressed in neutrophils, representing 40% of the cytosolic protein content [11]. These proteins are also found in monocytes, early differentiation stages of macrophages, and can be induced under inflammatory conditions in keratinocytes and epithelial cells [11]. Thirty minutes of injurious MV in rats resulted in pulmonary S100A9 mRNA up-regulation [16] and LPS stimulation of bronchial epithelial cells in vitro increased S100A8 and S100A9 protein release [29]. In addition, we here report that S100A8/A9 protein levels in BALF also elevate during an in vivo murine model of VILI and LPS-exposure. Our human data demonstrate high pulmonary S100A8/A9 levels in ICU patients suffering from ALI, which is in line with previous findings [17]–[19]. Although these results cannot be directly linked to our animal experiments since the settings that led to lung injury development were not similar, our patient study does illustrate once more that human lung injury is associated with a local rise in S100A8/A9 levels [17]–[19].
Studies in mice without pre-existing lung injury showed that VILI is in part dependent on TLR4 and the inflammasome [30]–[33]. It is currently unclear how TLR4 signaling is activated during VILI but evidence suggests an important role for endogenous ligands [30]. MV can result in the release of TLR4 activating DAMPs such as hyaluronan, high mobility group box-1, and heat shock proteins [34]. In addition, we observed that S100A8/A9 is released during MV. However, elimination of only S100A8/A9 had no effect on MV-induced VILI. In contrast, LPS-induced lung injury resulted in differences between WT and KO mice in reduction of several inflammatory parameters. IL-6, KC, MIP-2, and TNF-α in BALF were lower in KO mice, consistent with a current view that S100A8/A9 amplifies LPS-induced TLR4 activation [14].
The current theory for development of ARDS suggests a 2-hit mechanism [4]. The lung can be ‘primed’ by a direct insult such as pneumonia, aspiration, major surgery, and trauma, which sets the balance for an increased inflammatory response to a second insult such as MV. Previous experimental studies and the results presented in this manuscript indeed show synergistic interactions between innate immunity and MV: in the presence of microbial products the inflammatory response towards HVT MV is enhanced [7]–[9]. Presence of the DAMP S100A8/A9 highly increased upon the HVT MV/LPS double hit. We demonstrated here that these proteins are not only a marker of increased lung damage, they contributed to injury and inflammation in this 2-hit setting.
A hallmark of ARDS is loss of alveolar-capillary membrane barrier function resulting in increased vascular permeability [1]. A previous in vitro study reported S100A8/A9 proteins to induce endothelial disintegration and have cytotoxic effects contributing to endothelial damage [35]. In line, we observed reduced alveolar-capillary membrane permeability in S100A9 deficient mice, demonstrated by lower total protein content and IgM levels in BALF. S100A9KO mice undergoing the HVT MV/LPS double hit also demonstrated attenuated histopathological changes and lower cytokine and chemokine levels in BALF and lung tissue compared to WT mice.
To further characterize the extracellular contribution of these proteins in lung inflammation we administered exogenous proteins to healthy animals. In vitro it was previously demonstrated that S100A8 alone, in the absence of LPS, was capable of inducing TNF-α expression in bone marrow cells [14]. The S100A8/A9 complex had only additive effects in combination with LPS stimulation. Our in vivo experiments demonstrate that S100A8/A9 and S100A8 alone induce neutrophil recruitment in lungs of naive mice. These data are supported by other in vivo experiments where chemotactic features of the S100A8/A9 complex and S100A8 were shown in an arthritis air pouch model in mice [36]–[37]. In LPS-models however, the administration of S100A8/A9 proteins has led to conflicting results. It has been demonstrated that S100A8/A9 and S100A8 promote LPS-induced shock in mice [14], but a protective effect of extracellular S100A8/A9 on LPS-induced liver damage has also been reported [38]. Very recently it was shown that S100A8 administration attenuated inflammation and injury in a mouse model of endotoxemia [39]. These results suggest that S100A8/A9 proteins might have a dual role in inflammation depending on experimental setting (extent of inflammation, time or dosis of S100 exposure etc.). Our experiments demonstrated that in the absence of LPS, high levels of S100A8/A9 and S100A8 have synergistic effects with HVT MV and markedly enhance VILI. This provides evidence that the interaction between MV and innate immunity is not restricted for bacterial products and that in the inflamed lung, in the absence of infection, endogenous stimuli also amplify the inflammatory response to HVT MV.
In line with prior reports we observed that VILI is attenuated in TLR4 mutant mice [30]; [31]; [40]. Using TLR4 deficient mice it was previously clearly demonstrated that TLR4 mediates VILI in healthy animals [30]–[31] and also inflammation in HVT ventilated LPS challenged mice [40]. This manuscript also demonstrates that TLR4 plays a crucial role in S100A8/A9 induced aggravation of VILI, underscoring again the importance of this innate immune receptor in VILI. Animal studies have shown that MV increases the expression of pulmonary TLR4 [30]; [41]. It can be speculated that the up-regulated expression of TLR4 in overstretched lung tissue makes the lung more sensitive for high levels of S100A8/A9 proteins.
In a murine heart failure model it was demonstrated that S100A8/A9 proteins also activate the receptor for advanced glycation end products (RAGE) [15]. RAGE is an innate immune receptor that recognizes multiple ligands, including DAMPs like S100A8/A9, S100A12, and high mobility group box-1 [42]. RAGE is highly expressed by human and murine lung tissue and therefore an interesting receptor for future VILI research. In this manuscript we did not study the role of RAGE. However, since the S100A8/A9-induced aggravation of VILI was already significantly diminished in TLR4 mutant mice we believe that the influence of RAGE signaling was not major in our model. In the mouse there is no S100A12 and thus it might very well be possible that in the human situation, there is a role for either RAGE or S100A12. Hence, additional research is needed to study both the RAGE axis and S100A12.
The DAMP S100A12 is another member of the S100 family of proteins and human studies indicate that S100A12 levels are elevated during lung inflammation [17]; [19]. Moreover, it was demonstrated that despite similarly elevated levels of S100A8/A9, S100A12 was more increased in ARDS compared to levels in BALF obtained from cystic fibrosis patients [19]. Although this study was limited by the comparison of adult ARDS patients with pediatric cystic fibrosis patients and the fact that BALF was obtained from cystic fibrosis patients during a bronchoscopy performed because of increased respiratory symptoms suggestive of new infection, the difference in expression ratio suggest an important role for S100A12 in the onset of acute neutrophilic lung inflammation [19]. S100A12 can induce ROS, cytokines, and activate RAGE and is therefore considered to be a pro-inflammatory mediator [42]. Again, the function of S100A12 is however difficult to study in murine models since mice do not express S100A12. However, it was recently reported that transgenic mice expressing human S100A12 in a model of allergic pulmonary inflammation did not have increased lung inflammation [43]. These unexpected findings underscore the complexity of the role of S100 proteins and a more pleiotropic role of S100A12 in modulating inflammation was suggested [43]. Whether the lack of S100A12 in mice also influences the role of S100A8/A9 proteins is not known, research studying these proteins should therefore be interpreted with caution.
Increased lung injury may subsequently result in a further rise of S100A8/A9 levels as demonstrated here and it can be hypothesized that this uncontrolled loop of DAMP-mediated inflammation is relevant in ARDS development. Disrupting the S100A8/A9 signaling pathway with S100A8/A9 blocking antibodies would be an attractive approach to attenuate pulmonary inflammation. It has been demonstrated that passive immunization with anti-S100A8 and anti-S100A9 inhibits the accumulation of neutrophils in response to monosodium urate crystals in a murine air-pouch model [36]. In contrast, in a mouse model of lung inflammation induced by LPS inhalation had anti-S100A8 only a weak anti-neutrophil recruitment effect and anti-S100A9 had no effect at all [44]. These results plead against a major therapeutic potential. Others have speculated that the DAMP function of S100A8/A9 proteins is only elucidated in situations with sufficient cell death as S100A8/A9 proteins in the extracellular space function as a danger signal for the immune system [45]. LPS-induced lung inflammation would not have resulted in enough cell death and therefore the antibodies did not function. In line, in a mouse model of streptococcal pneumonia anti-S100A8 and anti-S100A9 caused neutrophil and macrophage recruitment to alveoli to diminish by 70–80% [45]. It has previously been demonstrated that VILI can lead to cell death [4]. Whether these antibodies work in VILI models and have the potential to attenuate the inflammatory response is an interesting subject for further research.
Different studies have indicated that S100A8/A9 proteins also have bactericidal activity [46]–[47]. The innate immune response may herein act as a double edged sword; it is important to fight intruding pathogens, but on the other hand, overwhelming inflammation can cause tissue damage. The presence of S100A8/A9 can severely increase the inflammatory response induced by HVT MV. Future research may study if the reduction in MV-induced injury comes at a cost of an increased susceptibility to live bacterial challenge.
Our study has limitations. First, the longer operation time, clamptime and pumptime of the cardiac surgery patients that developed ALI may also have influenced S100A8/A9 protein levels. For instance, it was previously shown that S100A8/A9 plasma levels are increased during cardiac surgery [48]. In our study S100A8/A9 levels were measured locally in BALF. We believe that this rise is a result of the inflammatory response within the lung and not due to systemic production. Second, since multiple factors may have influenced ALI development in patients a direct comparison between the mouse and the human studies is only partly possible. We used healthy young animals in our murine model and lung injury was solely induced by injurious ventilation combined with LPS-exposure. Third, we restricted our analysis to well-known parameters of lung inflammation and injury. Effects of S100A8/A9 proteins on lung mechanics were not measured.
Taken together, our data clearly demonstrate that S100A8/A9 proteins increase during lung injury and contribute to pulmonary inflammation in a 2-hit setting of HVT MV combined with LPS. Moreover, high levels of S100A8/A9 have synergistic effects with HVT MV, amplifying VILI via TLR4. These results may be translated into novel ARDS therapies in which dysregulated inflammation is attenuated by targeting endogenous S100A8/A9 proteins.
Supporting Information
Data S1 demonstrate blood pressures and heart rate throughout the experiment.
(DOC)
Data S2 demonstrate blood gas analysis.
(DOC)
Data S3 demonstrate PaO2/FiO2 ratios and outcome parameters of patients with ALI.
(DOC)
Data S4 demonstrate S100A8 stainings of lungs of S100A9 KO mice.
(DOC)
Data S5 demonstrate cytokine and chemokine concentrations in lung tissue homogenates.
(DOC)
Data S6 demonstrate S100A8/A9 levels in mice ventilated with low tidal volume.
(DOC)
Funding Statement
This work was supported by a grant to CW from The Netherlands Organization for Scientific Research, The Hague, The Netherlands, grant number 91676096, by the Interdisciplinary Center of Clinical Research grant Vo2/014/09 to TV as well as grant Ro2/004/10 to JR, by the German Research Foundation (DFG) CRC Transregio 128-A2, CRC 1009 B8 to TV and by the Federal Ministry of Education and Research, Germany, project AID-Net to JR and TV. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Matthay MA, Ware LB, Zimmerman GA (2012) The acute respiratory distress syndrome. J Clin Invest 122: 2731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The Acute Respiratory Distress Syndrome Network (2000) Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342: 1301–1308. [DOI] [PubMed] [Google Scholar]
- 3. Determann RM, Royakkers A, Wolthuis EK, Vlaar AP, Choi G, et al. (2010) Ventilation with lower tidal volumes as compared with conventional tidal volumes for patients without acute lung injury: A preventive randomized controlled trial. Crit Care 14: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dos Santos CC, Slutsky AS (2006) The contribution of biophysical lung injury to the development of biotrauma. Ann Rev Physiol 68: 585–618. [DOI] [PubMed] [Google Scholar]
- 5. Terragni PP, Rosboch G, Tealdi A, Corno E, Menaldo E, et al. (2007) Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med 175: 160–166. [DOI] [PubMed] [Google Scholar]
- 6. Martin TR (2008) Interactions between mechanical and biological processes in acute lung injury. Proc Am Thorac Soc 5: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, et al. (2005) Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol 175: 3369–3376. [DOI] [PubMed] [Google Scholar]
- 8. Moriyama K, Ishizaka A, Nakamura M, Kubo H, Kotani T, et al. (2004) Enhancement of the endotoxin recognition pathway by ventilation with a large tidal volume in rabbits. Am J Physiol Lung Cell Mol Physiol 286: L1114–L1121. [DOI] [PubMed] [Google Scholar]
- 9. Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS (1997) Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 99: 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81: 1–5. [DOI] [PubMed] [Google Scholar]
- 11. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, et al. (2012) Alarmins: awaiting a clinical response. J Clin Invest 122: 2711–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vogl T, Roth J, Sorg C (1999) Calcium-induced noncovalently linked tetramers of MRP8 and MRP14 detected by ultraviolet matrix-assisted laser desorption/ionization mass spectrometry. J Am Soc Mass Spectrom 10: 1124–1130. [DOI] [PubMed] [Google Scholar]
- 13. Vogl T, Ludwig S, Goebeler M, Strey A, Thorey IS, et al. (2004) MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 104: 4260–4268. [DOI] [PubMed] [Google Scholar]
- 14. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, et al. (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13: 1042–1049. [DOI] [PubMed] [Google Scholar]
- 15. Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, et al. (2012) S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol 107: 1–116. [DOI] [PubMed] [Google Scholar]
- 16. Copland IB, Kavanagh BP, Engelberts D, McKerlie C, Belik J, et al. (2003) Early changes in lung gene expression due to high tidal volume. Am J Respir Crit Care Med 168: 1051–1059. [DOI] [PubMed] [Google Scholar]
- 17. Chang DW, Hayashi S, Gharib SA, Vaisar T, King ST, et al. (2008) Proteomic and computational analysis of bronchoalveolar proteins during the course of the acute respiratory distress syndrome. Am J Respir Crit Care Med 178: 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Torre C, Ying SX, Munson PJ, Meduri GU, Suffredini AF (2006) Proteomic analysis of inflammatory biomarkers in bronchoalveolar lavage. Proteomics 6: 3949–3957. [DOI] [PubMed] [Google Scholar]
- 19. Lorenz E, Muhlebach MS, Tessier PA, Alexis NE, Hite RD, et al. (2007) Different expression ratio of S100A8/A9 and S100A12 in acute and chronic lung disease. Respiratory Medicine 102: 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vlaar AP, Hofstra JJ, Determann RM, Veelo DP, Paulus F, et al. (2011) The incidence, risk factors, and outcome of transfusion-related acute lung injury in a cohort of cardiac surgery patients: a prospective nested case-control study. Blood 117: 4218–4225. [DOI] [PubMed] [Google Scholar]
- 21. Passey RJ, Williams E, Lichanska AM, Wells C, Hu S, et al. (1999) A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. J. Immunol. 163: 2209–2216. [PubMed] [Google Scholar]
- 22. Manitz MP, Horst B, Seeliger S, Strey A, Skryabin BV, et al. (2003) Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol 23: 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wolthuis EK, Vlaar AP, Choi G, Roelofs JJTH, Juffermans NP, et al. (2009) Mechanical ventilation using non-injurious ventilation settings causes lung injury in the absence of pre-existing lung injury in healthy mice. Crit Care 13: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vogl T, Leukert L, Barczyk K, Strupat K, Roth J (2006) Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim. Biophys. Acta 1763: 1298–1306. [DOI] [PubMed] [Google Scholar]
- 25. Reijmers RM, Groen RW, Kuil A, Weijer K, Kimberley FC, et al. (2011) Disruption of heparan sulfate proteoglycan conformation perturbs B-cell maturation and APRIL-mediated plasma cell survival. Blood 2011 117: 6162–6171. [DOI] [PubMed] [Google Scholar]
- 26. Frosch M, Strey A, Vogl T, Wulffraat NM, Kuis W, et al. (2000) Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum 43: 628–637. [DOI] [PubMed] [Google Scholar]
- 27. Metnitz PG, Metnitz B, Moreno RP, Bauer P, Del Sorbo L, et al. (2009) Epidemiology of mechanical ventilation: analysis of the SAPS 3 database. Intensive Care Med 35: 816–825. [DOI] [PubMed] [Google Scholar]
- 28. Gattinoni L, Presenti A, Torresin A, Baglioni S, Riviolta M, et al. (1986) Adult respiratory distress syndrome profiles by computed tomography. J Thorac Imaging 1: 25–30. [DOI] [PubMed] [Google Scholar]
- 29. Henke MO, Renner A, Rubin BK, Gyves JI, Lorenz E, et al. (2006) Up-regulation of S100A8 and S100A9 protein in bronchial epithelial cells by lipopolysaccharide. Exp Lung Res 32: 331–347. [DOI] [PubMed] [Google Scholar]
- 30. Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ (2008) Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 109: 465–472. [DOI] [PubMed] [Google Scholar]
- 31. Li H, Su X, Yan X, Wasserloos K, Chao W, et al. (2010) Toll-like receptor 4-myeloid differentiation factor 88 signaling contributes to ventilator-induced lung injury in mice. Anesthesiology 113: 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuipers MT, Aslami H, Janczy JR, van der Sluijs KF, Vlaar AP, et al. (2012) Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology 116: 1104–1115. [DOI] [PubMed] [Google Scholar]
- 33. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, et al. (2012) Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 185: 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuipers MT, van der Poll T, Schultz MJ, Wieland CW (2011) Bench-to-bedside review: Damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care 15: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Viemann D, Barczyk K, Vogl T, Fischer U, Sunderkötter C, et al. (2007) MRP8/MRP14 impairs endothelial integrity and induces a caspase-dependent and -independent cell death program. Blood 109: 2453–2460. [DOI] [PubMed] [Google Scholar]
- 36. Ryckman C, McColl SR, Vandal K, de Medicis R, Lussier A, et al. (2003) Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum 48: 2310–2320. [DOI] [PubMed] [Google Scholar]
- 37. Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA (2003) Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 170: 3233–3242. [DOI] [PubMed] [Google Scholar]
- 38. Ikemoto M, Murayama H, Itoh H, Totani M, Fujita M, et al. (2007) Intrinsic function of S100A8/A9 complex as an anti-inflammatory protein in liver injury induced by lipopolysaccharide in rats. Clinica Chimica Acta 376: 197–204. [DOI] [PubMed] [Google Scholar]
- 39. Sun Y, Lu Y, Engeland CG, Gordon SC, Sroussi HY (2013) The anti-oxidative, anti-inflammatory, and protective effect of S100A8 in endotoxemic mice. Mol Immunol. 53: 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu G, Malik AB, Minshall RD (2010) Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 38: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Villar J, Cabrera NE, Casula M, Flores C, Valladares F, et al. (2010) Mechanical ventilation modulates TLR4 and IRAK-3 in a non-infectious, ventilator-induced lung injury model. Respir Res 11: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo WA, Knight PR, Raghavendran K (2012) The receptor for advanced glycation end products and acute lung injury/acute respiratory distress syndrome. Intensive Care med. 38: 1588–1598. [DOI] [PubMed] [Google Scholar]
- 43. Hofmann Bowman MA, Heydemann A, Gawdzik J, Shilling RA, Camoretti-Mercado B (2013) Transgenic expression of human S100A12 induces structural airway abnormalities and limited lung inflammation in a mouse model of allergic inflammation. Clinical & Experimental Allergy 41: 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bozinovski S, Cross M, Vlahos R, Jones JE, Hsuu K, et al. (2005) S100A8 chemotactic protein is abundantly increased, but only a minor contributor to LPS-induced, steroid resistant neutrophilic lung inflammation in vivo. J Proteome Res. 4: 136–145. [DOI] [PubMed] [Google Scholar]
- 45.Raquil MA, Anceriz N, Rouleau P, Tessier PA (2008) Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J immunol 180; 3366–3374. [DOI] [PubMed]
- 46. Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, et al. (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319: 962–965. [DOI] [PubMed] [Google Scholar]
- 47. Achouiti A, Vogl T, Urban CF, Röhm M, Hommes TJ, et al. (2012) Myeloid-related protein-14 contributes to protective immunity in gram-negative pneumonia derived sepsis. PLoS Pathog 8: e1002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boelke E, Storck M, Buttenschoen K, Berger D, Hannekum A (2000) Endotoxemia and mediator release during cardiac surgery. Angiology 51: 743–749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 demonstrate blood pressures and heart rate throughout the experiment.
(DOC)
Data S2 demonstrate blood gas analysis.
(DOC)
Data S3 demonstrate PaO2/FiO2 ratios and outcome parameters of patients with ALI.
(DOC)
Data S4 demonstrate S100A8 stainings of lungs of S100A9 KO mice.
(DOC)
Data S5 demonstrate cytokine and chemokine concentrations in lung tissue homogenates.
(DOC)
Data S6 demonstrate S100A8/A9 levels in mice ventilated with low tidal volume.
(DOC)