

Abstract
Tamoxifen is widely used to treat estrogen receptor (ER)-positive breast cancer. Recent findings that tamoxifen and its derivative 4-dehydroxy-tamoxifen (OHT) can exert ER-independent cytotoxic effects have prompted the initiation of clinical trials to evaluate its use in ER-negative malignancies. For example, tamoxifen and OHT exert cytotoxic effects in malignant peripheral nerve sheath tumors (MPNSTs) where estrogen is not involved. In this study, we gained insights into the ER-independent cytotoxic effects of OHT by studying how it kills MPNST cells. Although caspases were activated following OHT treatment, caspase inhibition provided no protection from OHT-induced death. Rather, OHT-induced death in MPNST cells was associated with autophagic induction and attenuated by genetic inhibition of autophagic vacuole formation. Mechanistic investigations revealed that OHT stimulated K-Ras degradation through autophagy induction, which is critical for survival of MPNST cells. Similarly, we found that OHT induced K-Ras degradation in breast, colon, glioma and pancreatic cancer cells. Our findings describe a novel mechanism of autophagic death triggered by tamoxifen and OHT in tumor cells that may be more broadly useful clinically in cancer treatment.
Keywords: autophagy, K-Ras, MPNST, tamoxifen, PKC
Introduction
The triphenylethylene compound tamoxifen is a non-steroidal selective estrogen receptor modulator (SERM) which is used as the first-line treatment for estrogen receptor (ER)-positive breast cancer (1). While its principal target is the ER (2), tamoxifen, at micromolar concentrations, exerts cytotoxic effects that are not reversed by estrogen addition (3). In fact, multiple non ER –mediated mechanisms have been implicated in death induced by tamoxifen and its hydroxylated derivative 4 – hydroxy tamoxifen (OHT). These mechanisms include changes in intracellular calcium (4); modulation of protein kinase C (PKC) (5;6); changes in calmodulin activity (7); and signaling though MAP kinases (8). The diverse target range of tamoxifen and its ER-independent effects have prompted its inclusion in clinical trials for multiple solid tumor types including ER-negative malignancies (9;10). In patients with largely inoperable and recurrent malignant gliomas, there seems to be a consistent relationship between higher doses of tamoxifen and higher radiographic response rates and longer survival (11). Tamoxifen has also been shown to augment the effects of cisplatin (12). However, a meta analysis of 6 randomized trials for metastatic melanoma reveals no significant change in the survival rates following tamoxifen treatment (13). Currently, there are ongoing trials using tamoxifen in combination with chemotherapy for stage 3 melanoma and metastatic bladder cancer.
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive sarcomas that have a poor prognosis, in part because no effective chemotherapeutic options are available (14;15). MPNSTs are the most common malignancy associated with neurofibromatosis type 1 (NF1) (16). As the NF1 gene codes for neurofibromin, a Ras GTPase activating protein (GAP), loss of this gene results in Ras hyperactivation in NF1-associated tumor types (17). Recently, we demonstrated that tamoxifen could potently inhibit malignant peripheral nerve sheath tumor (MPNST) growth in vivo (18). While MPNST cell lines express estrogen receptors, ablation of these receptors had no effect on OHT-induced cytotoxicity, indicating an ER-independent mechanism of action. Therefore, the overall goal of this study was to delineate the mechanism of OHT-induced cytotoxicity in MPNST cells.
OHT has been reported to induce apoptosis in tumor cells through activation of multiple upstream pathways. However, in addition to apoptotic features, cells dying in response to OHT treatment also display large scale autophagic vacuole (AV) accumulation, suggesting a possible role for autophagy in the regulation of OHT-induced death (19;20). Autophagy is a cellular catabolic pathway that targets long lived proteins and cellular content for degradation and mediates their recycling. Therapy-induced autophagy has been previously implicated in the regulation of cancer cell survival (21). It can play a pro-survival role and mediate resistance to therapy (22-25). Alternatively, autophagy can initiate and directly cause cell death (26). However, the precise mechanism by which therapy-induced autophagy causes cell death is poorly defined.
In this study, we demonstrate that OHT-induced death of MPNST cells is mediated by autophagy induction, not caspase-dependent apoptosis. This is achieved, at least in part, through degradation of K-Ras, a critical pro-survival protein previously identified as a regulator of tamoxifen sensitivity (27). Interestingly, the Ras pathway has been implicated as a determinant of the clinical effectiveness of tamoxifen therapy in breast cancer patients (28). Our findings identify a novel mechanism for autophagy mediated death and also a previously unreported mechanism for OHT-induced cytotoxicity in tumor cells. This study also sheds light on the role of Ras stability in mediating tumor cell response to tamoxifen treatment.
Materials and Methods
Antibodies and Other Reagents
Primary antibodies were obtained from the following sources: H-Ras and N-Ras (Santa Cruz Biotechnology Inc., Santa Cruz, CA); JNK, phospho-JNK (Thr183/Tyr185), p44/42, phospho-p44/42 (Thr202/Tyr204), S6 and phospho-S6 ribosomal protein, eIF4E, 4E-BP1 and GAPDH, (Cell Signaling, Danvers, MA); EGFR (Millipore, Billerica, MA); LC3 (Abgent, San Diego, CA); K-Ras (ABD Serotec, Raleigh, NC). Secondary antibodies were HRP-conjugated goat anti-rabbit antibody (Biorad, Hercules, CA) and horse anti-mouse (Cell Signaling, Danvers,MA) and IR dye anti-mouse and anti-rabbit antibodies (Li-Cor Biosciences, Lincoln, NE).
Cycloheximide (CHX), rapamycin and 3-methyladenine (3-MA) were purchased from Sigma (St. Louis, MO). BOC-aspartyl (Ome)-fluoromethyl ketone (BAF) was purchased from MP Biomedicals (Aurora, OH) and bafilomycin B1 (BafB1) was from A.G. Scientific (San Diego, CA). OHT was obtained from Enzo Life Sciences (Plymouth Meeting, PA), hygromycin B was from Cellgro (Manassas, VA) and doxycycline HCL (dox) was from Fisher Scientific (Pittsburgh, PA)
Cell Cultures
We have previously described the source of T265-2c, ST88-14 and the 90-8 cells, the human NF1- derived MPNST lines used in this study (29); (18). The identity of these cell lines was routinely verified according to the specifications outlined in the ATCC Technical Bulletin 8. Briefly, morphology and doubling times of cells was routinely assessed and the identity of cells was verified by short tandem repeat analysis. Cells were also regularly tested for Mycoplasma infection. SK-BR-3, MCF7 and MDA-MB-231 breast cancer cells and T84 colon cancer cells were obtained from the American Type Culture Collection. The colon cancer cell lines Caco2, HCT116, LoVo, HCT-15, RKO and DLD1 were kindly provided by Dr. Upender Manne (UAB Dept. of Pathology). The glioma cancer cell lines U87, LN229 and LN308 were a kind gift from Dr Yancey Gillespie (UAB Division of Neurosurgery) and the pancreatic cancer cell lines Panc1 and MiaPaCa2 were generously provided by Dr Boris Pasche (UAB Division of Hematology and Oncology). Pyrosequencing was performed to confirm the K-Ras mutational status of the cell lines.
All cell lines were cultured in DMEM10 [DMEM (Sigma, St. Louis, MO)] containing 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA), 1% L-glutamine (Sigma, St. Louis, MO), and 10% fetal bovine serum (FBS) (Hyclone, Logan, UT)] and incubated at 37°C in a humidified 5% CO2, 95% air atmosphere. Cells were plated onto uncoated 48 well plates at a density of 15,000/well and in 100mm dishes at a density of 800,000 cells/dish. Cultures were used in experiments 24 hours post-plating. Drug treatments were performed in the same media. Cell lines stably transduced with lentiviruses expressing shRNAs were maintained in DMEM supplemented with 10% tetracycline-free FBS (Serum Source International, Charlotte, NC) and hygromycin B (50μg/ml). During experiments requiring shRNA induction, cells were plated in media without hygromycin B. 24 hours after plating, 2 μg/ml dox was added to induce shRNA expression. 72 hours post- induction, cells were transferred to serum-free media and drug treatments initiated.
Construction of Cell Lines Stably Transduced with Lentiviral Vectors
Lentiviral vectors carrying cassettes encoding green fluorescent protein (GFP) and miR-like shRNAs under the control of a dox-inducible promoter were constructed using pSLIK vectors (30). Oligonucleotides encoding K-Ras shRNA sequences were designed using the RNAi Codex algorithm. Complementary oligonucleotides were annealed and ligated to BfuAI digested pen_TTGmiRc2. The resulting plasmids were recombined with pSLIK-hygro vector using LR Clonase per the manufacturer’s recommendations (Invitrogen; Carlsbad, CA). Three lentiviral vectors targeting distinct K-Ras sequences were constructed (pSLC751, pSLC752 and pSLC753 targeting nucleotides 508-530, 224-255 and 406-427 of NM_004985.2, respectively). To identify possible non-specific activation of RISC, a lentiviral vector was also constructed which encodes a shRNA targeting a sequence not present in the human genome (pSLC727). To make lentivirus, pSLIK vectors were transfected into 293FT cells together with helper plasmids (pPLP1, pPLP2, pVSVg) using Polyfect transfection reagent (Qiagen; Valencia, CA) per the manufacturer’s recommendations. 72 hours post-transfection, virus-containing media was collected and stored at -80°C until use.
To stably transduce lentiviral vectors into T265-2c cells, viral supernatant and 6 μg/ml Polybrene (Sigma-Aldrich) was added to cells plated in DMEM10 medium in 6 well plates. 24 hours later, virus-containing medium was removed and replaced with tetracycline-free DMEM10. 48-72 hours post-transduction, each well was split into three 100-mm dishes and selection with hygromycin begun. Colonies, which arose approximately 2 weeks later, were picked and individually expanded. Lines with appropriate regulation of the shRNA/GFP cassette were initially identified by finding that they had no GFP fluorescence in the absence of dox and strong GFP signals following dox induction. To verify that shRNA and GFP expression in the selected lines was appropriately regulated and to establish the optimal conditions for ablation of K-Ras expression, transformants were challenged with 0, 0.25, 0.5, 1.0 or 2.0 μg/ml dox for 72 hours. Lysates of cells stimulated with varying dox concentrations were then immunoblotted and probed with antibodies recognizing GFP and K-Ras.
Cell Viability and In Vitro Caspase Cleavage Assays
The calcein-AM conversion assay used to measure cell viability was performed as previously described (31). Caspase activation was assessed by the in vitro caspase-3 cleavage assay utilizing the chemical substrate DEVD-7-amino-4-methylcoumarin (AMC) (BIOMOL, Plymouth Meeting, PA).
Western Blotting and 7methyl-GTP Affinity Chromatography
Whole cell lysates were prepared by removing the media, washing the cells with PBS, scraping them off and pelleting the cells by centrifugation at 1750 rpm for 10 minutes. Cell pellets were resuspended in lysis buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, protease inhibitor cocktail (Sigma, St. Louis, MO), and phosphatase inhibitor cocktails 1 and 3 (Sigma, St. Louis, MO). Lysates were vortexed, clarified and stored at -80°C. 40 μg of protein was immunoblotted per our previously described protocol (32). All primary antibodies were diluted to a final concentration of 1:1000 except GAPDH and EGFR (1:5000). Immunoreactive species were detected by enhanced chemiluminescence.
7Methyl-GTP affinity chromatography was performed as previously described (33). Signals were detected using ECL western blotting analysis system (GE Healthcare, Piscataway, NJ) and the Odyssey Infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
RNAi
Atg7 siRNA was purchased from Thermo Scientific (Waltham, MA) and reconstituted according to manufacturers’ instructions. Cells were plated in DMEM10 and transfected 24 hours post-plating using X-tremeGene siRNA transfection reagent (Roche, Indianapolis, IN) with a ratio of 5:2 (transfection reagent:oligos). The next day, fresh media was added to cells and after 72 hours, cells were used in experiments.
Statistics
All data points represent mean ± S.D. n=6 wells for all experiments. All experiments were repeated at least 3 times unless stated otherwise. Representative data are shown. Statistical significance was determined by ANOVA followed by Bonferroni’s posthoc test. A p-value < 0.05 was considered significant.
Results
Caspase Activation Accompanies but does not Mediate OHT-Induced Death
4-Hydroxy-tamoxifen effects were examined in T265-2c cells, a human cell line derived from a NF1-associated MPNST. Micromolar concentrations of OHT have been reported to trigger apoptosis, marked by activation of effector caspases such as 3, 6 and 7 (34;34). Consequently, we first examined the possibility that the death induced by OHT in MPNST cells was apoptotic in nature. To determine whether OHT treatment activated effector caspases in MPNST cells, cells were treated with 8-12 μM OHT and, after 48 hours of treatment, the cleavage of DEVD-AMC, a pharmacological substrate for active effector caspases, was measured in cell lysates. These OHT concentrations were selected as they inhibit the proliferation and survival of breast carcinoma cells and are physiologically relevant to concentrations achieved in patients (35). We found that OHT-treated cells demonstrated a concentration-dependent increase in caspase 3-like enzymatic activity (Fig.1A). However, a one hour pretreatment with a broad spectrum caspase inhibitor failed to attenuate OHT-induced death (Fig.1B). We conclude that caspase-dependent apoptosis is not the sole mediator of OHT-induced cytotoxicity in MPNST cells.
Fig.1.
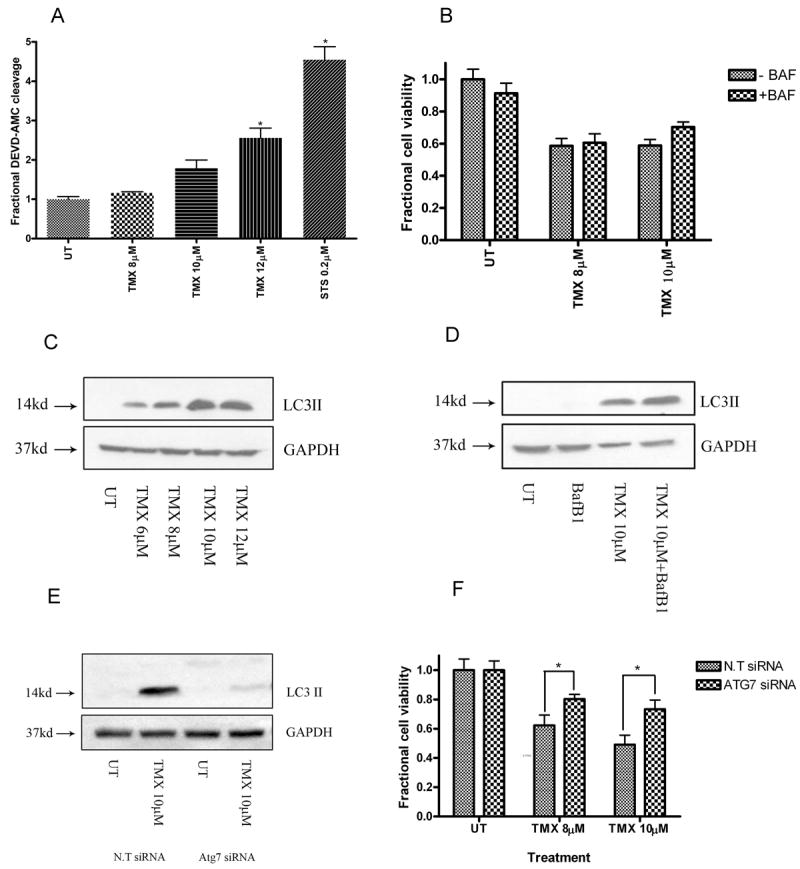
OHT triggers autophagic death in MPNST cells. (A) OHT-treated cells demonstrated a concentration-dependent increase in levels of caspase-3 like activity (48 hours). * indicates p<0.05 relative to untreated cells. However, broad caspase inhibition with BAF (50μM, 1hour pre-treatment) (B) did not provide protection from OHT-induced death (48 hours). Exposure to OHT (48 hours) triggered (C) a concentration-dependent increase in steady state levels of LC3-II, and (D) an increase in autophagic flux. Cells transfected with siRNA against Atg7 demonstrated (E) unchanged levels of LC3-II following OHT treatment (48 hours) and (F) partial protection from OHT-induced cytotoxicity (72 hours). * indicates p<0.05 relative to cells transfected with N.T siRNA.
OHT Induces an Autophagic Death in MPNST Cells
In other cell types, OHT treatment induces apoptotic features together with large scale accumulation of AVs (19;20). Further, a pro-death role has been ascribed to OHT-induced autophagy in breast cancer cells (19). We therefore asked whether OHT triggered the induction of autophagy in MPNST cells. Whole cell lysates prepared from OHT-treated cultures were probed for changes in levels of LC3 II, a surrogate marker for AV detection. LC3 II, the cleaved and lipidated form of the microtubule associated protein light chain 3 (LC3 or LC3 I) inserts itself in the outer membrane of AVs (36). Relative to untreated cells, OHT- treated cells demonstrated a dramatic increase in steady state levels of AVs (Fig.1C). LC3 II can accumulate in response to increased autophagy induction and/or decreased AV degradation. Therefore, autophagic flux was measured in control and OHT-treated cells using BafB1, which inhibits vacuolar ATPase, a molecule active in the late stage of autophagy (37;38). OHT- treated cells displayed increased autophagic flux, indicating that the increase in steady state AV levels was due, at least in part, to increased autophagy induction by OHT (Fig.1D). To assess the functional significance of this phenomenon, we next inhibited the initiation of autophagy by transfecting cells with siRNA targeting Atg7, a critical regulator of AV formation (Fig.1E). We found that Atg7 knockdown partially protected MPNST cells from OHT-induced death (Fig.1F). We conclude that OHT triggers autophagic death in MPNST cells.
OHT Triggers K-Ras Degradation
Autophagy plays a critical role in the turnover and recycling of long lived proteins. Our finding that OHT-induced autophagy mediates death raises the question of whether accelerated degradation of key pro-survival proteins through the autophagy-lysosomal degradation pathway mediates OHT-induced death. A recent genome wide screen in tamoxifen-treated breast cancer cells identified a number of candidate survival promoting genes present in these cells (27). These genes included the neurofibromin-regulated small GTP-binding protein K-Ras. Notably, K-Ras is the only neurofibromin-regulated Ras isoform that is targeted to lysosomes for degradation (39). Considered together, these observations led us to hypothesize that OHT triggers autophagic death in MPNST cells by decreasing levels of K-Ras.
We first examined the effects of OHT treatment on K-Ras protein levels in T265-2c cells. Immunoblot analyses demonstrated a concentration-dependent decrease in levels of K-Ras (Fig.2A). In contrast, levels of H- and N-Ras, which are degraded by proteasomes, remained unaltered following exposure to OHT (Figs.2B and 2C). To determine if the decrease in K-Ras levels was due to accelerated degradation, cells were pretreated for 1 hr with CHX, a protein synthesis inhibitor. In the absence of new protein synthesis, a time-dependent decrease in K-Ras levels was observed in cells treated with both CHX and OHT relative to those treated with CHX alone, indicating that OHT mediates accelerated degradation of K-Ras (Fig.2D). To determine whether OHT treatment globally inhibited protein translation, we examined the effect of this treatment on mTOR activity. Levels of phosphorylated S6 kinase, a downstream target of mTOR, did not significantly change 48 hours following OHT treatment (when K-Ras levels are found to decrease) whereas levels of phosphorylated S6 kinase were markedly decreased in cells treated with the mTOR inhibitor rapamycin (Fig.2E). Cap dependent translation can also be inhibited by an association between eukaryotic translation initiation factor (eIF4E) and another mTOR target, eIF4E binding protein (4EBP1) (40). Co-immunoprecipitation experiments in vehicle and OHT treated cells showed that there was no increase in binding of eIF4E and 4EBP1 in these cells, further indicating that OHT treatment did not affect global protein synthesis whereas synthesis was decreased by amino acid starvation (Fig.2F). In addition, steady state mRNA levels of K-Ras remained unchanged in OHT-treated cells, indicating that the decrease in K-Ras levels was not due to blocked transcription (data not shown). Considered jointly, these observations indicate that the decrease in K-Ras levels following OHT treatment is due to increased degradation rather than alterations in the transcription or translational machinery of the cells.
Fig.2.
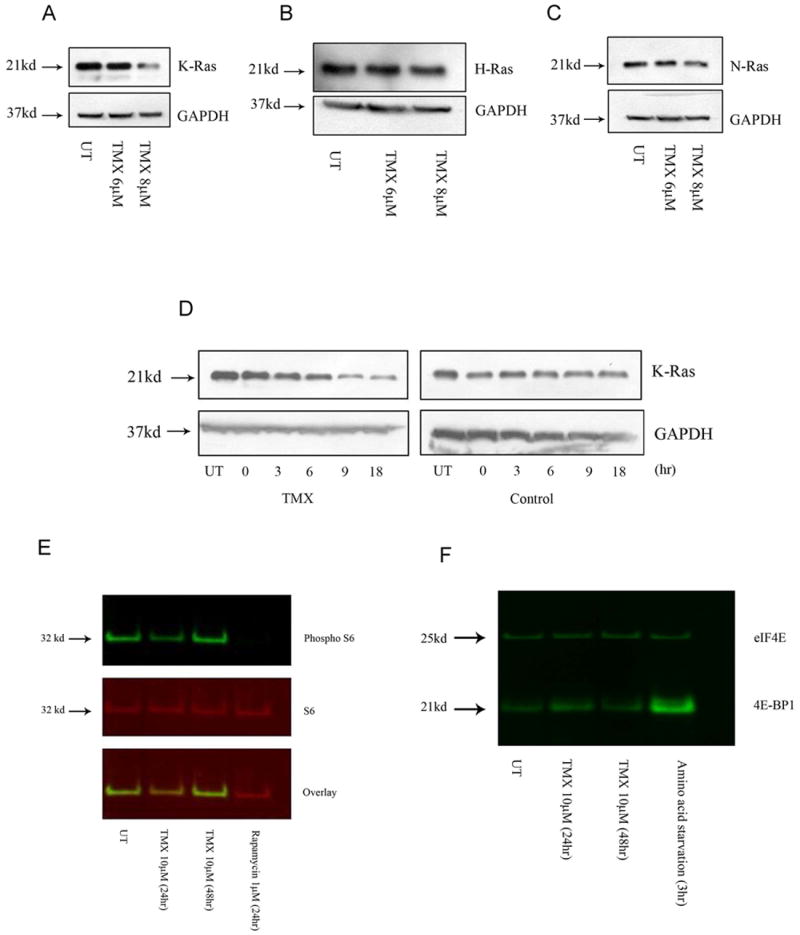
OHT triggers K-Ras degradation. Following OHT treatment (48 hours), (A) a concentration-dependent decrease was observed in K-Ras levels while (B) H- and (C) N-Ras levels remained unchanged. (D) Whole cell lysates from OHT-treated cells demonstrated a time-dependent decrease in K-Ras levels in the presence of the protein synthesis inhibitor CHX (100μM, 1hr pre-treatment). (E) OHT exposure caused a slight decrease (24hours) followed by an increase (48 hours) in levels of phosphorylated S6 protein. Rapamycin treatment was used as a positive control. (F) Co-immunoprecipitation experiments indicated no change in the interaction between eIF4E and 4EBP1 following OHT treatment. Amino acid starvation was used as a positive control.
OHT-triggered K-Ras degradation down regulates MAPK signaling
Several studies have indicated that activated Ras mediates tamoxifen resistance via MAPK activation. Therefore, we examined whether the level of K-Ras degradation observed in OHT-treated MPNST cells is sufficient to inhibit Ras-dependent MAPK activation in these cells. Immunoblot analyses showed that the levels of phosphorylated forms of Jun N-terminal Kinase (JNK) and p44/42 (Erk1/2) decreased in a time-dependent manner in OHT treated cells (Fig.3A and 3B). We conclude that the degree of K-Ras loss occurring in OHT-treated MPNST cells is sufficient to reduce the activation of at least some Ras-dependent MAPK signaling cascades.
Fig.3.
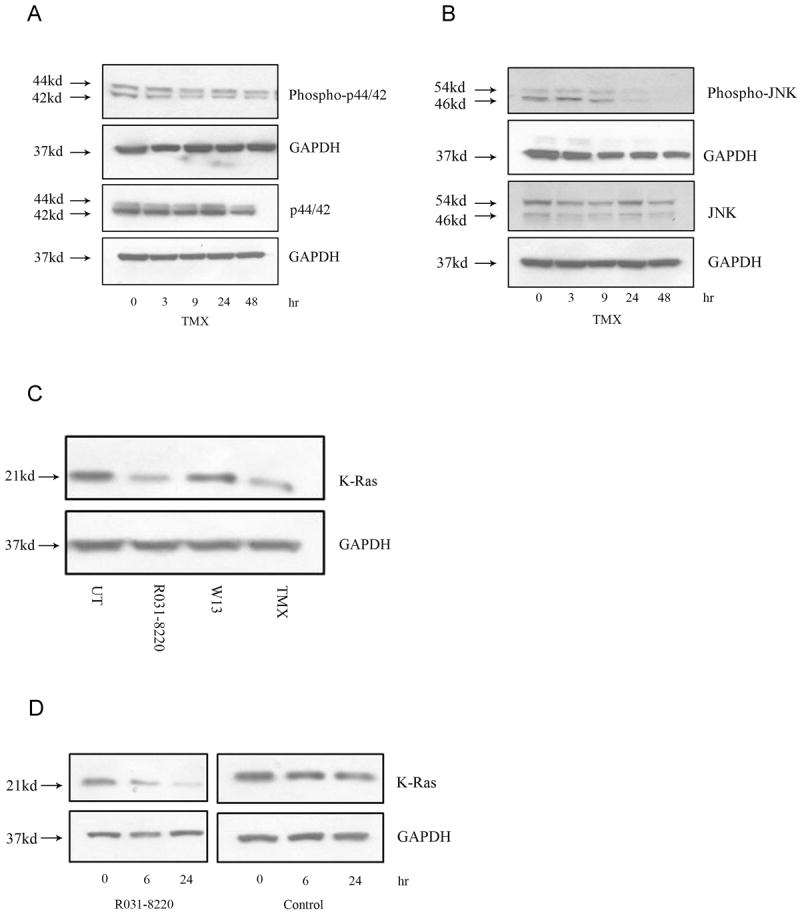
OHT treatment inhibits MAPK signaling. Whole cell lysates from OHT-treated cells demonstrate a time-dependent decrease in (A) levels of phospho-p44/42 and (B) phospho-JNK. (C) Treatment with the PKC inhibitor R031-8220 (10μM, 24 hours) and OHT (10μM, 24 hours) caused a decrease while the calmodulin inhibitor W13 (15mg/ml, 24 hours) had no effect on steady state K-Ras levels. (D) Whole cell lysates from R031-8220-treated cells demonstrated a time-dependent decrease in K-Ras levels in the presence of the protein synthesis inhibitor CHX (100μM, 1hr pre-treatment).
OHT primes K-Ras for degradation through PKC inhibition
Our previous study indicated that OHT-induced death of MPNST cells is ER-independent (18). Consequently, we next attempted to delineate an ER-independent mechanism by which OHT primes K-Ras for accelerated degradation. Earlier work has shown that tamoxifen directly binds to and antagonizes the calcium regulated proteins calmodulin and PKC (41-43). Further, calmodulin and PKC are known to differentially modulate the functionality and localization of K-Ras on the plasma membrane (44). These observations led us to hypothesize that inhibition of calmodulin and/or PKC promotes K-Ras degradation. We tested this hypothesis by assessing whether inhibitors of calmodulin or PKC could mimic the OHT-induced degradation of K-Ras. The calmodulin inhibitor W13 had no effect on steady state K-Ras levels (Fig.3C). However, PKC inhibition using Ro-31-8220 triggered a decrease in K-Ras levels under steady state as well as in the absence of new protein synthesis using CHX, indicating that similar to OHT, PKC inhibition also induces accelerated K-Ras degradation (Fig.3C and 3D). We conclude that OHT- triggered K-Ras degradation may be a function of its ability to antagonize PKC activity.
Inhibition of Autophagy Initiation Blocks OHT-Induced Decrease in K-Ras Levels
To determine if K-Ras degradation in OHT-treated MPNST cells was mediated by autophagy, we blocked the early stage of autophagy with 3-MA, a class III PI3K inhibitor, and Atg7 knockdown and examined the effect this had on K-Ras levels. A one hour pretreatment with 3-MA effectively blocked the OHT-induced decrease in K-Ras levels (Fig.4A). Similarly, siRNA-mediated knockdown of Atg7 prevented the OHT-mediated decrease in K-Ras levels whereas the nontargeting control siRNA had no effect (Fig.4B). Therefore, we conclude that OHT triggers K-Ras degradation through the autophagy pathway.
Fig.4.
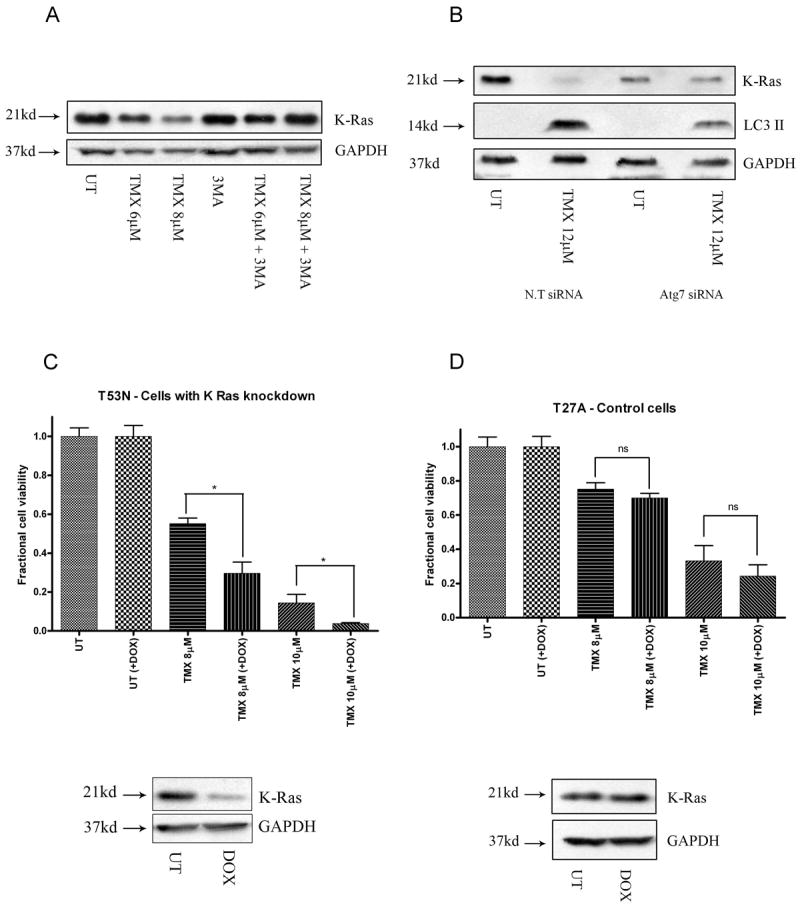
Autophagy mediates OHT-induced death through K-Ras degradation. Inhibition of autophagy initiation using (A) 3-MA (5mM, last 12 hours of treatment) and (B) siRNA mediated Atg7 knockdown blocks the decrease in levels of K-Ras following OHT-treatment (48 hours). Cells expressing shRNA under tetracycline-inducible promoter were treated with dox. Following dox induction, (C) cells expressing control shRNA demonstrated no change in levels of K-Ras and cell death while (D) cells expressing K-Ras shRNA demonstrated a drop in K-Ras levels accompanied by an increase in cytotoxicity following OHT- treatment. * indicates p<0.05 relative to cells not subjected to dox exposure.
OHT-Induces Cytotoxicity through Autophagy Mediated K-Ras Degradation
To establish that OHT triggers autophagic death in MPNST cells via K-Ras degradation, we assessed the effects of directly depleting K-Ras levels in OHT- treated cells. T265-2c MPNST cells stably transduced with lentiviral vectors expressing either K-Ras (T53N) or control (T27A) shRNAs under a doxycycline responsive promoter were generated. We found that cells expressing K-Ras shRNA demonstrated increased sensitivity to OHT in the presence of doxycycline (Fig.4C). In contrast, the addition of doxycycline had no effect on cells expressing control shRNA (Fig.4D). This is in keeping with previously published reports indicating that K-Ras plays a pro-survival role in the context of tamoxifen-induced death (27). Thus, we conclude that OHT triggers death in MPNST cells, at least in part, via autophagy mediated K-Ras degradation.
OHT also Triggers EGFR Degradation
Next, we examined whether OHT also affects the stability of membrane associated proteins such as EGFR that have been shown to co-localize with K-Ras en route to degradation (39). In response to growth factor stimulation, K-Ras and EGFR are internalized and occupy the same signaling scaffolds followed by degradation. Therefore, we tested the possibility that in addition to K-Ras, OHT might also trigger a decrease in EGFR stability and observed a time-dependent decrease in levels of EGFR in the presence of CHX indicating that this decrease was due to accelerated degradation (Fig.5A). Given the relevance of EGFR to malignant gliomas, we assessed the effects of OHT on EGFR levels in multiple glioma and MPNST cell lines and observed a concentration dependent decrease in all cell lines (Figs. 5B, 5C, 5D and 5E). Similar results were obtained for the breast cancer line MCF7 (Fig.5F). These results indicate that, in addition to K-Ras, OHT also influences EGFR stability.
Fig.5.
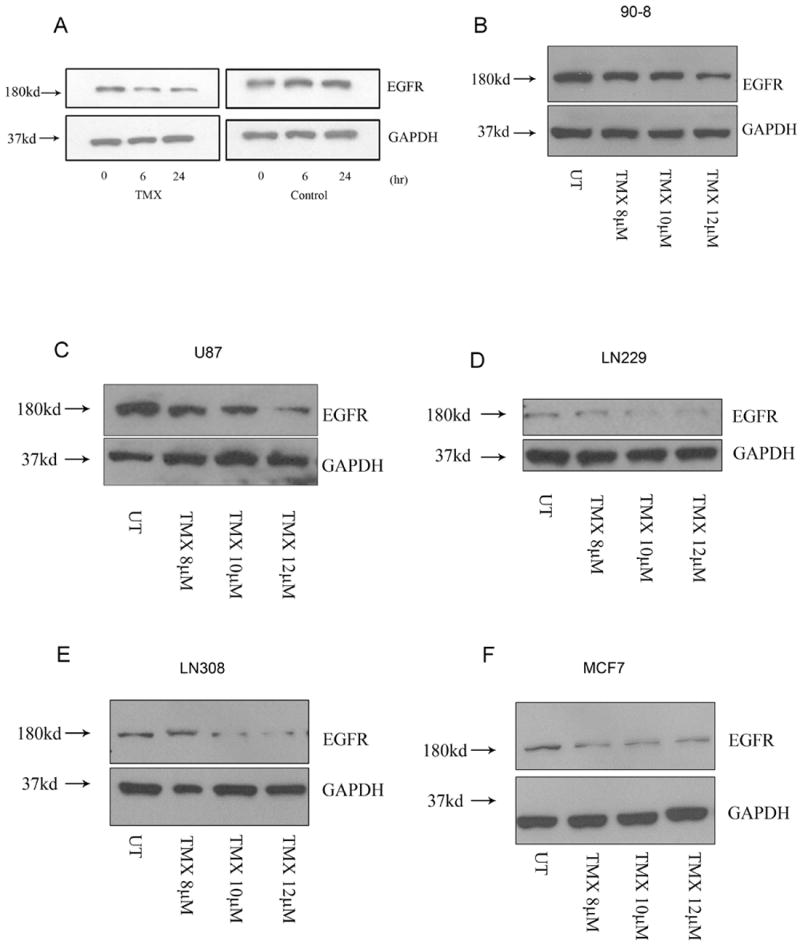
OHT triggers EGFR degradation. (A) Whole cell lysates from OHT-treated cells demonstrated a time-dependent decrease in EGFR levels in the presence of the protein synthesis inhibitor CHX (100μM, 1hr pre-treatment). (B) MPNST, (C, D and E) glioma and (F) breast cancer cell lines demonstrated a concentration-dependent decrease in EGFR levels following OHT treatment (48 hours).
OHT Triggers K-Ras Degradation in Multiple Tumor Types
Lastly, we wanted to determine whether the OHT effects on K-Ras that we observed in MPNST cells occurred more generally in other tumor types. We consistently observed an OHT-induced decrease in K-Ras levels across a panel of cell lines derived from multiple tumor types (Figs. 6A-I). Given the clinical relevance of tamoxifen to breast cancer, multiple breast cancer cell lines were also examined. We observed that OHT triggered a concentration-dependent decrease in K-Ras in the SK-BR-3 and MCF7 but not in MDA-MB-231 cells (Figs. 6H, 6I and 7A). Previously, mutations in Ras isoforms have been shown to influence their ubiquitination patterns and stability (45). This led us to test the hypothesis that OHT-induced K-Ras degradation may be affected by the mutational status of K-Ras.
Fig.6.
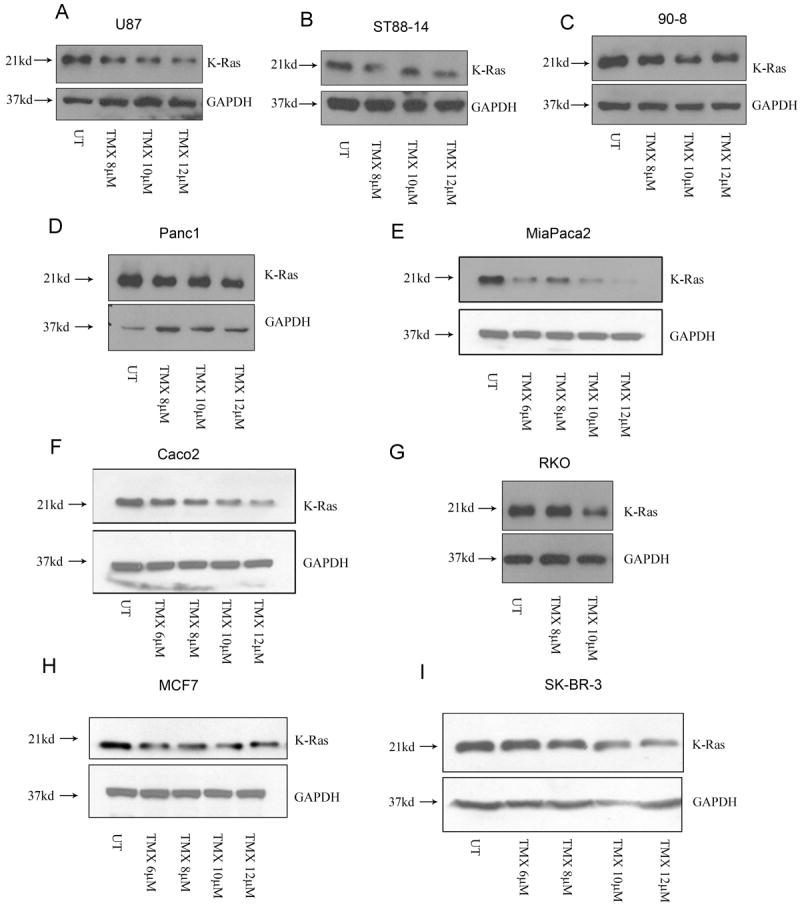
OHT triggers K-Ras degradation in multiple tumor types. Whole cell lysates from (A) glioma, (B, C) MPNST, (D, E) pancreatic, (F, G) colon and (H, I) breast cancer cell lines demonstrated a decrease in K-Ras levels following OHT treatment (48 hours)
Fig.7.
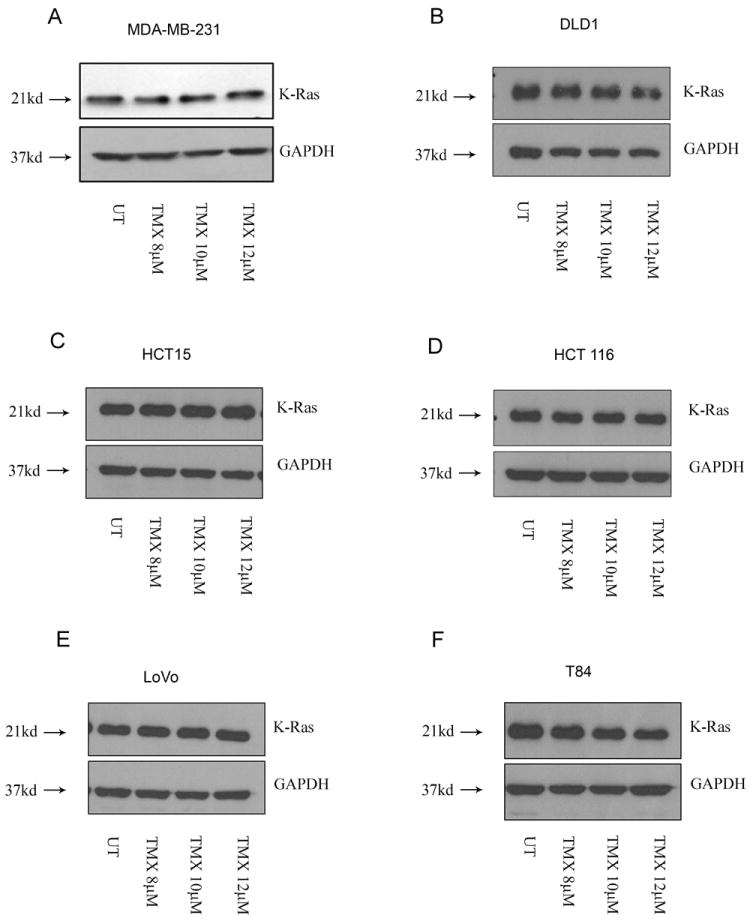
Mutant K-Ras resists OHT-induced degradation. Lysates from (A) breast and (B, C, D and E) colon cancer cell lines harboring a G13D mutation failed to demonstrate a decrease in K-Ras levels following OHT treatment (48 hours). (F) The colon cancer cell line T84 (G13D) was an exception.
Consistent with previously published studies, our pyrosequencing analysis revealed that while the SK-BR-3 and MCF7 cells harbor a wild type K-Ras, the MDA-MB-231 cells have a mutation in codon 13 (G13D). To assess whether cell lines harboring a mutant K-Ras (G13D) are relatively resistant to K-Ras degradation, we tested multiple colon cancer cell lines since the G13D mutation is commonly associated with colon cancer. We observed that OHT triggered a concentration-dependent decrease in K-Ras in the colon cancer cell lines Caco2 and RKO which have a wild type K-Ras (Figs. 6F and 6G). However, the cell lines DLD1, HCT-15, HCT 116 and LoVo that bear a G13D mutation were relatively resistant to K-Ras degradation (Figs. 7B-7E). These results indicate that while OHT can target K-Ras for degradation in multiple tumor types, the effects of OHT on K-Ras stability may be influenced by the mutation status of K-Ras. However, the colon cancer line T84 that also harbors a G13D mutation did demonstrate a decrease in K-Ras levels following OHT addition, indicating that additional factors might co-operate to determine the response of these cell lines to OHT treatment (Fig.7F).
Discussion
The goal of this study was to identify the molecular mechanisms that mediate ER-independent OHT-induced cytotoxicity in MPNST cells (18). This study offers evidence to support the hypothesis that the autophagic pathway is a critical mediator of OHT –induced death. Briefly, we demonstrate that caspases are activated following OHT treatment but are not the primary mediators of death. OHT triggers an increase in autophagic flux thereby impacting the autophagy pathway. Blocking autophagy initiation attenuates OHT-induced death, indicating a pro-death role for autophagy in this setting. Further, we have shown that through its inhibitory effects on PKC, OHT primes K-Ras for degradation in an autophagy-dependent manner. Hence, autophagy mediates death in OHT-treated cells, at least in part, through accelerated K-Ras degradation. This finding was extended to other tumor types and it was observed that OHT-mediated K-Ras degradation is affected by the mutational status of K-Ras. Thus, this study provides an insight into the role of altered protein turnover by autophagy as a death mechanism.
The role of therapy induced autophagy in cancer remains paradoxical. It is now well accepted that in response to various chemotherapeutic drugs, radiation and targeted therapeutics, dying cells display large scale accumulation of AVs (46). OHT is widely accepted as a potent autophagy inducer. In keeping with this, we observed an increase in autophagic flux following OHT treatment. This might result from the transient block in mTOR activity that was observed 24 hours following OHT treatment. We tested the role of OHT-induced autophagy by knocking down Atg7 and observed significant protection from OHT-induced death, thus establishing a pro-death role for autophagy.
Next, we attempted to delineate the underlying mechanism for autophagy-mediated death in the context of OHT. While the scope of autophagy has expanded dramatically to include diverse functions, the most basic role of autophagy remains the turnover of long lived proteins. There exists a delicate balance between pro-survival and death promoting proteins in the cell. We hypothesized that autophagy can regulate death by altering the rates of degradation of these proteins and disrupting the existing balance. This may offer an explanation for the context dependent roles of autophagy in response to different therapeutic agents. Accordingly, we proposed that OHT –induced increase in autophagic flux might accelerate the degradation of K-Ras, a protein shown to mediate resistance to TMX–induced death (27). We tested this hypothesis by examining the effects of OHT on the rate of K-Ras degradation and observed accelerated degradation in OHT-treated cells. Pharmacological inhibition using 3-MA and transient knockdown of Atg7 blocked the OHT-induced decrease in K-Ras levels indicating that K-Ras undergoes autophagic degradation. The functional relevance of this observation was established by assessing the effects of K-Ras degradation on downstream MAPK signaling. Modulation of protein turnover by drug-induced autophagy has not been previously established, thereby providing us with new insights on the role of autophagy in mediating death.
Next, we examined the possibility that OHT might specifically prime K-Ras for degradation through the autophagy pathway. TMX is known to bind to calmodulin and PKC and inhibit their functioning (47). Interestingly, PKC inhibition could simulate the effects of OHT on K-Ras degradation giving rise to the possibility that OHT might prime K-Ras for degradation through PKC inhibition. We also tested the possibility that OHT triggers the degradation of additional pro-survival proteins apart from K-Ras, especially other membrane associated proteins. EGFR and its family members are constitutively activated in MPNST cells and knockdown of EGFR signaling is associated with decreased proliferation and survival in MPNSTs cells thereby making it an attractive therapeutic target in this tumor type (29). We observed that similar to K-Ras, EGFR underwent accelerated degradation. However, it remains to be tested whether K-Ras and EGFR follow a similar degradation path and/ or are primed for degradation through a similar upstream stimulus that alters membrane dynamics.
To assess the relevance of our results to other tumor types, we tested the ability of OHT to induce K-Ras and EGFR degradation in cell lines derived from different tumor types. Additionally, we explored the possibility that mutations in K-Ras can influence its degradation. This is an important consideration since K-Ras mutations underlie the pathogenesis of several aggressive malignancies. We found that, unlike wild type K-Ras, K-RasG13D was resistant to OHT mediated degradation in nearly all cell lines tested. However, the results obtained for the colon cancer line T84 (K-RasG13D), which remained sensitive to OHT-induced K-Ras degradation, indicate that additional factors influence the stability of K-Ras following OHT treatment.
In summary, the findings of this study reveal yet another ER-independent mechanism for OHT-induced cell death. This paper also unveils a novel mechanism for autophagy mediated death and points towards the existence of a mechanism by which mutant K-Ras might resist autophagic degradation. Additional studies are now needed to examine the clinical relevance of these findings.
Acknowledgments
We wish to thank Dr Upender Manne and Dr Boris Pasche for generously providing us with the colon and pancreatic cancer cell lines and Dr Michael Crowley at the Heflin Center for Genomic Sciences at UAB for pyrosequencing.
Grant Support
This work was supported by NIH grants R01 NS041962 (K.A.R.), R01 CA134773 (K.A.R., S.L.C.), R01 CA122804 (S.L.C.), F30 NS063626 (N.M.B.) and P50CA890919 (T.C., M-A. B); and Department of Defense Grants X81XWH-09-1-0086 and X81XWH-12-1-0164 (S.L.C.).
Footnotes
Conflict of Interest: None exist
Reference List
- 1.Criscitiello C, Fumagalli D, Saini KS, Loi S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: overview of clinical use and molecular biomarkers for patient selection. Oncotargets and Therapy. 2011;4:1–11. doi: 10.2147/OTT.S10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997 Jun;11(6):657–66. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 3.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001 Dec;6(6):469–77. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Couldwell WT, Song H, Takano T, Lin JH, Nedergaard M. Tamoxifen-induced enhancement of calcium signaling in glioma and MCF-7 breast cancer cells. Cancer Res. 2000 Oct 1;60(19):5395–400. [PubMed] [Google Scholar]
- 5.Gundimeda U, Chen ZH, Gopalakrishna R. Tamoxifen modulates protein kinase C via oxidative stress in estrogen receptor-negative breast cancer cells. J Biol Chem. 1996 Jun 7;271(23):13504–14. doi: 10.1074/jbc.271.23.13504. [DOI] [PubMed] [Google Scholar]
- 6.Cabot MC, Zhang Z, Cao H, Lavie Y, Giuliano AE, Han TY, et al. Tamoxifen activates cellular phospholipase C and D and elicits protein kinase C translocation. Int J Cancer. 1997 Mar 4;70(5):567–74. doi: 10.1002/(sici)1097-0215(19970304)70:5<567::aid-ijc13>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 7.O’Brian CA, Ioannides CG, Ward NE, Liskamp RM. Inhibition of protein kinase C and calmodulin by the geometric isomers cis- and trans-tamoxifen. Biopolymers. 1990 Jan;29(1):97–104. doi: 10.1002/bip.360290114. [DOI] [PubMed] [Google Scholar]
- 8.Mandlekar S, Yu R, Tan TH, Kong AN. Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling pathways in tamoxifen-induced apoptosis of human breast cancer cells. Cancer Res. 2000 Nov 1;60(21):5995–6000. [PubMed] [Google Scholar]
- 9.Couldwell WT, Weiss MH, DeGiorgio CM, Weiner LP, Hinton DR, Ehresmann GR, et al. Clinical and radiographic response in a minority of patients with recurrent malignant gliomas treated with high-dose tamoxifen. Neurosurgery. 1993 Mar;32(3):485–9. doi: 10.1227/00006123-199303000-00034. [DOI] [PubMed] [Google Scholar]
- 10.Cocconi G, Passalacqua R, Foladore S, Carlini P, Acito L, Maiello E, et al. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen, or vindesine plus tamoxifen: a prospective randomized study. Melanoma Res. 2003 Feb;13(1):73–9. doi: 10.1097/00008390-200302000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Avgeropoulos NG, Batchelor TT. New treatment strategies for malignant gliomas. Oncologist. 1999;4(3):209–24. [PubMed] [Google Scholar]
- 12.Gelmann EP. Tamoxifen induction of apoptosis in estrogen receptor-negative cancers: new tricks for an old dog? J Natl Cancer Inst. 1996 Mar 6;88(5):224–6. doi: 10.1093/jnci/88.5.224. [DOI] [PubMed] [Google Scholar]
- 13.Beguerie JR, Xingzhong J, Valdez RP. Tamoxifen vs. non-tamoxifen treatment for advanced melanoma: a meta-analysis. Int J Dermatol. 2010 Oct;49(10):1194–202. doi: 10.1111/j.1365-4632.2010.04529.x. [DOI] [PubMed] [Google Scholar]
- 14.Hajdu SI. Peripheral nerve sheath tumors. Histogenesis, classification, and prognosis. Cancer. 1993 Dec 15;72(12):3549–52. doi: 10.1002/1097-0142(19931215)72:12<3549::aid-cncr2820721202>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 15.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002 Mar 1;62(5):1573–7. [PubMed] [Google Scholar]
- 16.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis Type 1 Revisited. Pediatrics. 2009 Jan;123(1):124–33. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 17.Gutmann DH, Wood DL, Collins FS. Identification of the neurofibromatosis type 1 gene product. Proc Natl Acad Sci U S A. 1991 Nov 1;88(21):9658–62. doi: 10.1073/pnas.88.21.9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byer SJ, Eckert JM, Brossier NM, Clodfelder-Miller BJ, Turk AN, Carroll AJ, et al. Tamoxifen inhibits malignant peripheral nerve sheath tumor growth in an estrogen receptor-independent manner. Neuro-Oncology. 2011 Jan;13(1):28–41. doi: 10.1093/neuonc/noq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis. 1996 Aug;17(8):1595–607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Malerva L, Park J, Zou LH, Hu YH, Moradpour Z, Pearlberg J, et al. High-throughput ectopic expression screen for tamoxifen resistance identifies an atypical kinase that blocks autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2011 Feb 1;108(5):2058–63. doi: 10.1073/pnas.1018157108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nature Reviews Cancer. 2005 Sep;5(9):726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 22.Mishima Y, Terui Y, Mishima Y, Taniyama A, Kuniyoshi R, Takizawa T, et al. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 2008 Nov;99(11):2200–8. doi: 10.1111/j.1349-7006.2008.00932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez G, Torres K, Lev D. Autophagy blockade enhances HDAC inhibitors’ pro-apoptotic effects: Potential implications for the treatment of a therapeutic-resistant malignancy. Autophagy. 2011 Apr 1;7(4) doi: 10.4161/auto.7.4.14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Z, Chang PC, Yang JC, Chu CY, Wang LY, Chen NT, et al. Autophagy Blockade Sensitizes Prostate Cancer Cells towards Src Family Kinase Inhibitors. Genes Cancer. 2010 Jan;1(1):40–9. doi: 10.1177/1947601909358324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007 Feb;117(2):326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006 Mar 28;103(13):4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendes-Pereira AM, Sims D, Dexter T, Fenwick K, Assiotis I, Kozarewa I, et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc Natl Acad Sci U S A. 2012 Feb 21;109(8):2730–5. doi: 10.1073/pnas.1018872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGlynn LM, Kirkegaard T, Edwards J, Tovey S, Cameron D, Twelves C, et al. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin Cancer Res. 2009 Feb 15;15(4):1487–95. doi: 10.1158/1078-0432.CCR-07-4967. [DOI] [PubMed] [Google Scholar]
- 29.Stonecypher MS, Byer SJ, Grizzle WE, Carroll SL. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene. 2005 Aug 25;24(36):5589–605. doi: 10.1038/sj.onc.1208730. [DOI] [PubMed] [Google Scholar]
- 30.Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI, et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proceedings of the National Academy of Sciences of the United States of America. 2006 Sep 12;103(37):13759–64. doi: 10.1073/pnas.0606179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geng Y, Kohli L, Klocke BJ, Roth KA. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro-Oncology. 2010 May;12(5):473–81. doi: 10.1093/neuonc/nop048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, Shacka JJ, et al. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem. 2010 Apr 2;285(14):10497–507. doi: 10.1074/jbc.M110.103747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnhart BC, Lam JC, Young RM, Houghton PJ, Keith B, Simon MC. Effects of 4E-BP1 expression on hypoxic cell cycle inhibition and tumor cell proliferation and survival. Cancer Biology & Therapy. 2008 Sep;7(9):1443–51. doi: 10.4161/cbt.7.9.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandlekar S, Hebbar V, Christov K, Kong ANT. Pharmacodynamics of tamoxifen and its 4-hydroxy and N-desmethyl metabolites: Activation of caspases and induction of apoptosis in rat mammary tumors and in human breast cancer cell lines. Cancer Research. 2000 Dec 1;60(23):6601–6. [PubMed] [Google Scholar]
- 35.Perry RR, Kang Y, Greaves B. Effects of tamoxifen on growth and apoptosis of estrogen-dependent and -independent human breast cancer cells. Ann Surg Oncol. 1995 May;2(3):238–45. doi: 10.1007/BF02307030. [DOI] [PubMed] [Google Scholar]
- 36.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000 Nov 1;19(21):5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006 Apr;69(4):1125–36. doi: 10.1124/mol.105.018408. [DOI] [PubMed] [Google Scholar]
- 38.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008 Oct 1;4(7):849–950. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 39.Lu A, Tebar F, Alvarez-Moya B, Lopez-Alcala C, Calvo M, Enrich C, et al. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. Journal of Cell Biology. 2009 Mar 23;184(6):863–79. doi: 10.1083/jcb.200807186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005 Feb 3;433(7025):477–80. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 41.Lopes MC, Vale MG, Carvalho AP. Ca2(+)-dependent binding of tamoxifen to calmodulin isolated from bovine brain. Cancer Res. 1990 May 1;50(9):2753–8. [PubMed] [Google Scholar]
- 42.Lam HY. Tamoxifen is a calmodulin antagonist in the activation of cAMP phosphodiesterase. Biochem Biophys Res Commun. 1984 Jan 13;118(1):27–32. doi: 10.1016/0006-291x(84)91062-3. [DOI] [PubMed] [Google Scholar]
- 43.O’Brian CA, Housey GM, Weinstein IB. Specific and direct binding of protein kinase C to an immobilized tamoxifen analogue. Cancer Res. 1988 Jul 1;48(13):3626–9. [PubMed] [Google Scholar]
- 44.varez-Moya B, Barcelo C, Tebar F, Jaumot M, Agell N. CaM interaction and Ser181 phosphorylation as new K-Ras signaling modulators. Small GTPases. 2011 Mar;2(2):99–103. doi: 10.4161/sgtp.2.2.15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4(163):ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meschini S, Condello M, Lista P, Arancia G. Autophagy: Molecular Mechanisms and their Implications for Anticancer Therapies. Current Cancer Drug Targets. 2011 Mar;11(3):357–79. doi: 10.2174/156800911794519707. [DOI] [PubMed] [Google Scholar]
- 47.Mccague R, Rowlands MG, Grimshaw R, Jarman M. Evidence That Tamoxifen Binds to Calmodulin in A Conformation Different to That When Binding to Estrogen-Receptors, Through Structure-Activity Study on Ring-Fused Analogs. Biochemical Pharmacology. 1994 Oct 7;48(7):1355–61. doi: 10.1016/0006-2952(94)90557-6. [DOI] [PubMed] [Google Scholar]