

Abstract
The major risk factors for hepatocellular carcinoma (HCC) are chronic liver diseases that include hepatitis B, hepatitis C, alcoholic liver disease and non-alcoholic steatohepatitis. However, the mechanisms of alcohol-associated HCC remain to be elucidated. The products of RNA Pol III (RNA polymerase III) dependent genes are elevated in both transformation cells and tumor cells. TBP (TATA-box binding protein) is a central transcription factor, which regulates Pol I, Pol II and Pol III gene activity. Our studies have demonstrated that alcohol increases TBP expression and Pol III gene transcription to promote liver tumor formation. We continue to investigate how ethanol mediates TBP expression. Here, we report that ethanol induces TBP promoter activity and the induction is ethanol dose dependent. Blocking the JNK1 pathway by a chemical inhibitor and siRNA reduce this ethanol-induced activity. Furthermore, mutating G>A at a −46bp Elk1 binding site of the TBP promoter or mutating AP-1 binding site at −37bp (A>G) and −38bp (C>T) reduces the TBP promoter activity. Mutation of both Elk1 and AP-1 binding sites dramatically represses this induction. Together, these studies demonstrate that, for the first time, alcohol increases Pol III gene transcription through a response element, which is composed of the overlapping the Elk1 and AP-1 binding sites of the TBP promoter. It suggests that these binding sites may play a critical role in alcohol-induced deregulation of Pol III genes in liver tumor development.
Keywords: Alcohol, TBP, Elk1, AP-1, Pol III genes, liver tumor
1. Introduction
Alcohol-induced liver injury, including liver steatosis, fibrosis, and cirrhosis, increases the risk of development of hepatocellular carcinoma (HCC) (El-Serag and Rudolph, 2007). Although small amounts of alcohol by itself are not considered a carcinogen, alcohol in combination with viruses (hepatitis C or B), carcinogens (aflatoxin), obesity, or diabetes mellitus, promotes liver cancer development (Maeda et al, 2005; Machida et al, 2009; Ha et al, 2001). However, the mechanisms of alcohol-associated HCC remain to be elucidated.
Cancer cells have a consistent cytological feature of nucleolar hypertrophy, where rRNAs are synthesized by RNA polymerase (Pol) I and III within a nucleolus. Pathologists have been using enlarged nucleoli as a strong diagnostic indicator of cell transformation and neoplasia. RNA Pol III transcribes a variety of untranslated RNAs, including tRNAs, 5S rRNAs, 7SL RNA, 7SK RNA and U6 RNA (Ullu and Tschudi, 1984; Dieci et al, 2007; Raha et al, 2010), whereas tRNA and 5S rRNA control the translational and growth capacity of cells (White RJ, 2001; Goodfellow et al, 2006)10, 14). Oncogenic proteins, such as Ras, c-Jun, and c-Myc stimulate RNA Pol III-dependent gene (Pol III gene) transcription (Zhong et al, 2004; Zhong et al, 2007; Johnson and Johnson, 2008). In contrast, tumor suppressors, such as pRb, p53, PTEN and Maf1 repress transcription of this class of genes (White, 2001; Johnson and Johnson, 2008; Woiwode et al, 2008). Studies have indicated that RNA Pol III transcription products are elevated in both transformed and tumor cells suggesting that they play a crucial role in tumorigenesis (White, 2001; Woiwode et al, 2008; Winter et al, 2007; Zhong et al, 2011). Enhanced Pol III transcription is required for oncogenic transformation (Johnson et al, 2008). The ability of these oncogenic and tumor suppressor proteins to alter Pol III transcription result from their capacity to regulate the TFIIIB complex, which is a component of the Pol III gene transcription machinery. The TFIIIB complex consists of TBP and its associated factors, Brf1 and Bdp1. TFIIIB, together with TFIIIC and RNA Pol III, are required to transcribe tRNA genes, whereas TFIIIB, together with TFIIIA, TFIIIC and RNA Pol III, are required to transcribe 5S rRNA genes. TBP is a central transcription initiation factor, which is required for directing transcription from all three nuclear RNA polymerases. Studies have indicated that TBP can be limiting for the transcription of RNA Pol I and Pol III genes (Zhong et al, 2004; Zhang et al, 2005).
Our recent studies have demonstrated that alcohol increased TBP expression and Pol III gene transcription in vivo and in vitro (Zhong et al, 2011). This induction of TBP and Pol III genes by ethanol was associated with liver tumor formation in HCV NS5A transgenic mice (Zhong et al, 2011). Increasing TBP expression promoted tumor formation in mice (Johnson et al, 2003). Further analysis indicates that alteration of cellular level of TBP affected Bdp1 expression, but did not affect Brf1 expression (Zhong and Johnson, 2009). This implies that ethanol-induced alteration of TBP expression is critically important for deregulation of Pol III genes in liver tumor development. However, it remains to be established how ethanol modulates TBP expression by identifying the alcohol-response elements at the TBP promoter. Our results indicate that ethanol activated JNK1 and induced TBP promoter activity. Inhibition of JNK1 by a chemical inhibitor (SP 600125) or JNK1 siRNA reduced the induction of TBP promoter by ethanol. Mutagenesis of Elk-1 (−46bp) and/or AP-1 (−37bp and −38bp) binding sites of TBP promoter eliminated this induction. These studies support the idea that ethanol mediates the ethanol-induced TBP expression and Pol III gene transcription through JNK1, Elk1 and AP-1 pathway. Elk1 and AP-1 sites, which overlap, may be an alcohol-response element at TBP promoter region. These novel findings will be of great interest both to the basic and clinical research communities and provide a potential approach of treatment for alcohol-associated HCC patients.
2. Materials and methods
2.1. Reagents and antibodies
Cell culture medium DMEM, Zeocin, Lipofectin reagent, Lipofectamine 2000, TRIzol reagent and OPTI-MEM were from Life Technologies. Ethanol was from Sigma-Aldrich. Antibodies against TBP and β-actin were obtained from San Cruz. JNK and phosphor-JNK antibodies were from Cell Signaling. JNK inhibitor, SP600125 was from A.G. Scientific, Inc. The sequences of TBP, tRNAleu and 5S rRNA primers and JNK1 and TBP siRNAs were as indicated (Zhong et al, 2004; Zhong et al, 2007). Plasmids of TBP expression and human TBP-luciferase reporter (TBP-Luc) constructs (p-84/+66hTBP, p-54/-1hTBP, p-54/-1ΔElk-1-hTBP, p-54/-1ΔAP-1-hTBP and p-54/-1ΔElk-1-ΔAP-1-hTBP) were kindly provided by Dr. Debbie Johnson (University of Southern California, USA). Engineered HepG2 (hepatocellular carcinoma G2) cell lines, stably expressing ADH (alcohol dehydrogenase, HepG2-ADH), or vector alone (HepG2-vector) were kindly provided by Dr. D.L. Clemens (Clement et al, 2002). Mice expressing the HCV NS5A gene under control of the apoE promoter were obtained from Ratna Ray (Mechida et al, 2009). Wild type and NS5A transgenic mice on the C57BL/6 strain were fed with Lieber–DeCarli diet containing 3.5% ethanol or isocaloric dextrin for long-term alcohol feeding ((Mechida et al, 2009). All animal experiments were performed with age- and sex-matched mice from same littermates and conducted in accordance with the approved Institutional Animal Care and Use Committee protocol at the University of Southern California.
2.1. RNA isolation and RT-qPCR
Total RNA was isolated from HepG2-ADH and HepG2-vector cells treated with alcohol using a TRIzol reagent (Life Technologies). Total RNA samples were quantified and reverse-transcribed in a 20 μl reaction containing 1 x RT (reverse transcription) buffer. After first-strand cDNA synthesis, the cDNAs were diluted in DNase-free water and real time qPCR (RT-qPCR) were performed with specific primers as indicated before (Zhong et al, 2004; Zhong et al, 2007) and PCR reagent kits (Bio-Rad Biotech) in the ABI prism 7700 Sequence Detection System. Precursor of tRNALeu and 5S rRNA transcripts and mRNAs of TBP and GAPDH were measured by RT-qPCT as described previously (Zhong et al, 2004; Zhong et al, 2007).
2.2. Transfection and TBP-luciferase reporter assays
For transient transfection assays, the cells were transfected with hTBP-Luc plasmids or plus siRNAs as described previously (Zhong et al, 2011). Serum-free medium was added to each dish with Lipofectin-DNA or Lipofectamine2000-siRNA complexes, and cells were further incubated for 4h. The medium was changed with 10% FBS/DMEM. Cells were incubated for 48h before harvesting. Protein concentrations of the resultant lysates were measured by the Bradford method. For TBP-Luc reporter activity assays, cells were transfected with 0.2 μg of the TBP-Luc constructs or plus JNK1 siRNA for 48 h. Cells were starved in serum-free DMEM for 3 h and treated with 50mM ethanol for 60min. Cell pellets were resuspended in Promega reporter lysis buffer. The lysates were analyzed for luciferase activity using a luminometer and the Promega Luciferase Assay System as described (Promega). Resultant luciferase activities were normalized to the amount of protein in each lysate as described (Zhong et al, 2004; Zhong et al, 2011). The fold change in luciferase activity was calculated by determining the level of luciferase activity in the absence of alcohol. This value will be set at 1 for each independent experiment. Values are means ± SE of at least three independent experiments.
2.3. SDS-PAGE and immunoblot analysis
HepG2 cells were incubated with 50mM ethanol for 60 min after starvation 3h. Cells were collected with lysis buffer and sonicated. The suspensions were centrifuged to save the supernatants. Protein concentrations were determined by the Bradford method. Lysates (50 μg of protein) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred from the SDS-PAGE gel to Hybond-P membrane and immunoblot analysis were performed with specific antibodies. Membranes were probed with either antibodies against TBP, JNKs, phosphor-JNK and β-actin as described (Zhong et al, 2011). Bound primary antibody was visualized using horseradish peroxidase-conjugated secondary antibody (Vector Laboratories) and enhancing chemiluminscence reagents (Amersham).
3. Results
3.1. Alcohol-induced TBP and RNA Pol III gene expression is ADH dependent
The products of Pol III genes, such as tRNAs and 5S rRNA, are elevated in both transformed and tumor cells suggesting that they play a crucial role in tumorigenesis. To investigate the mechanism of alcohol-associated HCC, HepG2-ADH and HepG2-vector cells were treated with ethanol and the amounts of precursor tRNALeu and 5S rRNA transcript were measured by RT-qPCR. Ethanol treatment increased pre-tRNALeu and 5S rRNA transcription at the ethanol concentration of 50mM for 60min in HepG2-ADH cells (Fig. 1A). The results indicate that the induction of Pol III genes by ethanol was in HepG2-ADH cells, but not in HepG2-vector cells (Fig. 1B), which suggests that alcohol-induced Pol III gene transcription was correlated with ADH expression. Primary mouse hepatocytes (PMH) treated with ethanol also displayed an increase in Pol III gene transcription (Fig. 1C). Ethanol treatment increased cellular levels of TBP protein and mRNA in HepG2-ADH cells (Fig. 2A). Animal experiments reveal that TBP protein and mRNA levels were increased in the livers of ethanol-fed mice (Fig. 2B upper panel). Interestingly, TBP mRNA level in liver tumor tissues was higher than in non-tumor liver tissue (Fig. 2B lower panel). Further analysis indicates that increasing cellular level of TBP by its expression construct enhanced Pol III gene transcription (Fig. 3A). In contrast, decreasing TBP expression by its siRNA reduced this transcription (Fig. 3B). These studies suggest that TBP is a limiting factor for Pol III gene activity, increasing TBP expression and Pol III gene transcription were associated with liver tumor formation in ethanol-fed mice (Zhong et al, 2011).
Fig. 1. Alcohol induces RNA Pol III-dependent transcription.

(A) Ethanol enhances the transcription in HepG2-ADH cells. The cells were starved in DMEM for 3h and treated with or without 50 mM ethanol for 1h. RNA was isolated from the cells and pre-tRNALeu, 5S rRNA, and GAPDH transcripts were measured by RT-qPCR. (B) Ethanol does not stimulate the transcription in HepG2-vector cells. HepG2-vector cells were treated with or without ethanol as described above. (C) Ethanol increases Pol III gene transcription in primary mouse hepatocytes (PMH). The hepatocytes were isolated from mouse and treated with ethanol as described in (A). RT-qPCR was performed to measure the amounts of pre-tRNALeu and 5S rRNA. The fold change was calculated by normalizing to the amount of GAPDH mRNA. The bars represent Mean ± SE of at least three independent determinations.
Fig. 2. Ethanol increases TBP expression in vivo and in vitro.

(A) Ethanol increased TBP expression in HepG2-ADH cells. HepG2-ADH cells were starved in DMEM for 3 h and treated with 50mM ethanol for 1 h. The total RNA and cell lysates from these cells were extracted to determine TBP mRNA by RT-qPCT. Resultant protein lysates were subjected to perform immunoblot analysis. The antibodies were indicated (A lower panel). (B) Chronic alcohol administration in mice induced TBP expression. C57BL/6 transgenic mice harboring the HCV NS5A gene that is selectively expressed in hepatocytes were chronically fed with control diet or 3.5% of ethanol in the Lieber-DeCarli liquid diet for 12 months. Liver tissues were harvested from these mice, Cell lysates and RNA were extracted from non-tumor or tumor portions of the livers. RT-qPCR was used to measure the amounts of TBP transcript relative to GAPDH. The values represent ± SE from three independent experiments. The lysates of the livers were subjected to immunoblot analysis with antibodies of TBP and β-actin. Each group of mouse includes at least three mice. The bars represent Mean ± SE of at least three independent determinations. **: p<0.01. The fold change was calculated in each group by normalizing to mice fed with control diet.
Fig. 3. Alteration of TBP expression affects RNA pol III-dependent transcription.
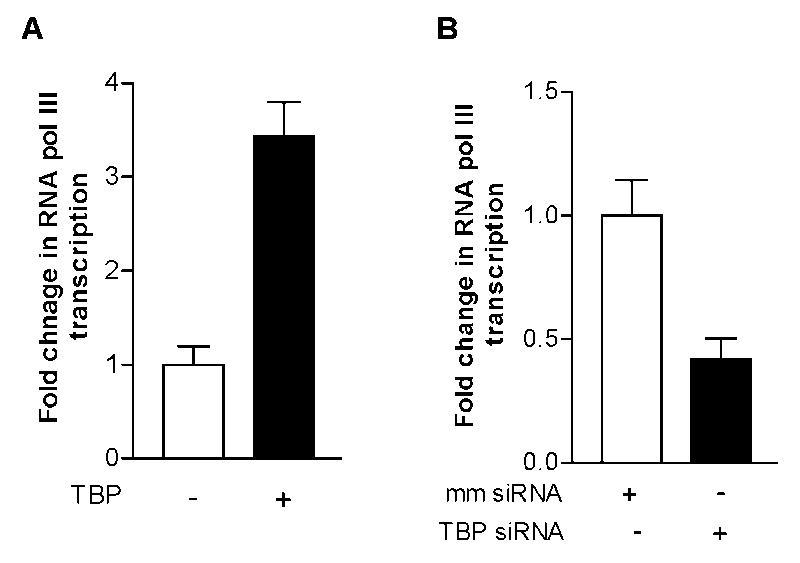
(A) overexpressing TBP enhanced Pol III gene transcription. HepG2-ADH cells were transiently transfected with a TBP expression plasmids or control vector for 48 hours. RT-qPCR was performed on RNA derived from these cells to measure pre-tRNALeu transcripts (B) Repression of TBP reduced RNA pol III-dependent transcription. HepG2-ADH cells were transfected with TBP siRNA or mismatch RNA (mm RNA) for 48 hours. RT-qPCR was performed on RNA isolated from these cells to measure pre-tRNALeu transcripts. The fold change was calculated by normalizing to the amount of transcript in cells transfected with vector control plasmid or mm RNA. The bars represent Mean ± SE of at least three independent determinations.
3.2. Alcohol induces TBP promoter activity and JNK1 activation
To determine how ethanol mediates TBP transcription, we examined potential changes in TBP promoter activity. Given that ethanol affected TBP expression and Pol III gene transcription (Fig. 1,2), we further investigated whether ADH expression was able to alter TBP promoter activity. HepG2-ADH or HepG2-vector cells were transiently transfected with TBP-Luc (p-84/+66hTBP) construct for 48h and treated with ethanol for another 1h. The results indicate that alcohol increased the TBP promoter activity in HepG2-ADH cells in a dose dependent manner (Fig. 4A). In contrast, there was not a significant induction of this activity in HepG2-vector cells after ethanol treatment (Fig. 4B). These results were consistent with the induction of Pol III gene by ethanol (Fig. 1). These studies indicate that ethanol-induce TBP and Pol III gene transcription are correlated with ADH expression.
Fig. 4. Alcohol induces TBP promoter activity in HepG2-ADH cells, but not hepG2-vector cells.

HepG2-ADH and HepG2-vector cells were transfected with TBP-Luc reporter construct for 48 h; the cells were starved in DMEM for 3 h. Cells were then treated with or without ethanol for another 1h. The cell lysates was extracted from these cells and luciferase activity assay was performed to measure the TBP-Luc activity as described in Materials and Methods. The change in TBP promoter activity was calculated relative to that with no ethanol treatment. The values represent mean ± SE from three independent experiments.
Given that alcohol has been shown to induce JNK activation (Zhong et al, 2011) and that the JNKs play an important role in regulating Pol III gene transcription (Zhong and Johnson, 2009), we examined the role of JNKs in alcohol-mediated TBP promoter activity. Ethanol induced a strong activation of JNK1 in the HepG2-ADH cells (Fig. 5A). Next, we assessed whether JNK1 mediated TBP expression. The results reveal that JNK1 siRNA repressed JNK1 expression and decreased cellular level of TBP protein (Fig. 5B). Therefore, we further determined whether alcohol-activated JNK1 mediates TBP promoter activity. The results indicate that either the JNK chemical inhibitor SP 600125 or JNK1 siRNA reduced alcohol-increased TBP promoter activity (Fig. 5C). Together, these studies support the idea that alcohol-mediated TBP expression requires the activation of JNK1. It suggests that alcohol-induced TBP expression and Pol III gene transcription in HepG2-ADH cells go through JNK1 pathway.
Fig. 5. Alcohol-mediated activation of JNK1 is required for induction of Pol III genes.

(A) Ethanol induces JNK1 activation. HepG2-ADH cells were treated with or without ethanol. Immunoblot analysis was performed using protein lysates derived from these cells and antibodies against phosphorylated JNK1 and 2, and JNK1 and JNK2 as designated. A representative blot from three independent determinations is shown. (B) Inhibition of JNK1 represses TBP expression. HepG2-ADH cells were transfected with JNK1 siRNA or mm RNA for 48h. Immunoblot analysis was performed using the lysates derived from the cells to measure the levels of JNK1, TBP and β-actin proteins. A representative blot from three independent determinations is shown). (C) JNK1 mediates TBP transcription. HepG2-ADH cells were transfected with TBP-Luc reporter construct or plus JNK1 siRNA (right) for 48 h; HepG2 cells were pretreated with 5μM SP600125 (JNK inhibitor) for 1 h (left). Cells were then treated with or without ethanol. The cell lysates was extracted from these cells and luciferase activity assay was performed to measure the TBP-dependent promoter activity. The values represent mean ± SE from three independent experiments.
3.3. Alcohol-mediates TBP transcription through Elk1 and AP-1 binding sites
Although our study has indicated that ethanol induced c-jun expression and Elk-1 phosphorylation and ethanol enhanced the activities of both AP-1- and Elk-1-dependent promoters (Zhong et al, 2011), it remains to be established whether there exists an alcohol response element in the TBP promoter region. The TBP promoter is regulated through the recruitment of either Elk-1 (Zhong et al, 2007; Fromm et al, 2008) to overlapping sites within the promoter (Fig. 6A). To identify a specific element affected by ethanol, we transiently transfected HepG2-ADH cells with TBP-Luc reporter constructs of wild type (p-54/-1hTBP), mutant Elk-1 site (p-54/-1ΔElk-1-hTBP), AP-1 mutant site (p-54/-1ΔAP-1-hTBP) or mutant both sites (p-54/-1ΔElk-1-ΔAP-1-hTBP) (Fromm et al, 2008). Comparable activities of the wild-type, mutant Elk-1 binding site (at −46bp G>A) and mutant AP-1 binding site (at −37 A>G, −38bp C>T) TBP promoters were observed in the absence of alcohol (Fig. 6B). The result indicates that the basic levels of TBP promoter activity were slightly decreased in mutant Elk-1 or AP-1 site of TBP-Lue reporters. Upon alcohol treatment, however, mutation of either the Elk-1 or AP-1 binding site significantly reduced alcohol-mediated induction of the promoter. Mutation of both Elk-1 and AP-1 sites resulted in the lowest basal activity of the promoter which could not be stimulated upon alcohol treatment (Fig. 6B). These results indicate that both Elk-1 and AP-1 binding sites are required for alcohol-mediated stimulation of the TBP promoter and they may be an alcohol-induced response element.
Fig. 6. Elk-1 and AP-1 binding sites at TBP promoter are required for alcohol-induced response.

(A) The nucleotide sequence of the most proximal TBP promoter depicting Elk-1 and AP-1 sites. (B) Ethanol-mediated stimulation of the TBP promoter requires both Elk-1 and AP-1 binding sites. The human TBP promoter sequence denoting the Elk-1 and AP-1 sites located between −54 and −1 of transcriptional start site (top). The TBP-Luc reporter constructs of the wild type (WT), mutant Elk-1 binding site (Elk-1 mt), mutant AP-1 binding site (AP-1 mt), or mutant both Elk-1 and AP-1 binding sites were transfected into HepG2-ADH cells. Where designated, cells were treated with 50 mM ethanol for 60 min. Luciferase activity was normalized to total protein levels, and changes were calculated based on untreated control. Fold change was calculated based on normalization to luciferase activity in WT TBP-Luc reporter transfected cells. All values shown are the means ± SEM of at least three independent experiments.
Discussion
This study presents a mechanistic analysis characterizing the effect of ethanol on TBP promoter. In this study, results demonstrate that alcohol increased TBP promoter activity and this induction is correlated with ADH expression. Repression of TBP decreases Pol III gene transcription, whereas overexpression of TBP enhances this transcription. Our studies identified a mechanism, by which alcohol activated JNK1 to mediate TBP promoter activity. Elk1 and AP-1 binding sites are critically important for ethanol-induced TBP transcription. Mutations of Elk-1 binding site at −46bp and/or AP-1 binding sites at −37bp and −38bp of TBP promoter abrogate the induction of TBP promoter activity. These findings support the notion that overlapping Elk-1 and AP-1 binding sites may be the response element of alcohol. Alcohol increases Elk-1 and AP-1 activity to enhance TBP expression, resulting in augmentation of Pol III gene transcription.
Alcohol consumption has consistently been associated with an increased risk for cancer development (El-Serag and Rudolph, 2007; Luedemann et al, 2008; Petri et al, 2004; singletary and Gapstur, 2001). Study by Wang et al have demonstrated that alcohol increased MCP-1 and CRR2 expression, which promoted mammary tumor growth in alcohol-fed mice (Wang et al, 2012). A recent study indicates that alcohol increased ERα expression to promote breast tumor formation in mice (Wong et al, 2012). Alcohol-fed HCV NS5A transgenic mice induced liver tumors in ~23% of these mice (Machida et al, 2009). These studies demonstrated alcohol consumption is associated with tumor development. However, the mechanism for alcohol-associated tumor formation remains to be established. Our studies have demonstrated that ethanol treatment augmented Pol III gene transcription in both liver cells (HepG2-ADH) or breast cells (MCF-7) (Zhong et al, 2011; Zhang et al, 2011), whereas increase in these gene activities are tightly linked to cell transformation and tumor formation (Zhong et al, 2011; Johnson et al, 2008; Zhang et al, 2011). This implies that increasing Pol III gene transcription is a potential mechanism for the formation of alcohol-associated tumors. Our earlier studies have demonstrated that alteration of TBP expression affected Pol III gene transcription (Zhong et al, 2004; Zhong et al, 2007). c-Jun and c-Fos are the subunits of AP-1. Our recent study indicates that alcohol induced TBP expression by enhancing Elk-1, c-Jun and c-Fos occupancy on the TBP promoter (Zhong et al, 2011). We have found that both Elk-1 and AP-1 activation are required for the TBP promoter induction (Zhong et al, 2004). However, it is not clear which region is affected by ethanol and whether there exists an alcohol-response element. In this present study by mutagenesis of the TBP promoter, we have demonstrated that mutations of Elk-1 binding site at −46bp G>A and/or AP-1 binding sites at −37bp A>G and −38bp C>T abrogate the induction of TBP promoter activity by ethanol. More interestingly, the Elk1 and AP-1 binding sites are overlapping at TBP promoter. It suggests that this overlapped region may be an alcohol-response element.
We have reported that JNK1 positively, whereas JNK2 negatively, mediated TBP expression and Pol III gene transcription (Zhong and Johnson, 2009). Our studies have revealed that ethanol induced JNK1 activation, but not JNK2 in both human liver cells and breast cells (Zhong et al, 2011; Zhang et al, 2011). The ethanol-induced activation of JNK1 was in HepG2-ADH cells; in contrast, ethanol could not activate JNK1 in HepG2-vector cells (Zhong et al, 2011). Previous analysis of these cells revealed that parental HepG2 cells did not exhibit detectable ADH activity, whereas the HepG2-ADH cells effectively metabolized ethanol to acetaldehyde (Clemens et al, 2002). This implies that alcohol metabolism induced the activation of JNK1. Inhibition of JNK1 reduced TBP expression and Pol III gene transcription (Zhong et al, 2007; Zhong and Johnson, 2009). Alteration of TBP expression affects Pol III gene transcription (Johnson et al, 2003; Zhong and Johnson, 2009). In the present study, we demonstrate that blocking JNK1 signaling inhibits TBP promoter induction by ethanol. As Elk-1 and AP-1 are the downstream components of JNK1 pathway, this indicates that alcohol activated JNK1 to enhance TBP transcription, resulted in increasing Pol III gene transcription. Thus, a possible mechanism of alcohol-induced liver tumor development is characterized by an alcohol-affected response element (Fig. 7).
Fig. 7. Schematic illustration of Alcohol mediating TBP promoter activity.
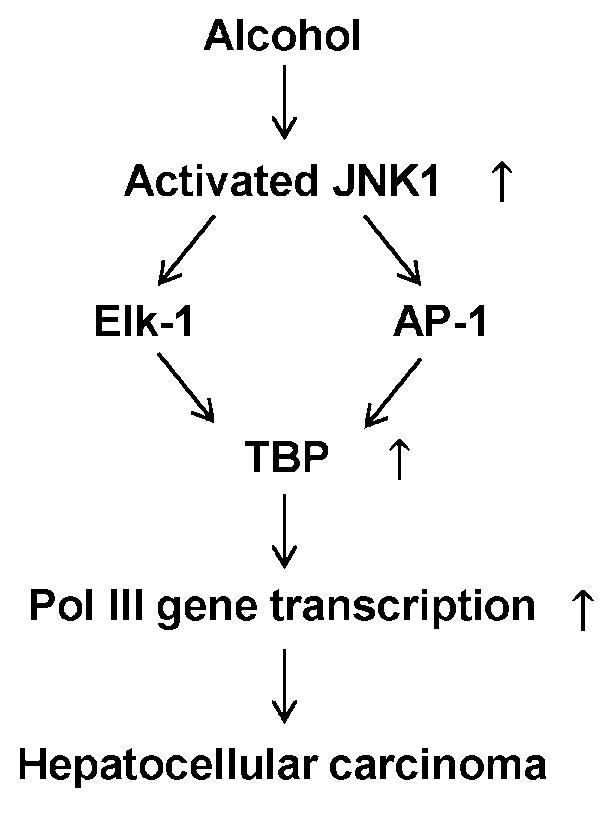
Ethanol activates JNK1 and increases Elk-1 and AP-1 activities to enhance TBP transcription, which in turn up-regulates Pol III gene transcription to promote alcohol-induced hepatocellular carcinoma.
In summary, the present study provides evidence that alcohol-induced JNK1 activation enhances TBP promoter activity through Elk-1 and AP-1 binding sites, which are critically important at this promoter in the alcohol-induced response (Fig. 7). The overlapping Elk-1 and AP-1sites may be an alcohol response element. It is the first report that this element mediates TBP transcription induced by alcohol. The novel findings suggest the possibility that inhibition of TBP expression may be a potential approach to repress alcohol-promoted HCC development.
Highlights.
Our studies demonstrate that JNK1 mediates alcohol-induces TBP promoter activity.
Alteration of TBP expression affects RNA Pol III-dependent gene transcription;
Mutation of Elk1 or AP-1 binding sites in TBP promoter region repress this induction by alcohol;
Alcohol affects Pol III gene transcription through the Elk1 and AP-1 binding sites in TBP promoter;
The binding sites may play a critical role in alcohol-induced deregulation of Pol III genes of HCC.
Acknowledgments
We would like to thank Dr. D.L., Johnson (University of Southern California), who provide TBP-luc reporter constructs. We would like to thank Dr. K. Machida (University of Southern California), who provide mouse liver tissues. This work was supported by National Institutes of Health grants AA017288 and AA021114 to S. Zhong.
Abbreviations
- JNKs
c-Jun N-terminal kinases
- AP-1
activator protein 1
- Elk-1
ETS domain-containing protein
- HCC
hepatocellular carcinoma
- Pol III
polymerase III
- ADH
Alcohol dehydrogenases
- Luc
luciferase
- HCV
hepatitis C virus
- NS5A
non-structure protein 5A
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Clemens DL, Forman A, Jerrells TR, Scorrell MF, Tuma DJ. Relationship between acetaldehyde levels and cell survival in ethanol-metabolizing hepatoma cells. Hepatology. 2002;35:1196–04. doi: 10.1053/jhep.2002.32668. [DOI] [PubMed] [Google Scholar]
- Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007;23:614–622. doi: 10.1016/j.tig.2007.09.001. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- Fromm JA, Johnson SA, Johnson DL. Epidermal growth factor receptor 1 (EGFR1) and its variant EGFRvIII regulate TATA-binding protein expression through distinct pathways. Mol Cell Biol. 2008;28:6483–95. doi: 10.1128/MCB.00288-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow SJ, Innes F, Derblay LE, MacLellan WR, Scott PH, White RJ. Regulation of RNA polymerase III transcription during hypertrophic growth. EMBO J. 2006;25:1522–1533. doi: 10.1038/sj.emboj.7601040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha WS, Kim CK, Song SH, Kang CB. Study on mechanism of multistep hepatotumorigenesis in rat: development of hepatotumorigenesis. J Vet Sci. 2001;2:53–58. [PubMed] [Google Scholar]
- Johnson DL, Johnson SA. Cell biology. RNA metabolism and oncogenesis. Science. 2008;320:461–462. doi: 10.1126/science.1158680. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Dubeau L, Johnson DL. Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation. J Biol Chem. 2008;283:19184–19191. doi: 10.1074/jbc.M802872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, et al. Increased expression of TATA-binding protein, the central transcription factor, can contribute to oncogenesis. Mol Cell Biol. 2003;23:3043–3051. doi: 10.1128/MCB.23.9.3043-3051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedemann CE, et al. Ethanol modulation of TNF-alpha biosynthesis and signaling in endothelial cells: synergistic augmentation of TNF-alpha mediated endothelial cell dysfunctions by chronic ethanol. Alcohol Clin Exp Res. 2005;29:930–938. doi: 10.1097/01.alc.0000171037.90100.6b. [DOI] [PubMed] [Google Scholar]
- Machida K, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci U S A. 2009;106:1548–1553. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Petri A, et al. Alcohol intake, type of beverage, and risk of breast cancer in pre- and postmenopausal women. Alcohol Clin Exp Res. 2004;28:1084–1090. doi: 10.1097/01.alc.0000130812.85638.e1. [DOI] [PubMed] [Google Scholar]
- Raha D, et al. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci U S A. 2010;107:3639–3644. doi: 10.1073/pnas.0911315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singletary KW, Gapstur SM. Alcohol and breast cancer: review of epidemiologic and experimental evidence and potential mechanisms, JAMA. 2001;286:2143–2151. doi: 10.1001/jama.286.17.2143. [DOI] [PubMed] [Google Scholar]
- Ullu E, Tschudi C. Alu sequences are processed 7SL RNA genes. Nature. 1984;312:171–172. doi: 10.1038/312171a0. [DOI] [PubMed] [Google Scholar]
- Wang S, et al. Ethanol promotes mammary tumor growth and angiogenesis: the involvement of chemoattractant factor MCP-1. Breast Cancer Res Treat. 2012;133:1037–48. doi: 10.1007/s10549-011-1902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ. RNA polymerase III transcription and cancer. Oncogene. 2001;23:3208–3216. doi: 10.1038/sj.onc.1207547. [DOI] [PubMed] [Google Scholar]
- Winter A, et al. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc Natl Acad Sci U S A. 2007;97:12619–12624. doi: 10.1073/pnas.230224097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woiwode A, et al. PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Mol Cell Biol. 2008;28:4204–4214. doi: 10.1128/MCB.01912-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AW, Dunlap SM, Holcomb VB, Nunez NP. Alcohol Promotes Mammary Tumor Development via the Estrogen Pathway in Estrogen Receptor Alpha-Negative HER2/neu Mice. Alcohol Clin Exp Res. 2012;36:577–587. doi: 10.1111/j.1530-0277.2011.01654.x. [DOI] [PubMed] [Google Scholar]
- Zhang C, Comai L, Johnson DL. PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol Cell Biol. 2005;25:6899–6911. doi: 10.1128/MCB.25.16.6899-6911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Jin J, Zhong Q, Yu XL, Levy D, Zhong SP* ERα mediates alcohol-induced deregulation of Pol III genes in breast cancer cells. Carcinogenesis. 2012 Oct 10; doi: 10.1093/carcin/bgs316. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Fromm J, Johnson DL. TBP is differentially regulated by JNK1 and JNK2 through Elk-1, controlling c-Jun expression and cell proliferation Mol. Cell Biol. 2007;27:54–64. doi: 10.1128/MCB.01365-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Johnson DL. The JNKs differentially regulate RNA polymerase III transcription by coordinately modulating the expression of all TFIIIB subunits. Proc Natl Acad Sci U S A. 2009;106:12682–12687. doi: 10.1073/pnas.0904843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Machida K, Tsukamoto H, Johnson DL. Alcohol induces RNA polymerase III-dependent transcription through c-jun by coregulating TBP and Brf1 expression. J Biol Chem. 2011;286:2393–2401. doi: 10.1074/jbc.M110.192955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Zheng C, Johnson DL. Epidermal Growth Factor enhances cellular TBP levels and induces RNA polymerase I- and III-dependent gene activity. Mol Cell Biol. 2004;24:5119–5129. doi: 10.1128/MCB.24.12.5119-5129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]