

Abstract
Glioblastoma multiforme (GBM) is the most common malignant brain tumor, which, despite combined modality treatment, reoccurs and is invariably fatal for affected patients. Recently, a member of the serine/threonine protein kinase D (PRKD) family, PRKD2, was shown to be a potent mediator of glioblastoma growth. Here we studied the role of PRKD2 in U87MG glioblastoma cell migration and invasion in response to sphingosine-1-phosphate (S1P), an activator of PRKD2 and a GBM mitogen. Time-lapse microscopy demonstrated that random cell migration was significantly diminished in response to PRKD2 silencing. The pharmacological PRKD family inhibitor CRT0066101 decreased chemotactic migration and invasion across uncoated or matrigel-coated Transwell inserts. Silencing of PRKD2 attenuated migration and invasion of U87MG cells even more effectively. In terms of downstream signaling, CRT0066101 prevented PRKD2 autophosphorylation and inhibited p44/42 MAPK and to a smaller extent p54/46 JNK and p38 MAPK activation. PRKD2 silencing impaired activation of p44/42 MAPK and p54/46 JNK, downregulated nuclear c-Jun protein levels and decreased c-JunS73 phosphorylation without affecting the NFκB pathway. Finally, qPCR array analyses revealed that silencing of PRKD2 downregulates mRNA levels of integrin alpha-2 and -4 (ITGA2 and -4), plasminogen activator urokinase (PLAU), plasminogen activator urokinase receptor (PLAUR), and matrix metallopeptidase 1 (MMP1). Findings of the present study identify PRKD2 as a potential target to interfere with glioblastoma cell migration and invasion, two major determinants contributing to recurrence of glioblastoma after multimodality treatment.
Keywords: Glioblastoma, Sphingosine-1-phosphate, PRKD2, MAPK, C-Jun, Invasion
Highlights
-
•
Sphingosine-1-phosphate induces glioma cell migration and invasion.
-
•
Part of the effects is mediated by protein kinase D2 (PRKD2) activation.
-
•
Inactivation of PRKD2 attenuates glioblastoma cell migration and invasion.
-
•
Both, RNAi and pharmacological inhibition of PRKD2 inhibits MAPK signaling.
-
•
PRKD2 regulates transcription of gene products implicated in migration and invasion.
Introduction
Glioblastoma multiforme (GBM; astrocytoma WHO grade IV) is the most common and malignant type of primary brain tumors. GBM is characterized by high proliferative rate, aggressive invasiveness and insensitivity to radio- and chemo-therapy. The median survival in patients with newly diagnosed GBM following treatment with surgery, radio- and chemo-therapy is approximately 15 months [1]. Comprehensive genetic analyses of GBMs identified a number of mutations affecting three major pathways: the receptor tyrosine kinase (RTK) pathway and its downstream oncogenic signaling by PI3K/AKT/mTOR and RAS/MAPK, the p53, and the retinoblastoma protein pathway [2,3]. Modifications of these pathways drive gliomagenesis by mediating proliferation, migration, angiogenesis, survival, and deregulation of apoptotic signaling.
Three major reasons account for therapy resistance: the presence of the blood–brain barrier that restricts drug distribution to the brain, the heterogeneity of the tumor that consists of cell populations with different drug sensitivities, and the propensity of the tumor cells to infiltrate the normal brain leading to recurrences [4]. Individual cells or groups of cells can detach from the primary tumor and migrate to distant sites within normal brain structures [5]. The invasive nature of GBM has been frequently implicated as a key feature of resistance to therapy [6]. GBM cell invasion is a multistep, but highly coordinated process that starts with the degradation of surrounding matrix proteins by matrix metalloproteinases (MMPs) [7]. Cells must then detach from their neighboring cells and matrix components in order to gain motility and move through healthy brain tissue. During this process cells must form new and release ‘old’ focal adhesions and constantly reorganize their cytoskeleton. This process is receptor mediated and involves complex interactions between integrins, receptor tyrosine kinases and (lipid-mediated) activation of downstream signaling cascades [7].
A core-signaling pathway in migration and invasion is the phospholipase C pathway catalyzing the formation of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) leading to activation of the PKC family [8]. One of the targets activated by DAG and different PKC isoforms is the protein kinase D (PRKD) family [9]. The PRKD family is located downstream of PKC and belongs to the calcium/calmodulin dependent protein kinase superfamily and comprises of three isoforms PRKD-1, -2, and -3. Being major players in cell proliferation, motility, and angiogenesis, PRKD members received considerable interest as potential anti-cancer targets. Although the evidence for PRKD1 in tumor development is somewhat ambiguous (for review see [10]), PRKD1 is overexpressed in pancreatic ductal adenocarcinoma [11,12] and pharmacological inhibition of PRKD1 was shown to impair pancreatic ductal adenocarcinoma proliferation [13]. Both, PRKD1 and PRKD3 are involved in the progression and invasion of prostate cancer [14,15]. PRKD2 mediates NFκB activation in leukemia cells [16] and expression levels correlate with the state of de-differentiation in malignant human lymphoma [17]. Only recently it was demonstrated that PRKD2 is a potent regulator of glioblastoma cell growth in vitro and in vivo [18].
There is ample evidence that PRKD members are regulators of cell migration and invasion. PRKD1 controls fibroblast motility by modulating membrane traffic from the trans-Golgi network to the plasma membrane [19]. PRKD1 is also involved in integrin transport to focal adhesions, thereby affecting cell migration [20]. PRKD members phosphorylate downstream targets that are involved in cell motility including cortactin [21], slingshot 1 like [22], and Par-1 [23]. PRKD1 regulates MMP expression in breast cancer cells [24] that control cell migration and invasion and play an important role in glioblastoma cell invasion [25]. PRKD signaling starts with ligand binding to seven-transmembrane or tyrosine kinase receptors that activate PLCβ or PLCγ. Activation of this pathway results in the generation of DAG and IP3. Membrane-associated DAG binds and activates PKC, which then phosphorylates two serine residues in the activation loop of PRKDs [9]. One of the G protein-coupled receptor (GPCR) ligands that activate PRKD is sphingosine-1-phosphate (S1P), a bioactive lipid that signals via five cognate G protein-coupled receptors termed S1P1–5 [26]. Exogenously added S1P is a potent glioblastoma mitogen and enhances glioblastoma invasiveness [27–31]. Accumulating evidence suggests that S1P is involved in cancer progression including cell transformation, cell survival, cell migration and metastasis, and neoangiogenesis of the tumor microenvironment [26]. In particular PRKD2 that is highly expressed in glioblastoma [18], has critical roles in vascular biology and angiogenesis [32]. PRKD2 mediates vascular endothelial growth factor (VEGF)-induced endothelial cell (EC) proliferation and migration and regulates the expression of VEGF receptor-2 and the production of cytokines in ECs [33]. Furthermore, PRKD2 controls hypoxia-induced VEGF synthesis and secretion by tumor cells and regulates VEGF signaling in tumor-associated endothelial cells [34]. In addition, strong PRKD2 expression in the tumor endothelium of microvascular proliferations in glioblastoma was reported [18]. Only recently a splice variant of human calcium and integrin-binding protein 1 was identified as a novel PRKD2 substrate and interactor that contributes to tumor growth in vivo by promoting angiogenesis [35].
In the present study we tested the hypothesis that PRKD2 regulates U87MG glioblastoma cell migration and invasion. Pharmacological inhibition and PRKD2 silencing impaired migratory, chemotactic, and invasive behavior of non-stimulated and S1P-stimulated U87MG cells and attenuated activation of MAPK pathways. Gene expression analysis by RT profiler PCR arrays revealed altered expression levels of several genes involved in GBM motility and migration.
Materials and methods
Cell culture supplies were from Gibco (Invitrogen, Vienna, Austria), PAA Laboratories (Linz, Austria) and Costar (Vienna, Austria). Horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG and mouse anti-actin were from Sigma (Vienna, Austria). Sphingosine-1-phosphate, mouse anti-PRKD2, rabbit anti-NFκB (p50 and p65), rabbit anti-lamin A/C and HRP-labeled goat anti-mouse IgG were from Santa Cruz Biotechnology (CA, USA). Rabbit anti-p44/42 MAPK, rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204), rabbit anti-p38 MAPK, rabbit anti-phospho-p38 MAPK (Thr180/Tyr182), rabbit anti-SAPK/JNK, rabbit anti-phospho-SAPK/JNK (Thr183/Tyr185), rabbit anti-c-Jun, rabbit anti-phospho-c-Jun (Ser63 and Ser73), and rabbit anti-GAPDH were from Cell Signaling Technology (MA, USA). Rabbit anti-phospho-PRKD2 (Ser876 in the autophosphorylation domain) was from Abcam (Cambridge, UK). SuperSignal Western blot detection reagent kit was from Pierce (Thermo Scientific, MA, USA) and ECL Plus Western Blotting Reagents were form Amersham Biosciences (Vienna, Austria). SuperScript II Reverse Transcriptase and Oligofectamine were from Invitrogen (Vienna, Austria). Random hexamer primer was from Thermo Scientific (MA, USA). RNeasy Plus Kit, QuantiFast SYBR Green PCR kit, QuantiTect primer assays Hypoxanthine phosphoribosyltransferase 1 (Hs_HPRT_1_SG; internal standard), Protein Kinase D2 (Hs_PRKD2_1_SG), Integrin alpha 2 (Hs_ITGA2_1_SG), Integrin alpha 4 (Hs_ITGA4_1_SG), Plasminogen activator, urokinase (Hs_PLAU_1_SG), Plasminogen activator, urokinase receptor (Hs_PLAUR_1_SG), Matrix metalloproteinase 1 (Hs_MMP1_1_SG) and the siRNA specifically targeting PRKD2 (Hs_PRKD2_5 and Hs_PRKD2_6) were from Qiagen (Hilden, Germany) and non-targeting siRNA (scrambled siRNA) was from Dharmacon (Thermo Scientific, MA, USA). BD Matrigel Invasion Chamber (8 µM pore size) and Control Inserts (8 µM pore size) were from BD Biosciences Europe (Erembodegem, Belgium). CRT0066101 was from Cancer Research Technology (London, UK).
Cell culture
U87MG cells were from ATCC (HTB 14) and maintained in Dulbecco´s modified Eagle medium (DMEM)/high glucose supplemented with 10% FCS and 2% penicillin/streptomycin. Cells were grown at 37 °C under 5% CO2.
RNA interference
U87MG cells were seeded at 40,000 cells per well into 12 well plates and grown for 24 h. Transfection of siRNA (20 nM) was performed using Oligofectamine according to the manufacturer's instructions (Invitrogen). Untreated cells (control) and cells transfected with Oligofectamine alone (mock) or non-targeting siRNA (scrambled siRNA) were used as controls. The siRNAs specifically targeting the four known transcript variants of PRKD2 (NM_001079880, NM_001079881, NM_001079882, NM_016457) were from Qiagen (Hs_PRKD2_5 and Hs_PRKD2_6) and scrambled siRNA (siSCR) was from Dharmacon. At the indicated time points cells were trypsinized, lysed and protein or RNA extracts were collected.
Motility assay
For time-lapse microscopy untreated cells and cells transfected with siSCR and siPRKD2_5 were seeded on 24-well tissues. Two days post transfection images were acquired every 20 min for 24 h at five different positions of each well using a Zeiss Cell Observer microscope. Cell motility of 50 cells per treatment group was assessed using the Manual Tracking Macro and the Chemotaxis plugin of ImageJ. Mean velocity±SEM, the mean accumulated distance±SEM, and the mean Euclidian distance±SEM of the analyzed cells were calculated. *p<0.05, **p<0.01 in comparison to siSCR
Chemotaxis assay
The migration assay was performed using cell culture inserts with PET membrane (8 µm pore size). U87MG cells were seeded at 50,000 cells per well into 12 well culture plates, grown for 24 h and transfected with specific siRNA (20 nM). Two days after transfection, cells were harvested and 50,000 cells in medium+0.5% FCS were added to the upper compartment of the cell culture inserts. Where specified, 1 µM CRT0066101 was added to the upper compartment of the cell culture inserts. The lower compartment was filled with medium+10% FCS, medium+0.5% FCS or serum free medium containing 1 µM S1P. After two days of incubation at 37 °C, the upper surface of the membrane was wiped with a cotton swab to remove non-migratory cells. Cells that migrated to the bottom side of the membrane were fixed with methanol, stained with toluidine blue for 2 min and the membrane was washed twice with water. Membranes were placed on a glass slide, and from each membrane six fields were counted using an optical microscope (20× magnification). Assays were performed in quadruplicate for each condition. Data are presented as mean±SEM. ***p<0.001, **p<0.01, *p<0.05 in comparison to control or siSCR.
Invasion assay
For the invasion assay cell culture inserts with PET membrane (8 µm pore size) coated with matrigel were used. U87MG cells were transfected with specific siRNA (20 nM) and two days post transfection, cells were harvested. After rehydration of the matrigel with medium, 50,000 cells in medium+0.5% FCS or 50,000 cells in medium+0.5% FCS+1 µM CRT0066101 were added to the upper compartment of the cell culture inserts. The lower compartment was filled with medium+10% FCS, medium+0.5% FCS or serum free medium containing 1 µM S1P or 2.5 µM S1P (2.5 µM S1P were readded after 24 h). After two days of incubation at 37 °C, cells that migrated through the matrigel were quantified as described above for the chemotaxis assay. Data are presented as mean±SEM. ***p<0.001, **p<0.01 in comparison to control or siSCR.
PRKD2 and MAPK activation
Prior to measurement of PRKD2 or MAPK activation, U87MG cells were serum-starved for 3 h. Cells were incubated in the absence or presence of S1P for the indicated times and concentrations. Where specified, cells were incubated with CRT0066101 over night before S1P treatment. After incubation cells were lysed and protein extracts were collected.
Western blot analysis
For immunoblotting, whole cell extracts and/or cytoplasmic/nuclear extracts (prepared with the NE-PER Nuclear and Cytoplasmic Extraction Reagents from Pierce) were used. Protein concentration was measured using the Bradford protein assay. Equal protein aliquots were loaded, separated on SDS-PAGE under reducing conditions, transferred to PVDF membranes and immunochemical detection of p44/42 MAPK, phospho-p44/42 MAPK, p38 MAPK, phospho-p38 MAPK, JNK, phospho-JNK, c-Jun, phospho-c-Jun, NFκB, PRKD2, and phospho-PRKD2 was performed with specific primary antibodies. Immunoreactive bands were visualized with HRP-conjugated goat anti-rabbit or goat anti-mouse IgG and the ECL detection system. Quantification of protein expression (relative optical density) was normalized to appropriate loading controls (lamin A/C, actin, GAPDH) and phosphorylation of MAPKs was referred to total MAPK protein.
Human Cancer PathwayFinder PCR array
Total RNA of mock and siPRKD2_5 transfected U87MG cells was isolated in triplicates on day three post transfection. cDNA synthesis and RT-PCR arrays were performed according to the manufacturer's instructions. For data analyses the web-based software package provided by SABiosiences was used. Raw data were normalized to the most stably expressed housekeeping gene (ribosomal protein L13a) and fold-changes in gene expression were calculated using the ΔCt method. Genes with at least 1.8 fold expression changes compared to mock transfected cells were considered for further analyses by qPCR.
qPCR
Cells were transfected with specific siRNAs, lysed and RNA extracts were collected two or four days post transfection. Total RNA was isolated using the RNeasy Kit. Aliquots of 3 µg of total RNA were reverse transcribed using SuperScript II Reverse Transcriptase and random hexamer primers according to the manufacturer's protocol (Invitrogen). Real-time PCRs were performed with an Applied Biosystems 7900HT Fast Real Time PCR System, the QuantiFast SYBR Green PCR kit and QuantiTect Primer Assays. Hypoxanthine phosphoribosyltransferase 1 (HPRT) was used as housekeeping gene. The following QuantiTect primer assays were used: Hypoxanthine phosphoribosyltransferase 1 (Hs_HPRT_1_SG; internal standard), Protein Kinase D2 (Hs_PRKD2_1_SG), Integrin alpha 2 (Hs_ITGA2_1_SG), Integrin alpha 4 (Hs_ITGA4_1_SG), Plasminogen activator, urokinase (Hs_PLAU_1_SG), Plasminogen activator, urokinase receptor (Hs_PLAUR_1_SG), Matrix metalloproteinase 1 (Hs_MMP1_1_SG).
Statistical analyses
Data are presented as mean±SEM. One-way ANOVA followed by Bonferroni post-hoc comparison test was used for analysis of statistical significance. ***p<0.001, **p<0.01, *p<0.05.
Statistical significance of differences in mRNA expression levels was analyzed using the relative expression software tool (REST©, http://www.gene-quantification.de/rest.html) using a pair-wise fixed reallocation test [36].
Results
PRKD2 silencing inhibits migration and invasion of U87MG glioma cells in response to S1P
Initially we determined the efficacy of PRKD2 (Gene ID: 25865) silencing in U87MG cells using two different 21mer siRNA constructs. As shown in Fig. 1A, PRKD2 silencing led to reduced mRNA expression. Maximum knockdown was reached at day 2 with relative mRNA expression levels of 0.28 for siPRKD2_5 and 0.12 for siPRKD2_6. On protein level, expression was efficiently decreased by 95% up to day four with both siRNAs. A scrambled control siRNA had no effect on PRKD2 protein expression (Fig. 1B).
Fig. 1.

Efficacy of PRKD2 silencing. Knockdown of PRKD2 expression in U87MG cells was performed by RNA interference using two different siRNA constructs (Oligofectamine was used to transfect 20 nM siRNA). Silencing efficacy was analyzed by (A) qPCR two and four days post transfection and (B) Western blotting three and five days post transfection. Results in (A) represent mean±SD of three different experiments. Results in (B) show one representative Western blot out of five independent experiments. Numbers represent relative optical densities of PRKD2 protein normalized to loading controls.
Next, we examined the effect of PRKD2 silencing on random 2D migration of U87MG cells by time-lapse video microscopy. By analyzing the tracks of 50 individual cells per group the velocity, accumulated distance, and Euclidian distance was calculated. The directionality of cell movement was determined as the Euclidian distance divided by the accumulated distance. As shown in Fig. 2A, silencing of PRKD2 resulted in a small, but statistically significant (p<0.05) reduction of migration speed as compared to cells transfected with scrambled siRNA (0.38 vs. 0.42 µm/h; siPRKD2_5 vs. siSCR). Both, accumulated (540 vs. 630 µm; siPRKD2 vs. siSCR) and Euclidian (125 vs. 195 µm; siPRKD2 vs. siSCR) distance was significantly reduced in response to RNAi (Fig. 2B and C). In addition, the directionality of cell movement was attenuated in PRKD2 silenced cells (0.23 vs. 0.31; siPRKD2 vs. siSCR).
Fig. 2.

Interference with PRKD2 expression impairs random migration of U87MG cells. For time-lapse microscopy untreated cells and cells transfected with siSCR or siPRKD2_5 were seeded on 24-well tissues. Two days post transfection images were acquired every 20 min for 24 h at five different positions of each well. Cell motility of 50 cells per treatment group was assessed using the Manual Tracking Macro and the Chemotaxis plugin of ImageJ. The (A) mean velocity, (B) mean accumulated distance and (C) the mean Euclidian distance was calculated. *p<0.05, **p<0.01 in comparison to siSCR (1way ANOVA).
To determine the impact of pharmacological inhibition (using the PRKD family inhibitor CRT0066101) or PRKD2 silencing on chemotactic migration, Transwell inserts were used. Directed migration was analyzed in medium containing FCS (10% or 0.5%) or S1P (1 µM complexed to BSA) in the lower chamber. Representative micrographs of transmigrated cells are shown in Fig. 3A. These microscopic pictures indicate that both, pharmacological inhibition and RNAi of PRKD2 attenuates cell migration across uncoated Transwell inserts. Quantitative evaluation of these experiments showed that a reduction of FCS from 10% to 0.5% reduced the number of migrated non-transfected cells by 80% (220 vs. 45 cells/field). The addition of S1P to the lower chamber increased the migration by 2-fold in comparison to 0.5% FCS (Fig. 3B).
Fig. 3.

Interference with PRKD2 expression reduces chemotactic migration and the invasive potential of U87MG cells. Untreated cells, cells transfected with siSCR and siPRKD2_5 and cells treated with 1 µM CRT0066101 were allowed to migrate across uncoated or matrigel-coated Transwell inserts. The lower chamber was loaded with medium containing FCS (10 or 0.5%; v/v) or S1P (1 µM or 2.5 µM; serum free). Cells that migrated to the bottom side of the membrane were fixed with methanol, stained with toluidine blue for 2 min and the membrane was washed twice with water. Cells in six fields from each membrane were counted using an optical microscope (20x magnifications). (A) Representative images of migrated cells. Quantitative analysis of cell migration across (B) uncoated or (C) matrigel-coated Transwell inserts. Data are presented as mean±SEM. Assays were performed in quadruplicate for each condition. ***p<0.001, **p<0.01 in comparison to control or siSCR (1way ANOVA).
CRT0066101 reduced transmigration in 10% FCS by 24% (167 vs. 220 cells/field) and in response to S1P by 23% (71 vs. 92 cells/field). Under unphysiological, serum free conditions migration in the presence of CRT0066101 was increased by 2.1-fold. Transfection with siSCR yielded essentially the same results as observed with non-transfected cells. In contrast, chemotactic migration was significantly attenuated in PRKD2 silenced cells under all experimental conditions applied. Compared to siSCR, PRKD2 silencing impaired chemotactic migration by 51, 30 and 45% (10% or 0.5% FCS, and 1 µM S1P, respectively; Fig. 3B).
We further assessed the impact of CRT0066101 and PRKD2 silencing on invasive properties of U87MG cells in matrigel-coated Transwell inserts. Under control conditions a reduction of FCS from 10% to 0.5% in the lower chamber completely abrogated cell invasion while in the presence of S1P 3 (1 µM) and 10 cells (2.5 µM) invaded per field (Fig. 3C). CRT0066101 inhibited invasion by 49% and 60% (10% FCS and 2.5 µM S1P, respectively). As shown for chemotactic migration, CRT0066101 increased invasion under serum free conditions (0 vs. 12 cells/field). Transfection with siSCR slightly diminished (6.6%) invasion. Silencing of PRKD2 significantly reduced the number of transmigrated cells by 69 (10% FCS), 95 (1 µM S1P) and 78% (2.5 µM S1P; Fig. 3C) in comparison to siSCR.
Exogenous S1P induces activation of PRKD2 and MAPK pathways
PRKD family members are activated in response to extracellular stimuli, including S1P, thereby affecting various downstream signaling cascades implicated in tumor biology [10]. Thus, we first analyzed activation of PRKD2 and MAPK family members in response to S1P. As shown in Fig. 4 the addition of exogenous, BSA-complexed S1P resulted in a concentration- and time-dependent activation of PRKD2, p44/42 MAPK, p54/46 JNK and p38 MAPK. Autophosphorylation levels of PRKD2 at S876 were continuously increasing in response to S1P (1 nM to 10 µM). Activation of the MAPK family members occurred at comparable S1P levels (10–100 nM) as observed for PRKD2 (Fig. 4A). Stimulation with S1P for increasing times revealed maximal activation of PRKD2 after 5 min decreasing to baseline after 60 min. Time-dependent activation of p44/42 and p38 MAPKs followed comparable kinetics as observed for PRKD2 (Fig. 4B). p54/46 JNK activation reached its maximum at later time points (15 and 30 min) and was still elevated at 60 min.
Fig. 4.

S1P activates mitogenic signaling cascades in U87MG cells. Cells were grown in 6 well plates and activated with BSA-complexed S1P in a concentration (A; 1 nM–10 µM; 10 min) or time-dependent (B; 1 µM; 5–60 min) manner. Whole cell lysates were separated by SDS-PAGE (10 or 30 µg protein/lane) and transferred to PVDF membrane. Protein levels of phospho-PRKD2 (Ser876), PRKD2, phospho-p44/42 MAPK (pp44/42), p44/42 MAPK, phospho-p38 MAPK (pp38), p38, phospho-p54/46 JNK (p54/46 JNK), and JNK were determined using specific antibodies. Results of one experiment out of three (that provided similar results) are shown.
Pharmacological inhibition and silencing of PRKD2 attenuates downstream MAPK pathways
In the next set of experiments the outcome of CRT0066101 and PRKD2 silencing on S1P-mediated activation of MAPK family members was studied. CRT0066101 (0.35 and 1 µM) attenuated basal and S1P-induced autophosphorylation of PRKD2, p44/42 MAPK and to a lesser extent p54/46 JNK and p38 MAPK (Fig. 5A). PRKD2 silencing significantly diminished phosphorylation of p44/42 MAPK and p54/46 JNK under basal or S1P-stimulated conditions (Fig. 5B), whereas phosphorylation of p38 MAPK was slightly impaired by scrambled siRNA. Neither CRT0066101 nor RNAi affected the levels of non-phosphorylated p54/46 JNK, p44/42, and p38 MAPKs. Thus, pharmacological inhibition and silencing of PRKD2 resulted in downregulation of signaling cascades that are implicated in glioblastoma migration and invasion.
Fig. 5.

Pharmacological inhibition and silencing of PRKD2 impairs MAPK signaling and c-Jun regulation but does not affect the NFκB pathway. Cells were incubated with (A) CRT0066101 at the indicated concentrations overnight or (B) transfected with scrambled siRNA (S) and siPRKD2_5 (P5) for two days and activated with 1 µM BSA-complexed S1P for the indicated times. Protein levels of PRKD2 and MAPK family member activation were analyzed as described in Fig. 4. Results of one experiment out of three (that provided similar results) are shown. (C) c-Jun expression and activation was analyzed in nontransfected and transfected (siSCR, siPRKD2_5) cells in the absence or presence of S1P (1 µM, 5 min). Nuclear lysates were separated by SDS-PAGE (10 µg protein/lane) and transferred to PVDF membrane. Total c-Jun expression and c-Jun protein activated by phosphorylation at Ser63 or Ser73 were analyzed using specific antibodies. Results of one experiment out of three are shown. (D) Cytosolic and nuclear PRKD2, pPRKD2, and NFκB (p50 and p65) expression was analyzed in nontransfected and transfected (siSCR, siPRKD2_5) cells under unstimulated and stimulated (1 µM S1P for 5 min) conditions. Cytosolic and nuclear protein fractions were separated by SDS-PAGE (10 µg protein/lane) and transferred to PVDF membrane. Immunoreactive bands were detected using rabbit antibodies. One experiment out of two is shown.
JNK and ERK direct distinct cellular activities even though they share a number of common substrates, including several transcription factors. In PC12 cells activation of the ERK pathway results in stimulation of both c-Jun synthesis and c-Jun phosphorylation, whereas the JNK pathway triggers phosphorylation [37]. Western blot analysis of nuclear cell extracts (c-Jun was undetectable in the cytosolic fraction, data not shown) demonstrated that PRKD2 silencing attenuates the level of total c-Jun protein under basal and S1P stimulated conditions (Fig. 5C). Analysis of phosphorylated c-Jun revealed that PRKD2 silencing was without effects on c-JunS63 but slightly downregulates c-JunS73 phosphorylation under basal and S1P stimulated conditions (Fig. 5C).
In prostate cancer cells PRKD-2 and -3 were shown to promote cell invasion via the NFκB pathway [15]. To investigate whether this pathway is affected by PRKD2 silencing in U87MG cells we have analyzed NFκB levels in cytosolic and nuclear fractions of unstimulated and S1P-stimulated cells. Data shown in Fig. 5D demonstrate that PRKD2 silencing was without effect on protein levels of the NFκB-associated subunits p65 and p50 in U87MG cells.
PRKD2 silencing in U87MG cells affects mRNA expression levels of genes involved in glioma migration and invasion
To get an indication about downstream targets of PRKD2 under basal conditions, pathway analysis by qPCR array analysis was performed. Out of 28 genes involved in adhesion, invasion and metastasis, 5 genes (ITGA2, ITGA4, MMP1, PLAU, and PLAUR) exhibited at least a 1.8-fold change in mRNA expression levels in response to PRKD2 silencing compared to mock transfected cells (Table 1). Altered expression levels of these five products were confirmed by qPCR (Fig. 6). The relative mRNA expression levels of ITGA2, ITGA4, PLAU, and PLAUR after transfection with siPRKD2_5 were reduced up to 0.36, 0.61, 0.53, and 0.70, respectively. The most prominent effect of PRKD2 silencing was seen for the expression of MMP-1 with a maximum decrease of mRNA levels to 0.22 on day 2. At day 4 post silencing most of these effects were reduced, with exception of PLAU, which was upregulated 1.9-fold.
Table 1.
Alphabetical list of human genes related to adhesion, invasion and metastasis from the RT Profiler Pathway Array (Human Cancer PathwayFinder) and their regulation in response to PRKD2 silencing using siPRKD2_5.
| Name of gene | Gene symbol | Accession no | Fold regulation |
|---|---|---|---|
| Integrin, alpha 1 | ITGA1 | NM_181501 | −1.11 |
| Integrin, alpha 2 | ITGA2 | NM_002203 | −3.11* |
| Integrin, alpha 3 | ITGA3 | NM_002204 | 1.35* |
| Integrin, alpha 4 | ITGA4 | NM_000885 | −1.93* |
| Integrin, alpha V | ITGAV | NM_002210 | 1.49* |
| Integrin, beta 1 | ITGB1 | NM_002211 | −1.01 |
| Integrin, beta 3 | ITGB3 | NM_000212 | 1.30* |
| Integrin, beta 5 | ITGB5 | NM_002213 | 1.48* |
| Melanoma cell adhesion molecule | MCAM | NM_006500 | −1.32* |
| Met proto-oncogene | MET | NM_000245 | −1.14 |
| Matrix metallopeptidase 1 | MMP1 | NM_002421 | −4.00* |
| Matrix metallopeptidase 2 | MMP2 | NM_004530 | 1.63* |
| Matrix metallopeptidase 9 | MMP9 | NM_004994 | low expression |
| Metastasis associated 1 | MTA1 | NM_004689 | −1.23* |
| Metastasis associated 1 family, member 2 | MTA2 | NM_004739 | −1.18* |
| Metastasis suppressor 1 | MTSS1 | NM_014751 | −1.32* |
| Non-metastatic cells 1, protein (NM23A) expressed in | NME1 | NM_000269 | −1.23* |
| Non-metastatic cells 4, protein expressed in | NME4 | NM_005009 | 1.00 |
| Plasminogen activator, urokinase | PLAU | NM_002658 | −2.69* |
| Plasminogen activator, urokinase receptor | PLAUR | NM_002659 | −1.92* |
| Pinin, desmosome associated protein | PNN | NM_002687 | −1.40* |
| S100 calcium binding protein A4 | S100A4 | NM_002961 | 1.11* |
| Serpin peptidase inhibitor, member 5 | SERPINB5 | NM_002639 | low expression |
| Serpin peptidase inhibitor, member 1 | SERPINE1 | NM_000602 | −1.34* |
| Spleen tyrosine kinase | SYK | NM_003177 | low expression |
| TIMP metallopeptidase inhibitor 1 | TIMP1 | NM_003254 | −1.09* |
| TIMP metallopeptidase inhibitor 3 | TIMP3 | NM_000362 | low expression |
| Twist homolog 1 | TWIST1 | NM_000474 | 1.45* |
The table lists the genes involved in adhesion and invasion that were analyzed by the RT² Profiler Array system. Data represent the fold-regulation of individual gene expression in PRKD2 silenced U87MG cells compared to mock transfected cells. Data were evaluated from three biological replicates and calculated by Student's t-test of the replicate 2^ (ΔCt) values for each gene (*p≤0.05). Positive values indicate upregulation, negative values downregulation of genes. Only target genes regulated >1.8 fold were considered for further qPCR analyses. Relative expression levels of four genes were too low for analyses.
Fig. 6.
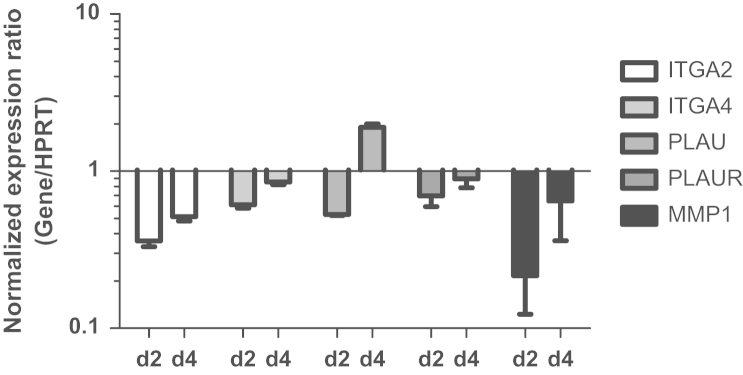
Silencing of PRKD2 impacts on U87MG gene expression. PRKD2 expression was silenced with siPRKD2_5. Two and four days post transfection target gene expression was analyzed by qPCR using validated primer pairs. Hypoxanthine phosphoribosyltransferase 1 (HPRT) was used as housekeeping gene. Relative gene expression of target genes is presented in relation to mock transfection. Results represent mean±SD from three independent experiments. Gene expression ratios were calculated by REST as described in Materials and methods.
Discussion
The acquisition of cell motility is a key property of glioblastoma cells contributing to the dismal perspectives for affected patients. Work presented here identifies a S1P/PRKD2/MAPK signaling pathway that contributes to GBM cell migration and invasion. Our results show that S1P activates PRKD2 and induces a migrational and invasive response, which is attenuated in response to PRKD2 silencing (Figs. 2–4). Accumulating evidence suggests that S1P and S1P receptors are regulators of GBM growth, migration, and invasion via outside-in or inside-out signaling [38]. S1P signal transduction is mediated via five G protein-coupled receptors (S1P1–5) [26]. Of these, S1P1–3 and S1P5 are expressed in glioblastoma cell lines and tissue [39,40]. S1P1–3 couple to the PLC/PKC pathway (via Gi and Gq) and are therefore candidates that are able to induce PRKD2 activation as observed during the present study (Fig. 4). Exogenously added S1P enhances glioblastoma invasiveness [30,31] and microarray analyses have shown that upregulation of proteases by S1P could contribute to invasive properties of GBM cells [41]. S1P directly regulates uPAR expression in GBM cells thereby increasing invasiveness [31].
During carcinogenesis alterations in cell morphology and gene expression and the disruption of cell–cell adhesions promote cellular motility and invasiveness [42]. We show that PRKD2 silencing resulted in decreased motility and invasiveness of U87MG cells (Fig. 3), which in principle can be a direct effect and/or result from signaling events downstream of PRKD2. Along this line we provide evidence that pharmacological inhibition (CRT0066101; a small molecule inhibitor of the PRKD family that blocks pancreatic cancer; Ref. [13]) or silencing of PRKD2 results in impaired activation of MAPK pathways (Fig. 5A, B). This is in agreement with other reports demonstrating MAPK activation in a PRKD-dependent manner [43–45] mediating pro-proliferative properties as specifically reported for PRKD2 in glioblastoma [18]. However, MAPK signaling is also centrally involved in several other steps of tumor development. Phorbolester-mediated activation of the p44/42 MAPK pathway was shown to induce migration and invasion of GBM cell [46]. p44/42 MAPK is essential for integrin expression [47] and p44/42-dependent phosphorylation of myosin light chain kinase, calpain, focal adhesion kinase, or paxillin promotes cancer cell migration (for review see Ref. [48]). p44/42 MAPK activation positively regulates uPAR expression [49], induces the expression of MMP-1 [25], and enhances 3D invasion of pancreatic cancer cells via MMP-1 expression [50]. Thus, decreased p44/42 MAPK signaling as observed here in response to PRKD2 silencing is compatible with altered gene expression patterns as described below.
The second MAPK pathway that was attenuated in response to CRT0066101 and PRKD2 silencing during the present study is the p54/46 JNK pathway (Fig. 5A, B). JNK signaling plays a pivotal role in GBM invasion as pharmacological inhibition reduces invasive properties [51]. Importantly, pharmacological inhibition of the JNK pathway in vivo depletes self-renewing and tumor-initiating glioblastoma stem cell population [52,53]. Such a targeted pharmacological approach could eliminate tumor initiating GBM cells that infiltrate deep into unresectable brain regions. In this context it is noteworthy that PRKD2 was identified in the phosphoproteome of EGF-activated human glioblastoma-initiating cells [54]. These findings make it tempting to speculate that PRKD2 not only plays a role in primary GBM [18] but also could contribute to tumor recurrence via GBM-initiating cells.
The c-Jun protein, a member of the AP-1 transcription factor family, activates several events required for tumor progression. In general, AP-1 proteins demand MAPK-derived phosphorylation to form transcriptionally active complexes. External stimuli, including growth factors, cytokines and cell stress, can cause a MAPK-dependent increase in c-Jun transcription as shown for p44/42 MAPK [37], p54/46 JNK [55], and p38 MAPK [56]. In addition to transcriptional activation, MAPK-induced phosphorylation contributes to the stability of c-Jun protein. Phosphorylation by JNK enhances protein stability via protection from ubiquitination [57], whereas constitutively active p44/42 MAPK increases c-Jun stability via inactivation of glycogen kinase 2 in melanoma cells [58]. Thus, it is reasonable to assume that sustained decrease of p44/42 MAPK and/or p54/46 JNK activation is (at least in part) responsible for decreased c-Jun levels as observed during the present study (Fig. 5C). Additionally, direct phosphorylation of c-Jun by PRKD2 may contribute to c-Jun regulation [59]. Of note, c-Jun accumulation is elevated in human glioblastoma and the migration and invasion capacity of U87MG cells is significantly reduced in response to c-Jun silencing [60]. In addition, c-Jun, as a component of the AP-1 transcription factor complex, regulates the expression of integrins [61], urokinase plasminogen activator receptor [62], and matrix metalloproteinases [63] that facilitate growth, invasion, and metastasis of GBM cells.
Array and qPCR analyses performed during the present study identified reduced expression levels of ITGA-2 and -4 (which encode integrin alpha2 and -alpha4, respectively) in response to PRKD2 silencing (Table 1, Fig. 6). The integrin family of cell adhesion receptors regulates a diverse array of cellular functions crucial to carcinogenesis [64] and mediates resistance towards radio- [65] and chemotherapy [66]. The importance of integrins in several cell types that affect tumor progression has made them an appealing target for cancer therapy including glioblastoma [67]. Results of a cilengitide phase II trial in patients with recurrent glioblastoma showed promising effects [68]. Of note, a splice variant of human calcium and integrin-binding protein 1a (CIB1a) was identified as a PRKD2 substrate that regulates tumor growth in vivo by promoting angiogenesis [35]. In light of the different signaling cascades that are elicited by PRKD2 the authors of that study suggested that targeting of PRKD2 phosphorylation substrates might be more specific than targeting the kinase itself [35].
Two other genes, namely PLAU (encoding urokinase-type plasminogen activator; uPA) and PLAUR (encoding uPA receptor; uPAR) were also downregulated after PRKD2 silencing. uPAR is frequently expressed at high levels in GBM [69] and promotes cancer invasion by uPA binding and activation of extracellular proteases [70]. Silencing of uPA and/or uPAR was shown to inhibit invasion of GBM cells and xenografts [71]. Attenuation of PRKD2 expression by RNAi during the present study also reduced MMP-1 expression, a matrix metalloproteinase, which is expressed in GBM but absent in normal brain or low grade astrocytomas [72]. Of note, MMP-1 expression has a pronounced negative impact on median patient survival times [72]. A recent report has shown that silencing of PRKD2 and PRKD3 downregulates uPA, uPAR and MMP-9 and decreases the invasive potential of prostate cancer cells [15], findings compatible with data reported during the present study. However, in contrast to Zou et al. [15], who have identified PRKD2 as a major regulator of the pIKK-IκB-p65 nuclear translocation pathway that controls prostate cancer invasiveness, this pathway was unaffected in our study (Fig. 5D).
Recently, Navarro and colleagues investigated T-cell function in PRKD2 null mice or mice expressing catalytically inactive PRKD2 [73]. These studies revealed that loss of PRKD2 increases thymic output resulting in lymphoid hyperplasia and splenomegaly indicating a role for the kinase in thymic homeostasis [73]. Whether and to what extent pharmacological inhibition of PRKD2 in glioblastoma models would interfere with a normal T-cell response and lead to altered properties of tumor-associated immune cells remains to be established. Ideally this issue should be tested with a blood–brain barrier permeable inhibitor of PRKD2 in a preclinical intracranial animal model of GBM.
Overall, data presented here identify PRKD2 as a potential kinase target to reduce glioblastoma cell motility and invasion, two key properties fostering dissemination and recurrence of GBM. Gene expression analyses point towards a beneficial effect of PRKD2 silencing leading to the formation of a less motile and less invasive cellular GBM phenotype.
Acknowledgments
Financial support was provided by the Austrian Research Promotion Agency (FFG; Grant no. Bridge P820107; co-funding provided by Cyathus Exquirere PharmaforschungsGmbH, Vienna, Austria), the Austrian Science Fund (FWF; Grant no. F3007 and DK-W1241), and the Austrian National bank (Anniversary Fund, project number 14534).
References
- 1.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U., Curschmann J., Janzer R.C., Ludwin S.K., Gorlia T., Allgeier A., Lacombe D., Cairncross J.G., Eisenhauer E., Mirimanoff R.O. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Parsons D.W., Jones S., Zhang X., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L., Olivi A., McLendon R., Rasheed B.A., Keir S., Nikolskaya T., Nikolsky Y., Busam D.A., Tekleab H., Diaz L.A., Jr., Hartigan J., Smith D.R., Strausberg R.L., Marie S.K., Shinjo S.M., Yan H., Riggins G.J., Bigner D.D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V.E., Kinzler K.W. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Cancer Genome Atlas Research Network, Comprehensive genomic characterization defines human glioblastoma genes and core pathways, Nature 455 (2008) 1061–1068. [DOI] [PMC free article] [PubMed]
- 4.Hadjipanayis C.G., Van Meir E.G. Tumor initiating cells in malignant gliomas: biology and implications for therapy. J. Mol. Med. (Berl) 2009;87:363–374. doi: 10.1007/s00109-009-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedl P., Locker J., Sahai E., Segall J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 6.Reardon D.A., Rich J.N., Friedman H.S., Bigner D.D. Recent advances in the treatment of malignant astrocytoma. J. Clin. Oncol. 2006;24:1253–1265. doi: 10.1200/JCO.2005.04.5302. [DOI] [PubMed] [Google Scholar]
- 7.Onishi M., Ichikawa T., Kurozumi K., Date I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011;28:13–24. doi: 10.1007/s10014-010-0007-z. [DOI] [PubMed] [Google Scholar]
- 8.Griner E.M., Kazanietz M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 9.Fu Y., Rubin C.S. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. EMBO Rep. 2011;12:785–796. doi: 10.1038/embor.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LaValle C.R., George K.M., Sharlow E.R., Lazo J.S., Wipf P., Wang Q.J. Protein kinase D as a potential new target for cancer therapy. Biochim. Biophys. Acta. 1806;2010:183–192. doi: 10.1016/j.bbcan.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trauzold A., Schmiedel S., Sipos B., Wermann H., Westphal S., Roder C., Klapper W., Arlt A., Lehnert L., Ungefroren H., Johannes F.J., Kalthoff H. PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene. 2003;22:8939–8947. doi: 10.1038/sj.onc.1207001. [DOI] [PubMed] [Google Scholar]
- 12.Ochi N., Tanasanvimon S., Matsuo Y., Tong Z., Sung B., Aggarwal B.B., Sinnett-Smith J., Rozengurt E., Guha S. Protein kinase D1 promotes anchorage-independent growth, invasion, and angiogenesis by human pancreatic cancer cells. J. Cell Physiol. 2011;226:1074–1081. doi: 10.1002/jcp.22421. [DOI] [PubMed] [Google Scholar]
- 13.Harikumar K.B., Kunnumakkara A.B., Ochi N., Tong Z., Deorukhkar A., Sung B., Kelland L., Jamieson S., Sutherland R., Raynham T., Charles M., Bagherzadeh A., Foxton C., Boakes A., Farooq M., Maru D., Diagaradjane P., Matsuo Y., Sinnett-Smith J., Gelovani J., Krishnan S., Aggarwal B.B., Rozengurt E., Ireson C.R., Guha S. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010;9:1136–1146. doi: 10.1158/1535-7163.MCT-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J., Deng F., Singh S.V., Wang Q.J. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008;68:3844–3853. doi: 10.1158/0008-5472.CAN-07-5156. [DOI] [PubMed] [Google Scholar]
- 15.Zou Z., Zeng F., Xu W., Wang C., Ke Z., Wang Q.J., Deng F. PKD2 and PKD3 Promote Prostate Cancer Cell Invasion via uPA by Shifting Balance Between NF-kappaB and HDAC1. J. Cell Sci. 2012;125:4800–4811. doi: 10.1242/jcs.106542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mihailovic T., Marx M., Auer A., Van Lint J., Schmid M., Weber C., Seufferlein T. Protein kinase D2 mediates activation of nuclear factor kappaB by Bcr-Abl in Bcr-Abl+ human myeloid leukemia cells. Cancer Res. 2004;64:8939–8944. doi: 10.1158/0008-5472.CAN-04-0981. [DOI] [PubMed] [Google Scholar]
- 17.Kovalevska L.M., Yurchenko O.V., Shlapatska L.M., Berdova G.G., Mikhalap S.V., Van Lint J., Sidorenko S.P. Immunohistochemical studies of protein kinase D (PKD) 2 expression in malignant human lymphomas. Exp. Oncol. 2006;28:225–230. [PubMed] [Google Scholar]
- 18.Azoitei N., Kleger A., Schoo N., Thal D.R., Brunner C., Pusapati G.V., Filatova A., Genze F., Moller P., Acker T., Kuefer R., Van Lint J., Baust H., Adler G., Seufferlein T. Protein kinase D2 is a novel regulator of glioblastoma growth and tumor formation. Neuro. Oncol. 2011;13:710–724. doi: 10.1093/neuonc/nor084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prigozhina N.L., Waterman-Storer C.M. Protein kinase D-mediated anterograde membrane trafficking is required for fibroblast motility. Curr. Biol. 2004;14:88–98. doi: 10.1016/j.cub.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Woods A.J., White D.P., Caswell P.T., Norman J.C. PKD1/PKCmu promotes alphavbeta3 integrin recycling and delivery to nascent focal adhesions. EMBO J. 2004;23:2531–2543. doi: 10.1038/sj.emboj.7600267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Kimpe L., Janssens K., Derua R., Armacki M., Goicoechea S., Otey C., Waelkens E., Vandoninck S., Vandenheede J.R., Seufferlein T., Van Lint J. Characterization of cortactin as an in vivo protein kinase D substrate: interdependence of sites and potentiation by Src. Cell Signal. 2009;21:253–263. doi: 10.1016/j.cellsig.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Eiseler T., Doppler H., Yan I.K., Kitatani K., Mizuno K., Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 2009;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watkins J.L., Lewandowski K.T., Meek S.E., Storz P., Toker A., Piwnica-Worms H. Phosphorylation of the Par-1 polarity kinase by protein kinase D regulates 14-3-3 binding and membrane association. Proc. Natl. Acad. Sci. USA. 2008;105:18378–18383. doi: 10.1073/pnas.0809661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eiseler T., Doppler H., Yan I.K., Goodison S., Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res.: 2009;11:R13. doi: 10.1186/bcr2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anand M., Van Meter T.E., Fillmore H.L. Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J. Neurooncol. 2011;104:679–687. doi: 10.1007/s11060-011-0549-x. [DOI] [PubMed] [Google Scholar]
- 26.Pyne N.J., Pyne S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 27.Van Brocklyn J., Letterle C., Snyder P., Prior T. Sphingosine-1-phosphate stimulates human glioma cell proliferation through Gi-coupled receptors: role of ERK MAP kinase and phosphatidylinositol 3-kinase beta. Cancer Lett. 2002;181:195–204. doi: 10.1016/s0304-3835(02)00050-2. [DOI] [PubMed] [Google Scholar]
- 28.Van Brocklyn J.R., Young N., Roof R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003;199:53–60. doi: 10.1016/s0304-3835(03)00334-3. [DOI] [PubMed] [Google Scholar]
- 29.Annabi B., Lachambre M.P., Plouffe K., Sartelet H., Beliveau R. Modulation of invasive properties of CD133+ glioblastoma stem cells: a role for MT1-MMP in bioactive lysophospholipid signaling. Mol. Carcinog. 2009;48:910–919. doi: 10.1002/mc.20541. [DOI] [PubMed] [Google Scholar]
- 30.Young N., Pearl D.K., Van Brocklyn J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009;7:23–32. doi: 10.1158/1541-7786.MCR-08-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryan L., Paugh B.S., Kapitonov D., Wilczynska K.M., Alvarez S.M., Singh S.K., Milstien S., Spiegel S., Kordula T. Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: implications for invasiveness. Mol. Cancer Res. 2008;6:1469–1477. doi: 10.1158/1541-7786.MCR-08-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guha S., Tanasanvimon S., Sinnett-Smith J., Rozengurt E. Role of protein kinase D signaling in pancreatic cancer. Biochem. Pharmacol. 2010;80:1946–1954. doi: 10.1016/j.bcp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao Q., Wang L., Zhao Z.J., Tang H. Identification of protein kinase D2 as a pivotal regulator of endothelial cell proliferation, migration, and angiogenesis. J. Biol. Chem. 2009;284:799–806. doi: 10.1074/jbc.M807546200. [DOI] [PubMed] [Google Scholar]
- 34.Azoitei N., Pusapati G.V., Kleger A., Moller P., Kufer R., Genze F., Wagner M., van Lint J., Carmeliet P., Adler G., Seufferlein T. Protein kinase D2 is a crucial regulator of tumour cell-endothelial cell communication in gastrointestinal tumours. Gut. 2010;59:1316–1330. doi: 10.1136/gut.2009.206813. [DOI] [PubMed] [Google Scholar]
- 35.M. Armacki, G. Joodi, S.C. Nimmagadda, L. de Kimpe, G.V. Pusapati, S. Vandoninck, J. Van Lint, A. Illing and T. Seufferlein. A novel splice variant of calcium and integrin-binding protein 1 mediates protein kinase D2-stimulated tumour growth by regulating angiogenesis, Oncogene, 10.1038/onc.2013.43 [DOI] [PubMed]
- 36.Pfaffl M.W., Horgan G.W., Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leppa S., Saffrich R., Ansorge W., Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 1998;17:4404–4413. doi: 10.1093/emboj/17.15.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim R.H., Takabe K., Milstien S., Spiegel S. Export and functions of sphingosine-1-phosphate. Biochim. Biophys. Acta. 1791;2009:692–696. doi: 10.1016/j.bbalip.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Brocklyn J.R., Jackson C.A., Pearl D.K., Kotur M.S., Snyder P.J., Prior T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005;64:695–705. doi: 10.1097/01.jnen.0000175329.59092.2c. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida Y., Nakada M., Sugimoto N., Harada T., Hayashi Y., Kita D., Uchiyama N., Yachie A., Takuwa Y., Hamada J. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int. J. Cancer. 2010;126:2341–2352. doi: 10.1002/ijc.24933. [DOI] [PubMed] [Google Scholar]
- 41.Natarajan J., Berrar D., Dubitzky W., Hack C., Zhang Y., DeSesa C., Van Brocklyn J.R., Bremer E.G. Text mining of full-text journal articles combined with gene expression analysis reveals a relationship between sphingosine-1-phosphate and invasiveness of a glioblastoma cell line. BMC Bioinf. 2006;7:373. doi: 10.1186/1471-2105-7-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalluri R., Weinberg R.A. The basics of epithelial–mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karam M., Legay C., Auclair C., Ricort J.M. Protein kinase D1 stimulates proliferation and enhances tumorigenesis of MCF-7 human breast cancer cells through a MEK/ERK-dependent signaling pathway. Exp. Cell Res. 2012;318:558–569. doi: 10.1016/j.yexcr.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Kim do Y., Jung M.S., Park Y.G., Yuan H.D., Quan H.Y., Chung S.H. Ginsenoside Rh2(S) induces the differentiation and mineralization of osteoblastic MC3T3-E1 cells through activation of PKD and p38 MAPK pathways. BMB Rep. 2011;44:659–664. doi: 10.5483/BMBRep.2011.44.10.659. [DOI] [PubMed] [Google Scholar]
- 45.Sinnett-Smith J., Zhukova E., Rey O., Rozengurt E. Protein kinase D2 potentiates MEK/ERK/RSK signaling, c-Fos accumulation and DNA synthesis induced by bombesin in Swiss 3T3 cells. J. Cell Physiol. 2007;211:781–790. doi: 10.1002/jcp.20984. [DOI] [PubMed] [Google Scholar]
- 46.Lin C.W., Shen S.C., Chien C.C., Yang L.Y., Shia L.T., Chen Y.C. 12-O-tetradecanoylphorbol-13-acetate-induced invasion/migration of glioblastoma cells through activating PKCalpha/ERK/NF-kappaB-dependent MMP-9 expression. J. Cell Physiol. 2010;225:472–481. doi: 10.1002/jcp.22226. [DOI] [PubMed] [Google Scholar]
- 47.Lai C.F., Chaudhary L., Fausto A., Halstead L.R., Ory D.S., Avioli L.V., Cheng S.L. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J. Biol. Chem. 2001;276:14443–14450. doi: 10.1074/jbc.M010021200. [DOI] [PubMed] [Google Scholar]
- 48.Huang C., Jacobson K., Schaller M.D. MAP kinases and cell migration. J. Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 49.Vial E., Sahai E., Marshall C.J. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 50.Botta G.P., Reginato M.J., Reichert M., Rustgi A.K., Lelkes P.I. Constitutive K-RasG12D activation of ERK2 specifically regulates 3D invasion of human pancreatic cancer cells via MMP-1. Mol. Cancer Res. 2012;10:183–196. doi: 10.1158/1541-7786.MCR-11-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eke I., Storch K., Kastner I., Vehlow A., Faethe C., Mueller-Klieser W., Taucher-Scholz G., Temme A., Schackert G., Cordes N. Three-dimensional Invasion of human glioblastoma cells remains unchanged by X-ray and carbon ion irradiation in vitro. Int. J. Radiat. Oncol. Biol. Phys. 2012;84:e515–523. doi: 10.1016/j.ijrobp.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Matsuda K., Sato A., Okada M., Shibuya K., Seino S., Suzuki K., Watanabe E., Narita Y., Shibui S., Kayama T., Kitanaka C. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sc. Rep. 2012;2:516. doi: 10.1038/srep00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon C.H., Kim M.J., Kim R.K., Lim E.J., Choi K.S., An S., Hwang S.G., Kang S.G., Suh Y., Park M.J., Lee S.J. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene. 2012;31:4655–4666. doi: 10.1038/onc.2011.634. [DOI] [PubMed] [Google Scholar]
- 54.Kozuka-Hata H., Nasu-Nishimura Y., Koyama-Nasu R., Ao-Kondo H., Tsumoto K., Akiyama T., Oyama M. Phosphoproteome of human glioblastoma initiating cells reveals novel signaling regulators encoded by the transcriptome. PLoS One. 2012;7:e43398. doi: 10.1371/journal.pone.0043398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 56.Han J., Jiang Y., Li Z., Kravchenko V.V., Ulevitch R.J. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997;386:296–299. doi: 10.1038/386296a0. [DOI] [PubMed] [Google Scholar]
- 57.Musti A.M., Treier M., Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- 58.Lopez-Bergami P., Huang C., Goydos J.S., Yip D., Bar-Eli M., Herlyn M., Smalley K.S., Mahale A., Eroshkin A., Aaronson S., Ronai Z. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waldron R.T., Whitelegge J.P., Faull K.F., Rozengurt E. Identification of a novel phosphorylation site in c-jun directly targeted in vitro by protein kinase D. Biochem. Biophys. Res. Commun. 2007;356:361–367. doi: 10.1016/j.bbrc.2007.02.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blau L., Knirsh R., Ben-Dror I., Oren S., Kuphal S., Hau P., Proescholdt M., Bosserhoff A.K., Vardimon L. Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc. Natl. Acad. Sci. USA. 2012;109:E2875–2884. doi: 10.1073/pnas.1203659109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eriksson M., Arminen L., Karjalainen-Lindsberg M.L., Leppa S. AP-1 regulates alpha2beta1 integrin expression by ERK-dependent signals during megakaryocytic differentiation of K562 cells. Exp. Cell Res. 2005;304:175–186. doi: 10.1016/j.yexcr.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 62.Amos S., Redpath G.T., Dipierro C.G., Carpenter J.E., Hussaini I.M. Epidermal growth factor receptor-mediated regulation of urokinase plasminogen activator expression and glioblastoma invasion via C-SRC/MAPK/AP-1 signaling pathways. J. Neuropathol. Exp. Neurol. 2010;69:582–592. doi: 10.1097/NEN.0b013e3181e008fe. [DOI] [PubMed] [Google Scholar]
- 63.Mittelstadt M.L., Patel R.C. AP-1 mediated transcriptional repression of matrix metalloproteinase-9 by recruitment of histone deacetylase 1 in response to interferon beta. PLoS One. 2012;7:e42152. doi: 10.1371/journal.pone.0042152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Desgrosellier J.S., Cheresh D.A. Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ou J., Luan W., Deng J., Sa R., Liang H. alphaV integrin induces multicellular radioresistance in human nasopharyngeal carcinoma via activating SAPK/JNK pathway. PLoS One. 2012;7:e38737. doi: 10.1371/journal.pone.0038737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naci D., El Azreq M.A., Chetoui N., Lauden L., Sigaux F., Charron D., Al-Daccak R., Aoudjit F. alpha2beta1 integrin promotes chemoresistance against doxorubicin in cancer cells through extracellular signal-regulated kinase (ERK) J. Biol. Chem. 2012;287:17065–17076. doi: 10.1074/jbc.M112.349365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chamberlain M.C., Cloughsey T., Reardon D.A., Wen P.Y. A novel treatment for glioblastoma: integrin inhibition. Expert Rev. Neurother. 2012;12:421–435. doi: 10.1586/ern.11.188. [DOI] [PubMed] [Google Scholar]
- 68.Reardon D.A., Fink K.L., Mikkelsen T., Cloughesy T.F., O’Neill A., Plotkin S., Glantz M., Ravin P., Raizer J.J., Rich K.M., Schiff D., Shapiro W.R., Burdette-Radoux S., Dropcho E.J., Wittemer S.M., Nippgen J., Picard M., Nabors L.B. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 2008;26:5610–5617. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]
- 69.Salajegheh M., Rudnicki A., Smith T.W. Expression of urokinase-type plasminogen activator receptor (uPAR) in primary central nervous system neoplasms. Appl. Immunohistochem. Mol. Morphol. 2005;13:184–189. doi: 10.1097/01.pai.0000138448.85231.da. [DOI] [PubMed] [Google Scholar]
- 70.Blasi F., Carmeliet P. uPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 71.Raghu H., Gondi C.S., Dinh D.H., Gujrati M., Rao J.S. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch -1 receptor. Mol. Cancer. 2011;10:130. doi: 10.1186/1476-4598-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stojic J., Hagemann C., Haas S., Herbold C., Kuhnel S., Gerngras S., Roggendorf W., Roosen K., Vince G.H. Expression of matrix metalloproteinases MMP-1, MMP-11 and MMP-19 is correlated with the WHO-grading of human malignant gliomas. Neurosci. Res. 2008;60:40–49. doi: 10.1016/j.neures.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 73.Navarro M.N., Sinclair L.V., Feijoo-Carnero C., Clarke R., Matthews S.A., Cantrell D.A. Protein kinase D2 has a restricted but critical role in T-cell antigen receptor signalling in mature T-cells. Biochem. J. 2012;442:649–659. doi: 10.1042/BJ20111700. [DOI] [PMC free article] [PubMed] [Google Scholar]