

Abstract
Productive T cell activation requires efficient reorganization of the actin cytoskeleton. We showed previously that the actin-regulatory protein, hematopoietic lineage cell-specific protein 1 (HS1), is required for the stabilization of F-actin and Vav1 at the immunological synapse and for efficient calcium responses. The Tec family kinase IL-2-inducible T cell kinase (Itk) regulates similar aspects of T cell activation, suggesting that these proteins act in the same pathway. Using video microscopy, we show that T cells lacking Itk or HS1 exhibited similar defects in actin responses, extending unstable lamellipodial protrusions upon TCR stimulation. HS1 and Itk could be coimmunoprecipitated from T cell lysates, and GST-pulldown studies showed that Itk’s Src homology 2 domain binds directly to two phosphotyrosines in HS1. In the absence of Itk, or in T cells overexpressing an Itk Src homology 2 domain mutant, HS1 failed to localize to the immunological synapse, indicating that Itk serves to recruit HS1 to sites of TCR engagement. Because Itk is required for phospholipase C (PLC)γ1 phosphorylation and calcium store release, we examined the calcium signaling pathway in HS1−/− T cells in greater detail. In response to TCR engagement, T cells lacking HS1 exhibited diminished calcium store release, but TCR-dependent PLCγ1 phosphorylation was intact, indicating that HS1’s role in calcium signaling is distinct from that of Itk. HS1-deficient T cells exhibited defective cytoskeletal association of PLCγ1 and altered formation of PLCγ1 microclusters. We conclude that HS1 functions as an effector of Itk in the T cell actin-regulatory pathway, and directs the spatial organization of PLCγ1 signaling complexes.
Effective T cell activation requires reorganization of the actin cytoskeleton. The interaction between a T cell and an APC bearing cognate Ag results in the activation of multiple actin-regulatory proteins, which promote the polymerization of actin filaments at the immunological synapse (IS)3 (1). T cells lacking these molecules or treated with pharmacological agents that disrupt actin dynamics exhibit functional defects in their responses to Ag. Commonly, diminished calcium (Ca2+) mobilization responses are observed, resulting in poor IL-2 production and proliferation. Although the molecular mechanisms by which actin dynamics influence T cell signaling are poorly understood, visualization of high-order signaling complexes using live cell imaging has begun to yield some insights. Proper actin dynamics are required for the formation of signaling microclusters at the cell periphery and for their movement toward the center, whereas the maintenance of centralized microclusters is largely independent of the actin cytoskeleton (2–4). Peripheral and central microclusters also appear to be functionally distinct with respect to their role in signaling. Although details differ depending on experimental conditions (5), newly formed, peripheral microclusters are thought to be the predominant sites for early phosphorylation events, whereas central microclusters are usually associated with signal down-regulation (6). Thus, actin dynamics are required to assemble active signaling complexes, and to drive the mechanism that ultimately extinguishes signaling.
Hematopoietic lineage cell-specific protein 1 (HS1) is an actin-regulatory protein expressed throughout the hematopoietic system (7). Like its more widely expressed homologue, cortactin, HS1 binds to the Arp2/3 complex and actin filaments, and is thought to stabilize branched actin filaments (8, 9). We have shown that T cells lacking HS1 can generate actin-rich lamellipodial protrusions in response to TCR engagement, but these protrusions are disordered and rapidly collapse (10). Consistent with this observation, accumulation of F-actin at the IS is abnormally short-lived. HS1-deficient T cells also exhibit defects in Ca2+ signaling leading to diminished IL-2 production. The regulation of HS1 activity is not fully understood. TCR engagement leads to rapid phosphorylation of HS1 at tyrosines 378 and 397. This is required for recruitment of HS1 to the IS, and for its binding to numerous key signaling proteins, including the Rac-1/Cdc42 guanine nucleotide exchange factor Vav1. Although T cells lacking HS1 exhibit defects in the maintenance of Vav1 at the IS, Vav1 is dispensable for recruitment of HS1 to the IS (10). How HS1 is recruited to the IS is unknown.
TCR ligation-induced activation of actin-regulatory proteins, including HS1, depends on signaling through tyrosine kinases and adapter proteins (reviewed in Ref. 1). Although these molecules do not directly influence the actin cytoskeleton, cells lacking them exhibit severe defects in actin dynamics. We and others have shown that T cells lacking the Tec family kinase IL-2-inducible T cell kinase (Itk) fail to polymerize actin properly in response to TCR ligation (11–13), and fail to recruit Vav1 to the IS (11). Interestingly, the actin-regulatory role of Itk is independent of kinase activity, and depends instead on adapter functions conducted by its Src homology (SH)2 domain (11, 12).
To begin to understand how the interactions between actin-regulatory proteins and upstream regulators lead to productive T cell activation, we examined the possibility of an interaction between HS1 and Itk. Using live cell imaging, we now show that cells lacking Itk exhibit similar defects in actin dynamics to those lacking HS1. The SH2 domain of Itk binds to phosphotyrosines in HS1, and this interaction is required for the recruitment of HS1 to the IS. Recruitment of HS1 to the IS then promotes productive Ca2+ signaling by modulating the formation of phospholipase C (PLC)γ1 microclusters. Taken together, these studies show that Itk-HS1 interactions play an important role in controlling actin-driven dynamics of TCR signaling microclusters leading to productive T cell activation.
Materials and Methods
Reagents and Abs
All reagents are from Sigma-Aldrich, unless otherwise specified. Rabbit anti-human HS1 has been previously described (10), and rabbit anti-mouse HS1 was generated against aa 226–351 (Cocalico Biologicals). Anti-Itk (2F12) and anti-phosphotyrosine (4G10) were from Millipore, anti-PLCγ1 was from ECM Biosciences, rabbit anti-PLCγ1 phosphorylated at Y783 was from Cell Signaling Technology, anti-actin (C-2) and anti-lamin B (M-20) were from Santa Cruz Biotechnology, and anti-GAPDH was from Calbiochem. Anti-mouse CD3ε (2c11) and anti-mouse CD28 (PV1) were prepared in the University of Chicago monoclonal facility. Anti-mouse CD3ε (500.A2) and mouse anti-human HS1 were from BD Pharmingen, and anti-human CD3 (OKT3; Orthoclone) was from the Children’s Hospital of Philadelphia pharmacy. Goat anti-mouse IgG-IRDye800 was from Rockland Immunochemicals, and goat anti-rabbit IgG Alexa Fluor 680 was from Molecular Probes. Goat anti-mouse IgG1 Alexa Fluor 555 and goat anti-mouse IgG Alexa Fluor 594 were from Molecular Probes, and donkey anti-mouse IgG Cy3 was from Jackson ImmunoResearch Laboratories.
Mice
HS1−/− mice on the C57BL/6J background have been previously described (14). To generate T cells specific for moth cytochrome c 88–103 (MCC88–103) presented on I-Ek, HS1−/− mice were crossed to AND TCR transgenic mice (The Jackson Laboratory) (15–17), and maintained as heterozygotes for the AND transgene. C57BL/6J mice were obtained from The Jackson Laboratory. All mice were housed under pathogen-free conditions in the Children’s Hospital of Philadelphia animal facility. All studies involving animals were reviewed and approved by the Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee.
Cell culture
All tissue culture reagents were from Invitrogen. The human T cell line Jurkat E6.1, the human B cell line Raji, and the murine B cell line CH27 were maintained, as described previously (18). Jurkat T cells stably expressing GFP-actin (10), or PLCγ1-enhanced yellow fluorescent protein (eYFP) (19), gift of L. Samelson (National Institutes of Health, Bethesda, MD), were maintained in RPMI 1640 containing 5% FBS, 5% newborn calf serum, penicillin/streptomycin, glutamine, and 1 mg/ml G418. Murine CD4+ T cells were isolated from lymph nodes and spleens by negative selection using a mixture of anti-MHC class II (M5/114.15.2) and anti-CD8 (2.43), followed by magnetic bead-conjugated goat anti-rat Ig (Qiagen). Where specified, total T cells were isolated using anti-MHC II alone. T cell blasts were prepared from CD4+ T cells by stimulation on plates coated with anti-CD3 (2c11) and anti-CD28 (PV1) for 3 days, followed by 3–8 days of resting culture. Cells were maintained using DMEM supplemented with 10% FBS, nonessential amino acids, penicillin, streptomycin, HEPES, and 2-ME (Sigma-Aldrich), supplemented with 50 U/ml human rIL-2 (obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health; human rIL-2 from M. Gately, Hoffmann-LaRoche, Nutley, NJ).
DNA constructs, transfection, and RNA interference
The Itk-myc expression constructs were gifts of L. Berg (University of Massachusetts, Worcester, MA). The short hairpin RNA vectors pFRT. H1p and pCMS3.eGFP.H1p and the shHS1f targeting sequence have been described (10, 20, 21). Small interfering RNA duplexes against human Itk (11) were synthesized by Qiagen. For some studies, Itk suppression sequences were cloned into pFRT.H1p and pCMS3.eGFP.H1p, as previously described (20, 21). The following targeting sequences were used: shItkc, GAAGAAACGAGGAATAATA and shItke, GCACTATCCGATCCT CATC. All results were confirmed using both targeting constructs; results with only one construct are shown for simplicity. Electroporation was done using an ECM 830 square wave electroporator (BTX) in 500 μl of antibiotic-free RPMI 1640 using a 310 V, 10 ms pulse. Cells were used 24 h after transfection with 40 μg of Itk-myc expression constructs or 1.5 μg of small interfering RNA duplexes, or 72 h after transfection with 40 μg of short hairpin RNA vectors. In each case, suppression efficiency in the bulk population of cells was at least 60%.
Immunofluorescence microscopy
Jurkat T cells were conjugated to Raji B cells at 37°C, as described previously (22), and stained for F-actin using Alexa Fluor 647-phalloidin (Molecular Probes) and for HS1, followed by anti-mouse IgG Alexa Fluor 594 or anti-mouse IgG Cy3. For experiments involving CD4+ T cells from AND transgenic mice, CH27 B cells were labeled with 7-amino-4-chloromethyl coumarin (CMAC) (cell tracker blue; Molecular Probes) and incubated with 5 μM MCC88–103. Equal numbers of T and B cells were centrifuged together, incubated at 37°C for various times, and plated on poly(L-lysine)-coated coverslips, followed by fixation in 3% paraformaldehyde. Conjugates were stained for PLCγ1 or Itk, followed by anti-mouse IgG1 Alexa Fluor 555, and for F-actin using Alexa Fluor 647-phalloidin. Conjugates were analyzed on a Zeiss Axiovert 200M microscope equipped with a 63× planapo 1.4 NA objective. Images were collected using a Photometrics Coolsnap FX-HQ (Roper Scientific) camera, and deconvolution was performed using a constrained iterative algorithm (Slidebook version 4.2; Intelligent Imaging Innovations). Alternatively, conjugates were imaged on a Zeiss Axiovert 200 equipped with a PerkinElmer Ultra-view ERS6 spinning disk confocal system and a 63× planapo 1.4 NA objective. Images were collected using an Orca ER camera (Hamamatsu) and analyzed using Volocity version 5 (Improvision). For analysis, conjugates were identified at random as a blue B cell in contact with a T cell, and scored for the presence of a bright band of protein at the cell-cell contact site. All experiments were analyzed by an individual blinded to the experimental conditions. At least 50 conjugates were scored per condition in each experiment.
Live cell imaging
Eight-well Lab-Tek II chambered coverglasses (Nalge Nunc International) were cleaned with 70% ethanol containing 1 M HCl before incubation for 15 min with 0.1 mg/ml poly(L-lysine). Before use, coverglasses were coated with 10 μg/ml OKT-3 for 2 h at 37°C or overnight at 4°C. Coverglasses were rinsed in PBS and covered with 400 μl of RPMI 1640 without phenol red immediately before imaging and equilibrated to 37°C on the microscope stage within a Solent environmental chamber. Cultured cells were resuspended in phenol red-free RPMI 1640 at 2 × 106/ml, and spreading was initiated by adding 5–10 μl of cell suspension to the coverglass chamber. Time-lapse images were collected at ×63 using a PerkinElmer ERS6 Ultraview spinning disk confocal system. Stacks of three to seven images were collected at 0.5-μm spacing every 3–5 s for ~6 min. GFP-actin dynamics were analyzed using Volocity Quantitation for area measurements and Slidebook for radial variance, as previously described (10). At least 40 cells were analyzed per condition. To analyze PLCγ1 microclusters, maximum intensity projections were generated, and individual objects were identified based on pixel intensity and tracked using Volocity. Microcluster area was measured from a single time point, collected 90 s after contact with the coverslip for each individual cell. At least 20 cells were analyzed per condition. To visualize microcluster formation, objects were pseudocolored based on time of appearance, relative to the total duration of each time-lapse sequence.
Ca2+ assays
For single-cell Ca2+ measurements, freshly isolated CD4+ T cells were loaded with the cell-permeant Ca2+ indicator fura 2-AM (3.0 μM; Molecular Probes) for 15 min in normal bath solution (155 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4)). Cell suspensions were placed into the recording chamber on an inverted fluorescence microscope (Nikon) and allowed to adhere to poly(L-lysine)-treated (100 μg/ml; Sigma-Aldrich) coverslips for 5 min. Extracellular fura 2-AM was removed by perfusion with additional normal bath solution. Before stimulation, the chamber was perfused with Ca2+-free bath solution containing 155 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 0.5 mM EGTA, 10 mM glucose, and 10 mM HEPES (pH 7.4). Intracellular Ca2+ mobilization was initiated by addition of anti-CD3 (500.A2) in Ca2+-free bath solution; consequently, the initial Ca2+ transient observed in Ca2+-free solution is due to Ca2+ release from stores. The activation state of calcium entry channels was then examined by replacing the Ca2+-free solution with normal (Ca2+-containing) bath solution. Where indicated, 1 μM thapsigargin was added to block SERCA pump activity, thereby depleting intracellular Ca2+ stores. Fura 2-AM was alternately excited at 340 and 380 nm, and fluorescence emission (plotted as the 340/380 nm ratio) of individual cells was measured by digital imaging microscopy using Metafluor software (Molecular Devices).
Western blotting
Lysates were separated on Tris-glycine SDS-PAGE gels, transferred to immobilon-FL (Millipore) or nitrocellulose (Bio-Rad) membranes, blocked in 3% nonfat dry milk in TBS, and probed with primary Abs in 3% milk in TBST or 3% BSA in TBST for phospho-specific Abs. Blots were probed with goat anti-mouse IgG-IRDye800 or goat anti-rabbit IgG-Alexa Fluor 680 and imaged on a Licor Odyssey infrared fluorescence scanner.
GST pulldowns
Fragments comprised of the SH3-SH2 domains (aa 154–352) or the SH2 domain alone (aa 237–352) of human Itk were cloned into pGEX-KG, and mutations to inactivate the SH3 (W208/209Y) and/or SH2 (R265A) domains were incorporated using standard molecular biology techniques. The SH3 domain of HS1 (aa 415–486) was cloned into pGEX-KG, and inactivating mutations (W455Y, W456Y) were incorporated. rGST fusion proteins were generated in bacteria and bound to glutathione resin (Amersham). Jurkat cells were harvested, resuspended in PBS with Ca2+ and Mg2+, allowed to rest at 37°C for 20 min, and treated with pervanadate, as described (18), or left untreated. Cells were collected by centrifugation and lysed in radioimmunoprecipitation assay buffer (50 mM Tris-HCl (pH 8), 1% Nonidet P-40, 100 mM NaCl, 5 mM EDTA, 0.5 mM CaCl2, protease inhibitors, 1 mM Na3VO4, and 5 mM NaF). Lysates were clarified by centrifugation and exposed to glutathione resin-bound GST fusion proteins for 2 h. Beads were washed three times in Nonidet P-40 lysis buffer and analyzed by Western blotting and Coomassie staining. Maltose-binding protein (MBP) fusion proteins and HS1 phosphopeptides were prepared as described (10).
Coimmunoprecipitations
Cultured CD4+ T cell blasts were harvested and resuspended at 1 × 108/ml in serum-free DMEM for Ab stimulation or in PBS with Ca2+ and Mg2+ for pervanadate treatment. Cells were allowed to rest for 30 min at 37°C, and then labeled on ice with 10 μg/ml biotinylated anti-CD3 (2c11) for 20 min. Signaling was initiated by the addition of 2.5 μg/ml streptavidin at 37°C. At the indicated time points, cells were collected by centrifugation and lysed in Nonidet P-40 lysis buffer. Postnuclear lysates from 4 × 107 cells were incubated with anti-mouse HS1 bound to protein A-Sepharose for 2 h at 4°C, washed in lysis buffer, eluted in 2× SDS-PAGE sample buffer, and analyzed by Western blotting for HS1, Itk, and phosphotyrosine.
Analysis of PLCγ1 phosphorylation and cytoskeletal association
Total T cells isolated from the spleen and lymph nodes of wild-type or HS1−/− C57BL/6 mice were resuspended at 1 × 108/ml in DMEM and rested at 37°C for 30 min. Cells were stimulated by the addition of 5 μg/ml anti-CD3 (500.A2). Stimulation was stopped by transferring an aliquot of cells to a tube containing ice-cold PBS. Cells were collected by centrifugation and lysed in lysis buffer (50 mM Tris (pH 8), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, protease inhibitors, 5 mM NaF, 1 mM Na3VO4, and 5 mM EDTA). Lysates were incubated on ice for 20 min, and then clarified by centrifugation at 16,000 × g. Samples were analyzed by Western blotting. For isolation of insoluble F-actin-rich fractions, cells were lysed in cytoskeletal stabilizing lysis buffer (80 mM PIPES (pH 6.9), 1% Triton X-100, 1 mM EGTA, 1 mM MgCl2, protease inhibitors, 5 mM NaF, and 1 mM Na3VO4), and centrifuged immediately at 5,000 × g for 5 min (23). The F-actin-rich pellet was resuspended in high salt buffer (20 mM HEPES (pH 7.9), 0.4 M NaCl, 1 mM EDTA, protease inhibitors, 5 mM NaF, and 1 mM Na3VO4) and vortexed for 1 h at 4°C to extract proteins. Samples were centrifuged at 16,000 × g for 10 min, and supernatants were used as the insoluble fraction for Western blot analysis. In addition to probing for PLCγ1 and phospho-PLCγ1, Western blots were probed for GAPDH as a loading control for soluble fractions, and the nuclear protein lamin B as a loading control for F-actin-rich insoluble fractions. Fluorescence intensity for each band was determined using Licor Odyssey software, taking care to remain within the linear range of the instrument. The amount of PLCγ1 or phospho-PLCγ1 in each lane was quantified by normalizing to the relevant loading control for each lane, and then to the normalized value for unstimulated cells.
Results
Itk-deficient T cells show unstable lamellipodial protrusions
Previous studies have shown that Itk is required for proper actin responses in a manner dependent on its SH2 domain, but not its kinase activity (11–13). To better understand how Itk regulates T cell actin responses, we generated Itk-deficient T cells using RNA interference (Fig. 1E), and visualized actin dynamics in these cells during spreading on coverslips coated with anti-CD3. As previously shown (24), control Jurkat T cells expressing GFP-actin extended stable actin-rich lamellipodia and exhibited retrograde actin flow from the periphery of the cell toward the center (Fig. 1A and supplemental Videos 1 and 2).4 Itk-deficient T cells also extended actin-rich lamellipodia, but these structures were disordered and retracted frequently (Fig. 1B and supplemental Videos 3 and 4). The overall spreading area of Itk-suppressed cells was less than that of wild-type cells (Fig. 1C). Furthermore, whereas control cells maintained a regular, round profile, Itk-suppressed cells were irregularly shaped. This difference is illustrated by an increased radial variance in the Itk-suppressed cells relative to controls (Fig. 1D). The phenotype of Itk-deficient T cells in this assay is strikingly similar to that of T cells lacking HS1, an actin-binding protein that is thought to stabilize branched actin filaments generated in response to TCR engagement. Like Itk-suppressed cells, HS1-suppressed cells exhibit diminished spreading and irregular and unstable lamellipodial protrusions (10) (supplemental Videos 5 and 6).
FIGURE 1.

Itk-deficient T cells exhibit unstable lamellipodial protrusions. A, Jurkat T cells stably expressing GFP-actin were transfected with empty suppression vector. After 72 h, cells were plated on coverslips coated with anti-CD3 and imaged by confocal microscopy for the indicated times (seconds). Selected images from one time-lapse series are shown; these correspond to supplemental Video 1. B, Jurkat cells stably expressing GFP-actin were transfected with Itk suppression vector and analyzed as in A. Selected images from one time-lapse series are shown; these correspond to supplemental Video 3. C, The contact area of each cell at each time point was determined, and the average was calculated for each cell population at each 5-s time point (control = 46 cells; shItk = 49 cells). D, Irregularity of cell shape was assessed by measuring the radial variance for each cell at each time point and calculating the average values. E, Western blot analysis showing suppression of Itk or HS1 in GFP-actin Jurkat cells transfected with the indicated suppression vectors.
The SH2 domain of Itk mediates binding to HS1
To test the possibility that HS1 is an effector for Itk in the T cell actin-regulatory pathway, we assessed interaction between these two proteins by coimmunoprecipitation. As shown in Fig. 2, Itk was found in association with HS1 in both activated and resting mouse CD4+ T cell blasts. Similar results were obtained using Jurkat T cells; moreover, HS1 was present in Itk immunoprecipitates (data not shown).
FIGURE 2.
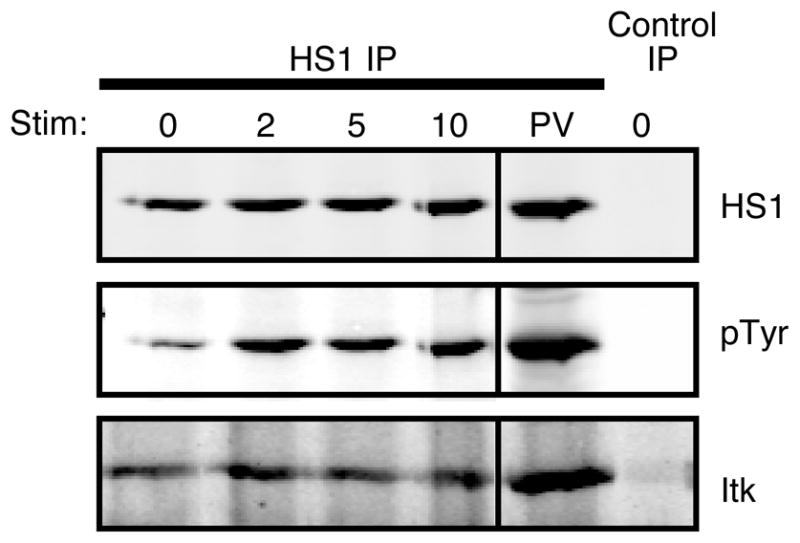
HS1 and Itk interact in T cells. CD4+ T cell blasts were stimulated with anti-CD3 for the indicated times (minutes), treated with pervanadate (PV), or left untreated. HS1 was immunoprecipitated from lysates, and immunoprecipitates were analyzed by Western blotting for HS1, phosphorylated HS1 (by blotting for total phosphotyrosine, pTyr), or Itk. Control immunoprecipitation was performed using preimmune rabbit serum.
To map the site(s) of interaction between the two proteins, we used a GST-pulldown approach. Domain maps of HS1 and Itk are shown in Fig. 3A. Itk has SH3 and SH2 domains that could bind to proline-rich sequences or phosphotyrosines in HS1, respectively. Conversely, the SH3 domain of HS1 could bind to the proline-rich region of Itk. To test the role of the Itk SH3 and SH2 domains, we generated a panel of GST-fusion constructs encoding the wild-type SH3-SH2 domain fragment of Itk, or with inactivating point mutations in the SH3 domain, the SH2 domain, or both. These fragments were used to probe Jurkat T cell lysates for binding of HS1. As shown in Fig. 3B, HS1 bound to the wild-type Itk fragment in an activation-dependent manner. This binding was unaffected by mutation of the Itk SH3 domain, but was abolished upon mutation of the SH2 domain, consistent with binding mediated by SH2 domain-phosphotyrosine interactions. In some experiments, we detected modest HS1 binding to the wild-type construct in the absence of pervanadate treatment (data not shown). However, this was also abolished upon mutation of the SH2 domain, suggesting that this interaction is attributable to basal tyrosine phosphorylation of HS1. To ask whether the SH3 domain of HS1 provides an additional means of interaction with Itk, we conducted reciprocal pulldown experiments using the HS1 SH3 domain. However, Itk binding to the HS1 SH3 domain could not be detected, even under conditions in which interactions with other binding partners could be verified (Fig. 3C). We conclude that HS1 binding to Itk depends predominantly on interactions between the SH2 domain of Itk and tyrosine phosphorylation sites in HS1.
FIGURE 3.

The SH2 domain of Itk binds directly to phosphorylated HS1. A, Domain maps of HS1 and Itk. B, A panel of rGST fusion proteins comprised of the wild-type SH3-SH2 fragment of Itk and inactivating point mutants in either domain were coupled to glutathione resin and incubated with lysates from Jurkat cells that had been treated with pervanadate (PV) or left untreated. The pulldown reactions were washed and analyzed by Western blotting for HS1 (top) or Coomassie staining to monitor the GST-Itk proteins (bottom). C, The wild-type GST-HS1-SH3 domain or an inactivating point mutant was coupled to glutathione resin and incubated with lysates from Jurkat cells that had been treated with pervanadate (PV) or left untreated. The pulldown reactions were washed and analyzed by Western blotting for Itk or Dynamin 2. D, Jurkat cells were transfected with expression vectors encoding wild-type FLAG-tagged HS1 or the indicated point mutants. These cells were treated with pervanadate, lysed, and probed with GST-Itk SH2 domain. Left, Bound proteins were analyzed by Western blotting for FLAG or Coomassie staining to monitor the GST-Itk proteins. Right, FLAG immunoprecipitates were probed for phosphotyrosine and FLAG to monitor phosphorylation status of HS1 mutants. E, Purified rGST-Itk SH2 domain was incubated with rHS1 phosphopeptide fused to MBP. Left, Bound proteins were analyzed by Western blotting and Coomassie staining. Right, Purified MBP fusion peptides were analyzed by Western blotting for phosphotyrosine and MBP.
Phosphorylation at Y378 and Y397 of HS1 mediates binding to many SH2 domains (10), making these sites the likely points of interaction between HS1 and Itk. To test this hypothesis, we transfected Jurkat cells with vectors expressing wild-type FLAG-tagged HS1, or HS1 point mutants at one or both tyrosines. Transfected T cells were treated with pervanadate, and lysates were probed for binding of FLAG-HS1 to the SH2 domain of Itk. As shown in Fig. 3D, wild-type HS1 bound efficiently. Mutation of either tyrosine substantially reduced binding, and mutation of both sites abolished the interaction altogether. This shows that both tyrosines 378 and 397 contribute to the interaction with Itk.
To determine whether HS1 and Itk interact directly, we asked whether the rGST-Itk SH2 domain could bind to phosphorylated rHS1 fusion protein in the absence of other components of the T cell signaling machinery. As Fig. 3E shows, HS1 and Itk interacted directly, and efficient binding required both Y378 and Y397. We conclude that HS1 and Itk interact in T cells, and that this interaction involves direct binding between the SH2 domain of Itk and tyrosines 378 and 397 of HS1. Because we have shown previously that these sites on HS1 are phosphorylated transiently upon TCR engagement (10), direct interaction between HS1 and Itk is predicted to be transient, and the observed constitutive association between these proteins (Fig. 2) is likely to involve additional interactions with mutual binding partners.
Itk is required for HS1 recruitment to the IS
We have shown previously that phosphorylation of HS1 at Y378 and Y397 is required for its recruitment to the IS (10). Because these sites mediate direct binding to the SH2 domain of Itk, we asked whether Itk is required for recruitment of HS1 to the IS. As shown in Fig. 4A, control Jurkat cells conjugated to superantigen-pulsed B cells recruited HS1 to the IS, but this recruitment was impaired in cells transfected with an Itk suppression vector. In contrast, T cells lacking HS1 showed no defect in recruitment of Itk to the IS (Fig. 4B), indicating that the requirement for recruitment is unidirectional. To quantitate the requirement for Itk in recruiting HS1 to the IS, conjugates were prepared using control and Itk-suppressed Jurkat cells, and the frequency of conjugates showing a bright band of HS1 at the IS was scored. As shown in Fig. 4C, Itk suppression diminished HS1 recruitment to frequencies similar to those observed in the absence of superantigen. Using a similar approach, we asked what portions of Itk are required to mediate HS1 recruitment. Jurkat T cells were transfected with wild-type Itk, the kinase-dead (K390R) mutant, or the SH2 domain (R265A) mutant, and HS1 recruitment to the IS was assessed. As shown in Fig. 4D, overexpression of wild-type or kinase-dead Itk had no effect on HS1 localization, but overexpression of the Itk SH2 domain mutant efficiently inhibited HS1 recruitment to the IS. The requirements for HS1 recruitment with respect to Itk parallel those for actin recruitment (11). We conclude that Itk, through its SH2 domain, recruits HS1 to the IS to promote TCR-induced actin responses.
FIGURE 4.

The SH2 domain of Itk is required for recruitment of HS1 to the IS. A, Jurkat cells transfected with Itk suppression or control vectors were conjugated to CMAC-stained, staphylococcal enterotoxin E-loaded Raji B cells (blue) for 15 min. Conjugates were fixed and stained for HS1 (red) or F-actin (green). B, CD4+ T cells from HS1+/− or HS1−/− AND TCR transgenic mice were conjugated to CMAC-stained, MCC peptide-loaded CH27 B cells for 3 min and stained for Itk (red). C, Itk-suppressed or control Jurkat T cells conjugated to Raji B cells were scored for recruitment of HS1 to the cell-cell contact site. D, Jurkat cells were transfected with expression vectors for wild-type, SH2 domain mutant (SH2m), or kinase-dead (KD) Itk. Conjugates were prepared and analyzed, as in C. Data in C and D represent analysis of 50–100 conjugates obtained from two independent experiments.
HS1 is required for efficient Ca2+ store release
In addition to its role in regulating actin responses, Itk is a key signaling intermediate in TCR signaling-induced Ca2+ responses (25, 26). Although there is some evidence that Itk is involved in activation of Ca2+ release activated Ca2+ (CRAC) channels (25), it is thought that Itk’s primary role is to phosphorylate PLCγ1, leading to inositol 1,4,5-triphosphate (IP3) production and release of Ca2+ from intracellular stores. HS1 is also required for normal T cell Ca2+ responses (10), but the level at which HS1 functions in this pathway has not been addressed. To test this, HS1−/− T cells were stimulated with anti-CD3 in the absence of extracellular Ca2+ to visualize endoplasmic reticulum (ER) store release, and then exposed to extracellular Ca2+ to permit Ca2+ influx via CRAC channels. As we reported previously, HS1−/− T cells showed a significant blunting of Ca2+ responses (Fig. 5A). Expansion of the y-axis to better show the response in the absence of extracellular Ca2+ (Fig. 5A, right panel) reveals that HS1-deficient T cells fail to show significant release of Ca2+ from ER stores. To confirm this finding, T cells were treated with the SERCA pump inhibitor thapsigargin to pharmacologically induce store depletion, and Ca2+ responses were assessed. As shown in Fig. 5B, treatment with thapsigargin rescued the Ca2+ defect in HS1-deficient T cells, indicating that HS1, like Itk, is required for Ca2+ store release.
FIGURE 5.

HS1 is required for release of Ca2+ from intracellular stores. A, CD4+ T cells from HS1+/+ or HS1−/− mice were loaded with the fura 2-AM and plated on poly(L-lysine)-coated coverslips. Immediately before TCR stimulation, the cells were superfused with Ca2+-free bath solution. At the indicated time, cells were stimulated by the addition of anti-CD3 (500.A2) and analyzed by fluorescence microscopy to visualize a rise in cytoplasmic Ca2+ indicative of release from ER stores. Extracellular Ca2+ was then added to visualize Ca2+ influx. Right, An expansion of the y-axis to visualize release from ER stores. B, Cells prepared as in A were treated with 1 μM thapsigargin (Tg) and imaged as in A.
Previous studies have demonstrated that at least one Itk-binding partner, murine SHZ domain-containing leukocyte protein of 76 kDa (SLP-76), can activate Itk kinase activity toward PLCγ1 (27). To ask whether HS1 plays a similar role, T cells from wild-type and HS1−/− mice were stimulated with anti-CD3 for various times and the phosphorylation of PLCγ1 was analyzed by Western blotting using site-specific Abs. As shown in Fig. 6A, phosphorylation of PLCγ1 at tyrosine 783 proceeded normally in the absence of HS1. Phosphorylation at tyrosine 775 was also intact (data not shown). Thus, we conclude that HS1 is not required for phosphorylation-dependent activation of PLCγ1. PLCγ1 activation is characterized not only by phosphorylation, but also by localization to the IS, in association with other components of the signalosome (19). We therefore analyzed PLCγ1 localization in wild-type and HS1−/− AND TCR transgenic T cells conjugated to MCC-pulsed CH27 B cells. As shown in Fig. 6B, PLCγ1 localized efficiently to the IS at early time points in both control and HS1-deficient T cells. Time course analysis showed that wild-type cells were able to maintain PLCγ1 at the IS for 20 min, whereas HS1−/− cells showed significant loss of IS-associated PLCγ1 by 10 min (Fig. 6C). This mirrors the defects we have previously observed for F-actin and Vav1 (10). Nonetheless, given the rapid kinetics of Ca2+ store release relative to the defects we observed in PLCγ1 localization, the Ca2+ signaling defects that we observe in HS1-deficient T cells cannot be explained by a failure to recruit PLCγ1 to the IS at the relevant early time points.
FIGURE 6.

HS1 is dispensable for PLCγ1 phosphorylation and recruitment to the IS. A, T cells from HS1+/+ or HS1−/− mice were stimulated as indicated, and lysates were analyzed by Western blotting for PLCγ1 phosphorylated at Y783, total PLCγ1, and HS1. B, CD4+ T cells from HS1+/− or HS1−/− AND TCR transgenic mice were conjugated to CMAC-stained, MCC peptide-loaded CH27 B cells (blue) and stained for PLCγ1 (red). C, Conjugates prepared as in B were scored for recruitment of PLCγ1 to the cell-cell contact site.
The finding that sustained PLCγ1 localization to the IS depends on HS1 led us to examine earlier actin-driven aspects of PLCγ1 signaling in greater detail. To ask whether HS1 functions to modulate PLCγ1 association with the cytoskeleton, we prepared insoluble actin-rich fractions from wild-type or HS1−/− T cells, and assessed the association of total and phospho-PLCγ1 with these fractions by Western blotting. In wild-type T cells, a fraction of PLCγ1 became associated with the cytoskeleton within 1 min of TCR engagement (Fig. 7A). PLCγ1 continued to accumulate in the cytoskeletal fraction over the next 5 min (Fig. 7B). Importantly, this pool was enriched in phospho-PLCγ1 such that the vast majority of phospho-PLCγ1 was cytoskeletally associated (Fig. 7, A and C). In contrast, little or no activation-induced cytoskeletal association of PLCγ1 was observed in lysates from HS1−/− T cells. Although the experiment shown in Fig. 7A shows increased phospho-PLCγ1 in the cytoskeletal fraction at 3 min in HS1−/− T cells, this result was not reproducible. The significance of the insoluble pool of PLCγ1 remains to be defined; however, given that the phosphorylated pool of the molecule is thought to represent the active pool, it seems likely that cytoskeletal association is intimately linked with PLCγ1 function. These results support the idea that HS1 affects PLCγ1 organization during early phases of TCR signaling.
FIGURE 7.

HS1 is required for PLCγ1 cytoskeletal association. A, T cells from HS1+/+ or HS1−/− mice were stimulated as indicated, lysed in cytoskeletal stabilizing lysis buffer, and separated into cytosol-enriched supernatant and F-actin-rich pellet fractions. Fractions were analyzed by Western blotting, as indicated. pPLCγ1 was detected using an Ab specific for PLCγ1 phosphorylated at Y783. B, The amount of PLCγ1 in the pellet fractions at each time point was quantified and normalized to the amount of PLCγ1 in the pellet in unstimulated cells. Data represent averages from four independent experiments ± SEM. *, p < 0.05. C, The amount of PLCγ1 phosphorylated at Y783 in the pellet fractions at each time point was quantified and normalized to the amount of PLCγ1 phosphorylated at Y783 in the pellet in unstimulated cells. Data represent averages from four independent experiments ± SEM. *, p < 0.05; **, p < 0.01.
To observe directly the effects of HS1 on PLCγ1 dynamics, we analyzed the formation of signaling microclusters in Jurkat cells expressing PLCγ1-eYFP during spreading on anti-CD3-coated coverslips. In control cells, as reported previously (19), PLCγ1 assembled into microclusters at the T cell-coverslip interface. As spreading progressed, new microclusters were formed within actin-rich lamellipodial protrusions, near the edge of the spreading cell (Fig. 8A and supplemental Videos 7 and 8). HS1-suppressed cells also formed microclusters, but these clusters were often disorganized and formed large aggregates (Fig. 8B and supplemental Videos 9 and 10). To assess this phenotype, individual microclusters were identified in video sequences, and pseudocolored in order of appearance. In control cells, microclusters were formed at distinct sites in a concentric fashion, with newer microclusters in the periphery (Fig. 8C). In contrast, HS1-deficient T cells exhibited formation of new microclusters that overlapped with older ones, resulting in aggregates of mixed age (Fig. 8D). We assessed the size of these mixed aggregates in each cell at 90 s after contact with the coverslip. As shown in Fig. 8E, suppression of HS1 resulted in a 2-fold increase in average microcluster size. This increase in average size was attributable to an abnormally disparate size distribution. Although vast majority of microclusters in control cells was less than 0.5 μm in diameter, nearly half of the microclusters in HS1-deficient cells exceeded 0.5 μm, with some as large as 6 μm (Fig. 8F). Taken together, these results show that HS1 is required for the processive formation of discrete PLCγ1 microclusters, consistent with the view that microcluster formation is linked to actin-dependent T cell spreading. Because newly formed microclusters are key sites for early TCR signaling events (2, 3, 28), the defect in Ca2+ responses in HS1-deficient T cells may reflect a role for HS1 in controlling PLCγ1 signaling at the level of microcluster dynamics.
FIGURE 8.

HS1 regulates PLCγ1 microcluster dynamics. A, Jurkat cells stably expressing PLCγ1-eYFP were transfected with empty vector, plated on coverslips coated with anti-CD3, and imaged by confocal microscopy. Selected images from one time-lapse series are shown; these correspond to supplemental Video 7. B, Jurkat cells stably expressing PLCγ1-eYFP were transfected with HS1 suppression vector and analyzed as in A. Selected images from one time-lapse series are shown; these correspond to supplemental Video 9. C and D, PLCγ1 microclusters in empty vector (C) or HS1-suppressed (D) T cells were identified and color coded by order of appearance, with newer microclusters colored orange and red, and older microclusters colored blue and green. Left-hand panels, Cells from the sequences shown in A and B. Right-hand panels, Additional examples. E, The area of individual microclusters was determined at 90 s after the initiation of contact with the coverslip. At least 20 cells were analyzed per condition, yielding ~400 microclusters each. Data represent mean ± SEM. **, p < 0.0001. F, The areas of individual microclusters from E were grouped into the indicated bins. The percentage of total microclusters falling into each size category is shown.
Discussion
Regulation of actin dynamics in T cells involves coordination of multiple actin-regulatory proteins by upstream kinases, including Lck, ZAP70, and c-Abl (29–31). Itk has also been demonstrated to play a role in regulating T cell actin responses, but this function is independent of its kinase activity and instead requires its SH2 domain (11–13, 32). We show in this study that one role of the SH2 domain of Itk is to recruit HS1 to the IS. Video analysis shows that T cells lacking Itk or HS1 have similar defects in actin reorganization downstream of TCR engagement. In contrast to T cells lacking WAVE2 or Arp2/3 complex components, which fail to extend actin-rich lamellipodial protrusions (33, 34), T cells deficient for HS1 or Itk can extend these structures, but they are unstable and retract frequently. This phenotype is consistent with in vitro studies showing that HS1 functions to stabilize existing branched actin filaments (8, 9). In addition to stabilizing actin filaments directly, HS1, like Itk, has been implicated in regulating the localization of Vav1 at the IS (10). Localization of Vav1 to the IS requires an intact Itk SH2 domain (11). Because Vav1 binds directly to HS1, this suggests that HS1 links Vav1 to Itk during T cell activation. It is important to point out, however, that these proteins also interact with other signalosome components, including LAT, SLP-76, and PLCγ1, such that loss of any one of these molecules disrupts interactions among the others and perturbs T cell actin responses (35). Finally, there is evidence that the actin scaffolds generated by these signaling molecules function to stabilize newly formed signaling complexes (2), thereby generating a positive feedback loop to facilitate T cell activation.
We have mapped binding of the Itk SH2 domain to phosphotyrosines 378 and 397 of HS1. These residues are phosphorylated upon TCR engagement in a ZAP70-dependent manner and are required for recruitment of HS1 to the IS and for its functions in actin remodeling and signaling to the Il2 promoter (10). Interestingly, these two tyrosines also serve as docking sites for Vav1 binding. It is unclear whether one molecule of HS1 can bind simultaneously to Itk and Vav1. This need not be the case, because multiple molecules of HS1 would be present in any given actin-associated signaling complex. Although our in vitro data clearly indicate that HS1 and Itk associate via activation-dependent phosphotyrosine-based interactions, these two molecules can be coimmunoprecipitated constitutively from T cell lysates. This constitutive interaction does not appear to involve the SH3 domain of either molecule. This interaction can be explained, in part, by basal tyrosine phosphorylation of HS1, but it probably also involves mutual interactions of these proteins with other T cell signaling molecules. Similar conclusions have been reached for other signalosome proteins, e.g., Itk and SLP-76 (36).
In addition to their similarities with respect to actin dynamics, T cells lacking Itk or HS1 both have defects in Ca2+ store release (10, 25, 26). In the case of Itk-deficient T cells, the defects in Ca2+ store release are well studied and stem from the requirement for Itk-dependent phosphorylation of PLCγ1. Because HS1 binds to Itk, we considered the possibility that HS1 binding promotes Itk kinase activity, as has been documented for SLP-76 and Itk (27). However, HS1 appears to be dispensable for Itk kinase activity, because HS1-deficient cells show normal kinetics and magnitude of PLCγ1 phosphorylation. Based on these findings, we conclude that HS1 and Itk function to promote Ca2+ store release through distinct mechanisms. Although Itk phosphorylates PLCγ1 (27), HS1 appears to regulate PLCγ1 function by linking it to the actin cytoskeleton. Interestingly, because Itk functions upstream of HS1 in the actin-regulatory pathway, this suggests that Itk may promote PLCγ1 activation in two ways: via its kinase activity and through SH2 domain-mediated adaptor functions. Further analysis will be required to test this idea and to address the interdependence of these two mechanisms.
Consistent with this dual mode of PLCγ1 activation, Braiman et al. (19) have shown that proper PLCγ1 function involves a complex interplay between phosphorylation and association with other signalosome components in signaling microclusters. There is evidence in other cell types that PLCγ1 associates with the actin cytoskeleton, and this association can modulate PLCγ1 activity (37–40). We now show that T cell activation induces partitioning of PLCγ1 into an F-actin-rich fraction, and that the phosphorylated pool of PLCγ1 is enriched in this fraction. HS1−/− T cells show impaired cytoskeletal association of PLCγ1. Moreover, whereas control T cells spreading on anti-CD3-coated surfaces show processive assembly of PLCγ1 microclusters near the periphery, HS1-deficient cells show disorganized microcluster assembly, with newly formed microclusters overlapping with older ones. The simplest explanation for these observations is that formation of new, active PLCγ1 microclusters is linked to formation of branched actin filaments at the periphery of spreading T cells. Thus, in HS1-deficient cells, defects in stabilization of lamellipodial protrusions result in perturbation of processive microcluster assembly.
It seems likely that the defects that we observe in PLCγ1 dynamics underlie the defects in Ca2+ store release, and it will be interesting to explore the molecular mechanisms through which this occurs. Recent studies have led to a paradigm in which microcluster dynamics are mechanistically linked to signal generation and extinction. According to this model, early TCR signaling events mostly take place in newly formed peripheral microclusters. Depending on the quality of the Ag, signaling can continue in centralized signaling complexes, but centralized complexes are ultimately subject to signal extinction via internalization and degradation (5, 41–43). Thus, the defects that we observe in PLCγ1 microcluster dynamics might result in aberrant signaling due to premature mixing with proteins responsible for signal extinction. Alternatively, efficient TCR signaling may require the forces associated with myosin-based microcluster centralization (4, 44). If so, the spreading defects in HS1-deficient T cells may result in reduced PLCγ1 signaling as a result of diminished tension. Finally, the observed defects in Ca2+ store release may arise as a result of altered cell geometry. In a well-spread T cell, much of the ER is localized within a short distance of the cell surface, whereas in a poorly spread T cell, the bulk of the ER is situated much further away. Because PLCγ1-induced IP3 production is very short-lived, typically peaking within 1 min of TCR engagement (45), and activation of IP3 receptors is highly cooperative (46), T cell spreading could serve to promote Ca2+ store release by bringing ER-associated IP3 receptors into proximity with sites of IP3 production. Additional investigation will be required to test these possibilities and to explore in more general terms the importance of actin polymerization and cell spreading for T cell signaling.
Supplementary Material
Acknowledgments
We thank Dr. P. Zhu for assistance with Ca2+ studies, B. Chang for technical assistance, Dr. L. Samelson and Dr. L. Berg for generously sharing reagents, and members of the Burkhardt and Oliver laboratories for helpful discussions and comments on the manuscript.
Footnotes
This work was supported by Grant R01AI065644 to J.K.B., Grant F31AI071385 to E.C., and Grant R01AI065474 to D.D.B. D.D.B. is a Leukemia and Lymphoma Scholar.
Abbreviations used in this paper: IS, immunological synapse; ER, endoplasmic reticulum; eYFP, enhanced yellow fluorescent protein; HS1, hematopoietic lineage cell-specific protein 1; IP3, inositol 1,4,5-triphosphate; Itk, IL-2-inducible T cell kinase; MBP, maltose-binding protein; MCC, moth cytochrome c; PLC, phospholipase C; SH, Src homology; CMAC, 7-amino-4-chloromethyl coumarin; CRAC, Ca2+-release activated Ca2+; SLP-76, SHZ-domain containing protein of 76 kDa.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 2.Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cemerski S, Das J, Giurisato E, Markiewicz MA, Allen PM, Chakraborty AK, Shaw AS. The balance between T cell receptor signaling and degradation at the center of the immunological synapse is determined by antigen quality. Immunity. 2008;29:414–422. doi: 10.1016/j.immuni.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seminario MC, Bunnell SC. Signal initiation in T-cell receptor microclusters. Immunol Rev. 2008;221:90–106. doi: 10.1111/j.1600-065X.2008.00593.x. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura D, Kaneko H, Miyagoe Y, Ariyasu T, Watanabe T. Isolation and characterization of a novel human gene expressed specifically in the cells of hematopoietic lineage. Nucleic Acids Res. 1989;17:9367–9379. [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370–374. doi: 10.1016/s0960-9822(01)00098-7. [DOI] [PubMed] [Google Scholar]
- 9.Uruno T, Zhang P, Liu J, Hao JJ, Zhan X. Haematopoietic lineage cell-specific protein 1 (HS1) promotes actin-related protein (Arp) 2/3 complex-mediated actin polymerization. Biochem J. 2003;371:485–493. doi: 10.1042/BJ20021791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, Zhu P, Freedman BD, Clark MR, Rawlings DJ, et al. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity. 2006;24:741–752. doi: 10.1016/j.immuni.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dombroski D, Houghtling RA, Labno CM, Precht P, Takesono A, Caplen NJ, Billadeau DD, Wange RL, Burkhardt JK, Schwartzberg PL. Kinase-independent functions for Itk in TCR-induced regulation of Vav and the actin cytoskeleton. J Immunol. 2005;174:1385–1392. doi: 10.4049/jimmunol.174.3.1385. [DOI] [PubMed] [Google Scholar]
- 12.Grasis JA, Browne CD, Tsoukas CD. Inducible T cell tyrosine kinase regulates actin-dependent cytoskeletal events induced by the T cell antigen receptor. J Immunol. 2003;170:3971–3976. doi: 10.4049/jimmunol.170.8.3971. [DOI] [PubMed] [Google Scholar]
- 13.Labno CM, Lewis CM, You D, Leung DW, Takesono A, Kamberos N, Seth A, Finkelstein LD, Rosen MK, Schwartzberg PL, Burkhardt JK. Itk functions to control actin polymerization at the immune synapse through localized activation of Cdc42 and WASP. Curr Biol. 2003;13:1619–1624. doi: 10.1016/j.cub.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taniuchi I, Kitamura D, Maekawa Y, Fukuda T, Kishi H, Watanabe T. Antigen-receptor induced clonal expansion and deletion of lymphocytes are impaired in mice lacking HS1 protein, a substrate of the antigen-receptor-coupled tyrosine kinases. EMBO J. 1995;14:3664–3678. doi: 10.1002/j.1460-2075.1995.tb00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaye J, Hsu ML, Sauron ME, Jameson SC, Gascoigne NR, Hedrick SM. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 1989;341:746–749. doi: 10.1038/341746a0. [DOI] [PubMed] [Google Scholar]
- 16.Kaye J, Vasquez NJ, Hedrick SM. Involvement of the same region of the T cell antigen receptor in thymic selection and foreign peptide recognition. J Immunol. 1992;148:3342–3353. [PubMed] [Google Scholar]
- 17.Vasquez NJ, Kaye J, Hedrick SM. In vivo and in vitro clonal deletion of double-positive thymocytes. J Exp Med. 1992;175:1307–1316. doi: 10.1084/jem.175.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaffer MH, Dupree RS, Zhu P, Saotome I, Schmidt RF, McClatchey AI, Freedman BD, Burkhardt JK. Ezrin and moesin function together to promote T cell activation. J Immunol. 2009;182:1021–1032. doi: 10.4049/jimmunol.182.2.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braiman A, Barda-Saad M, Sommers CL, Samelson LE. Recruitment and activation of PLCγ1 in T cells: a new insight into old domains. EMBO J. 2006;25:774–784. doi: 10.1038/sj.emboj.7600978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez TS, Hamann MJ, McCarney S, Savoy DN, Lubking CM, Heldebrant MP, Labno CM, McKean DJ, McNiven MA, Burkhardt JK, Billadeau DD. Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol. 2005;6:261–270. doi: 10.1038/ni1168. [DOI] [PubMed] [Google Scholar]
- 21.Trushin SA, Pennington KN, Carmona EM, Asin S, Savoy DN, Billadeau DD, Paya CV. Protein kinase Cα (PKCα) acts upstream of PKCθ to activate IκB kinase and NF-κB in T lymphocytes. Mol Cell Biol. 2003;23:7068–7081. doi: 10.1128/MCB.23.19.7068-7081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cannon JL, Labno CM, Bosco G, Seth A, McGavin MH, Siminovitch KA, Rosen MK, Burkhardt JK. Wasp recruitment to the T cell: APC contact site occurs independently of Cdc42 activation. Immunity. 2001;15:249–259. doi: 10.1016/s1074-7613(01)00178-9. [DOI] [PubMed] [Google Scholar]
- 23.Gatfield J, Albrecht I, Zanolari B, Steinmetz MO, Pieters J. Association of the leukocyte plasma membrane with the actin cytoskeleton through coiled coil-mediated trimeric coronin 1 molecules. Mol Biol Cell. 2005;16:2786–2798. doi: 10.1091/mbc.E05-01-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity. 2001;14:315–329. doi: 10.1016/s1074-7613(01)00112-1. [DOI] [PubMed] [Google Scholar]
- 25.Liu KQ, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J Exp Med. 1998;187:1721–1727. doi: 10.1084/jem.187.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaeffer EM, Debnath J, Yap G, McVicar D, Liao XC, Littman DR, Sher A, Varmus HE, Lenardo MJ, Schwartzberg PL. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 1999;284:638–641. doi: 10.1126/science.284.5414.638. [DOI] [PubMed] [Google Scholar]
- 27.Bogin Y, Ainey C, Beach D, Yablonski D. SLP-76 mediates and maintains activation of the Tec family kinase ITK via the T cell antigen receptor-induced association between SLP-76 and ITK. Proc Natl Acad Sci USA. 2007;104:6638–6643. doi: 10.1073/pnas.0609771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 29.Huang Y, Comiskey EO, Dupree RS, Li S, Koleske AJ, Burkhardt JK. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood. 2008;112:111–119. doi: 10.1182/blood-2007-10-118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowin-Kropf B, V, Shapiro S, Weiss A. Cytoskeletal polarization of T cells is regulated by an immunoreceptor tyrosine-based activation motif-dependent mechanism. J Cell Biol. 1998;140:861–871. doi: 10.1083/jcb.140.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan MM, Labno CM, Van Seventer GA, Denny MF, Straus DB, Burkhardt JK. Superantigen-induced T cell: B cell conjugation is mediated by LFA-1 and requires signaling through Lck, but not ZAP-70. J Immunol. 2001;167:5708–5718. doi: 10.4049/jimmunol.167.10.5708. [DOI] [PubMed] [Google Scholar]
- 32.Donnadieu E, Lang V, Bismuth G, Ellmeier W, Acuto O, Michel F, Trautmann A. Differential roles of Lck and Itk in T cell response to antigen recognition revealed by calcium imaging and electron microscopy. J Immunol. 2001;166:5540–5549. doi: 10.4049/jimmunol.166.9.5540. [DOI] [PubMed] [Google Scholar]
- 33.Gomez TS, Kumar K, Medeiros RB, Shimizu Y, Leibson PJ, Billadeau DD. Formins regulate the actin-related protein 2/3 complex-independent polarization of the centrosome to the immunological synapse. Immunity. 2007;26:177–190. doi: 10.1016/j.immuni.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, Burkhardt JK, Freedman BD, Billadeau DD. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bunnell SC, Singer AL, Hong DI, Jacque BH, Jordan MS, Seminario MC, Barr VA, Koretzky GA, Samelson LE. Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Mol Cell Biol. 2006;26:7155–7166. doi: 10.1128/MCB.00507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jordan MS, Smith JE, Burns JC, Austin JE, Nichols KE, Aschenbrenner AC, Koretzky GA. Complementation in trans of altered thymocyte development in mice expressing mutant forms of the adaptor molecule SLP76. Immunity. 2008;28:359–369. doi: 10.1016/j.immuni.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bar-Sagi D, Rotin D, Batzer A, Mandiyan V, Schlessinger J. SH3 domains direct cellular localization of signaling molecules. Cell. 1993;74:83–91. doi: 10.1016/0092-8674(93)90296-3. [DOI] [PubMed] [Google Scholar]
- 38.Nojiri S, Hoek JB. Suppression of epidermal growth factor-induced phospholipase C activation associated with actin rearrangement in rat hepatocytes in primary culture. Hepatology. 2000;32:947–957. doi: 10.1053/jhep.2000.18662. [DOI] [PubMed] [Google Scholar]
- 39.Pei Z, Yang L, Williamson JR. Phospholipase C-γ1 binds to actin-cytoskeleton via its C-terminal SH2 domain in vitro. Biochem Biophys Res Commun. 1996;228:802–806. doi: 10.1006/bbrc.1996.1735. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki K, Takahashi K. Actin filament assembly and actin-myosin contractility are necessary for anchorage- and EGF-dependent activation of phospholipase Cγ. J Cell Physiol. 2001;189:64–71. doi: 10.1002/jcp.1134. [DOI] [PubMed] [Google Scholar]
- 41.Balagopalan L, V, Barr A, Sommers CL, Barda-Saad M, Goyal A, Isakowitz MS, Samelson LE. c-Cbl-mediated regulation of LAT-nucleated signaling complexes. Mol Cell Biol. 2007;27:8622–8636. doi: 10.1128/MCB.00467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barr VA, Balagopalan L, Barda-Saad M, Polishchuk R, Boukari H, Bunnell SC, Bernot KM, Toda Y, Nossal R, Samelson LE. T-cell antigen receptor-induced signaling complexes: internalization via a cholesterol-dependent endocytic pathway. Traffic. 2006;7:1143–1162. doi: 10.1111/j.1600-0854.2006.00464.x. [DOI] [PubMed] [Google Scholar]
- 43.Lee KH, Dinner AR, Tu C, Campi G, Raychaudhuri S, Varma R, Sims TN, Burack WR, Wu H, Wang J, et al. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302:1218–1222. doi: 10.1126/science.1086507. [DOI] [PubMed] [Google Scholar]
- 44.Varma R. TCR triggering by the pMHC complex: valency, affinity, and dynamics. Sci Signal. 2008;1:pe21. doi: 10.1126/stke.119pe21. [DOI] [PubMed] [Google Scholar]
- 45.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 46.Meyer T, Holowka D, Stryer L. Highly cooperative opening of calcium channels by inositol 1,4,5-trisphosphate. Science. 1988;240:653–656. doi: 10.1126/science.2452482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.