

Abstract
Genetic disorders that present with a high incidence of autism spectrum disorders (ASD) offer tremendous potential both for elucidating the underlying neurobiology of ASD and identifying therapeutic drugs and/or drug targets. As a result, clinical trials for genetic disorders associated with ASD are no longer a hope for the future but rather an exciting reality whose time has come. Tuberous sclerosis complex (TSC) is one such genetic disorder that presents with ASD, epilepsy, and intellectual disability. Cell culture and mouse model experiments have identified the mTOR pathway as a therapeutic target in this disease. This review summarizes the advantages of using TSC as model of ASD and the recent advances in the translational and clinical treatment trials in TSC.
Introduction
Autism spectrum disorders (ASD) affect approximately 1% of children in the United States, and are characterized by defects in social interaction, language delay, and repetitive interests or behaviors. ASD is a major public health problem that disrupts families and leads to significant disability, resulting in a total annual societal cost of ∼$35 billion to care for and treat autistic individuals [1]. When initially described by Kanner in the 1940s, researchers believed that this disorder of behavior and cognition resulted from emotional deprivation in infancy [2]. This notion has long since been discarded, and rather it has been recognized that neuronal dysfunction occurs in early brain development in individuals affected with ASD [3–5]. In the last decade or so, we have started to undertake the studies needed to understand the underlying etiology of ASD. Genes play a greater role in the risk of ASD than in any other common neurodevelopmental disorder, with estimates of heritability as high as 60–90% [6•,7•]. However, the genetic cause is unknown in most cases. Collectively, rare copy number variants (CNVs) account for the largest category (6–8%) of known genetic causes of ASD. Combining recurrent CNVs and single gene Mendelian disorders, known genetic causes currently account for less than 15% of ASD cases [8•,9]. Since each of the rare CNVs associated with autism is identified in a very small number of individuals with ASD, leads to alterations in dosage of a relatively large number of genes, and demonstrates phenotypic variability of expression, it is not yet clear how we will be able to utilize this knowledge to develop therapeutics. Identifying the key gene(s) in CNVs is likely to take some time. In contrast, single-gene disorders associated with ASD appear to provide us with a critical opportunity to understand and develop treatments for ASD since they provide both a much larger number and more homogenous group of patients to study. Thus, ASD studies that leverage findings from these single-gene disorders have the potential to elucidate both the underlying neurobiology of ASD and to identify therapeutic drugs and/or drug targets.
Until recently, Mendelian etiologies with high penetrance of ASD such as fragile X syndrome, tuberous sclerosis complex (TSC), and Rett syndrome had been relatively ignored in autism research. For several decades, most of the research in the ASD field was focused on ‘pure’ or ‘idiopathic’ autism. In fact, much of the imaging and cognitive neuroscience studies were performed on a relatively small segment of the ASD population with ‘high functioning autism’ and without known genetic syndromes. This was an unusual approach in neuroscience since research on diseases such as Alzheimer's, Parkinson's and amyotrophic lateral sclerosis have benefited enormously from the study of familial cases with known genetic causes. However, ASD researchers have recently turned to the opportunities provided by the Mendelian disorders strongly associated with autism. A case in point is TSC. This review summarizes the recent progress in TSC translational and clinical research, as an example of how a Mendelian disorder can be used to investigate early detection and development of effective treatments for ASD.
The case for tuberous sclerosis complex in ASD research
TSC is a multisystem genetic disorder in which 90–95% of the affected individuals have CNS involvement. Epilepsy occurs in 80–90% of these patients and can be medically refractory [10,11]. Approximately 45% of TSC patients have mild-to-profound intellectual disability, and ASD occurs in up to 50% of TSC individuals [10,12]. The neuropathological findings in the TSC brain typically include subependymal nodules (SENs), subependymal giant cell astrocytomas (SEGAs) and cortical tubers [13]. Accumulating evidence suggests that TSC patients have non-tuber abnormalities that significantly contribute to the development of the neurological phenotype, including disorganization of axon tracts, aberrant myelination, and defects in synaptic plasticity (see below).
The proteins encoded by the TSC1 and TSC2 genes, respectively known as hamartin and tuberin, bind together to form a protein complex that plays a critical role in the regulation of a serine-threonine kinase mammalian target of rapamycin (mTOR), a key regulator of protein synthesis [14]. Thus, without a functional TSC complex, mTOR is hyperactive, resulting in disinhibited protein synthesis and subsequent cell growth [15,16]. Importantly, mutations in at least two other genes that regulate protein translation (FMR1 and PTEN) are also associated with ASD. Taken together these findings strongly suggest that defects in translational regulation may represent one common mechanism leading to ASD phenotypes [17].
Multiple factors make TSC patients a unique population to study the early development of ASD. First, many TSC patients may be diagnosed before birth or at the time of birth due to the presence of rhabdomyomas in the heart. A fetus or a newborn with multiple cardiac tumors has a 95% chance of having TSC [18]. Increased use of prenatal ultrasounds has significantly increased the detection of TSC in the fetus such that cardiac manifestations have become the most common presenting sign of TSC [19], providing a ‘newborn imaging screen’ to detect individuals with TSC very early in life. Second, approximately 50% of TSC patients are affected with ASD symptoms. This is a much higher incidence than other high-risk groups that can be identified early in life, such as those who are siblings of affected individuals. Third, significant understanding of the aberrant cellular mechanisms in TSC already exists. Among single gene disorders frequently associated with ASD, TSC is one of the best characterized at the cellular level. Finally, the mTOR pathway has been studied from various medical perspectives, including those of oncology, immunology, and transplantation. As a result, there are now three FDA-approved mTOR specific inhibitors. Taken together, these four factors make TSC a unique genetic disorder for the study of the early development of ASD.
Basic cell biology of TSC in neurons
Studies performed both in vitro and in vivo using mouse models have demonstrated that the Tsc1 and Tsc2 proteins play crucial roles not only in cell growth, but also in axonal, dendritic and synaptic development and function. On the axonal side, TSC1/2 proteins regulate axon specification, guidance, myelination and regeneration. In cultured hippocampal neurons undergoing early neuronal polarity determination, TSC pathway components are expressed in a polarized manner, much higher in nascent axons than dendrites [20–22]. Over-expression of Tsc1 and Tsc2 suppresses axon formation while loss of either gene results in increased axon number [20]. In addition, Tsc2 haploinsufficiency in retinal ganglion cells leads to aberrant neuronal projections, due to the crucial role TSC1/2 proteins play in ephrin/Eph receptor signaling [23•]. Finally, neuronal expression of Tsc1 or Tsc2 is crucial for the proper myelination of axons. Loss of Tsc1 in neurons causes a block in myelination, consistent with crucial developmental interaction between neurons and oligodendrocytes [24].
On the dendritic side, the TSC/mTOR pathway also regulates dendritic arborization and spine morphogenesis [25–27]. Loss of either Tsc1 or Tsc2 expression increases spine length and head width and reduces spine density in hippocampal slice cultures [27] and in vivo[28]. In addition, the mTOR pathway plays a role in postsynaptic AMPA receptor expression [29]. In Tsc1 deficient hippocampal neurons, the AMPA/NMDA receptor current ratio is increased, suggesting a relative enhancement of synaptic AMPA receptors [27]. Furthermore, loss of Tsc1 or Tsc2 in the mouse hippocampus abolishes mGluR-de-pendent long-term depression (LTD), but not NMDA receptor-dependent LTD [30••,31••]. Tsc2+/− rats also display increased paired-pulse facilitation and reduced long-term potentiation [32]. Together, the roles that the TSC1/2 proteins play in axonal, dendritic development and synaptic function strongly indicate that loss of TSC1/2 is likely to result in defects in synapse formation and plasticity, likely correlating with the neurocognitive and behavioral symptoms of TSC. Since the ASD are starting to emerge as ‘developmental disconnection syndromes,’ [33,34] TSC is especially suited to understand the contribution of axonal, dendritic and synaptic defects in ASD.
Mouse models of TSC can be treated with mTOR inhibitors
Several mouse models of TSC have been generated and all of them display neurocognitive deficits [24,28,35–37], many of which can be reversed with treatment with mTOR inhibitors [28,35]. For example, Tsc1+/− mice show impaired learning in hippocampal-dependent learning tasks and impaired social behavior, supporting a model in which haploinsufficiency for the TSC genes leads to aberrations in neuronal functioning resulting in impaired learning and social behavior [36]. Similarly, Tsc2+/− mice show deficits in synaptic plasticity, learning, and memory [35]. These deficits emerge in the absence of gross structural brain abnormalities or seizures, demonstrating that other disease mechanisms are involved. Notably, a brief 5-day treatment with rapamycin in adult mice rescued not only the synaptic plasticity, but also learning and memory deficits in this animal model [35]. More severe mouse models of TSC have been generated by homozygous inactivation of the Tsc1 gene in neurons or astrocytes [24,28]. In both cases, mice develop epilepsy and premature death, which can be prevented or reversed by rapamycin. These experiments demonstrate that postnatal mTOR inhibition prevents the neurological and behavioral defect in TSC-deficient mice.
Another brain pathology in TSC disease is the presence of SENs and SEGAs. SENs are lesions found along the wall of the lateral ventricles in the brain. These benign lesions do not cause any medical problems for patients; however, in 5–10% of cases, a SEN can grow into SEGA. SEN-like structures in mice were only recently achieved by loss of Tsc1 specifically in postnatal neural stem/progenitor cells [38•]. Recently, huge progress has been achieved in testing the efficacy of mTOR inhibitors on SEGA volume in patients (see below).
Human clinical trials with mTOR inhibitors in TSC
In parallel with the basic neurobiology research in the role of TSC1/2 in brain development, there has been major progress in the use of mTOR inhibitors in several aspects of medical care (Figure 1). Following the discovery of rapamycin in the 1970s, rapamycin and related mTOR inhibitors have been developed for several clinical indications; first to prevent solid organ transplant rejection, and later to prevent stenosis of artificial coronary stents and treatment for several types of cancer. While TSC1 and TSC2 were identified in the 1990s as causative genes in TSC, it was not until 2002 that evidence demonstrating a role for TSC1/2 in mTOR regulation emerged [39–43]. At that time, David Franz and colleagues began to consider the possibility that rapamycin and related mTOR inhibitors might have clinical benefit for various clinical features in TSC [44]. They initially treated SEGAs in patients who had failed previous surgical resections or were otherwise unsuitable for conventional surgery. For these studies, the size of the SEGA on brain imaging provided an objective primary outcome measure, as commonly used in oncology treatment trials. The initial case series, in which all 5 patients responded to treatment with rapamycin [44], was followed up with a prospective, phase I/II clinical trial with the mTOR inhibitor everolimus. In that study 21 of 28 (75%) TSC patients showed a reduction in SEGA volume of at least 30%, and none progressed [45••]. The latter trial led to the approval of everolimus for SEGAs in TSC by the FDA in 2010 (US) and EMEA in 2011 (Europe). Other studies have reported on the efficacy of mTOR inhibitors for treatment of additional manifestations of TSC, including renal angiomyolipomas [46,47•,48•], pulmonary lymphangioleiomyomatosis [46,47•,48•,49•], cardiac rhabdomyoma [50•] and facial angiofibromas [47•,51–55]. In addition to providing clinical evidence of benefit for these TSC-related tumors, these trials provided crucial safety data on the use of mTOR inhibitors in TSC patients. Consequently, evidence of major benefit of rapamycin and related drugs for neurobehavioral phenotypes in mouse models and evidence of both benefit and overall tolerance in human clinical trials on tumor manifestations of TSC have led to the design of clinical treatment trials aimed at neurocognitive deficits in TSC.
Figure 1.
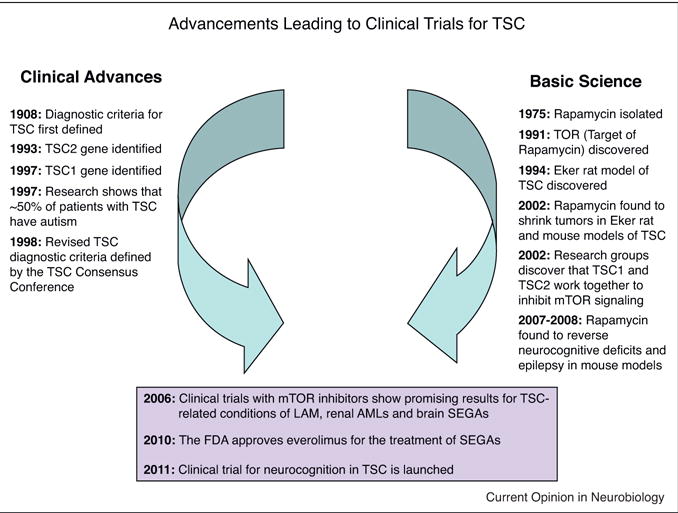
Some milestones in defining the pathophysiology of tuberous sclerosis complex (TSC). The current therapeutic efforts in the neurocognitive phenotype of TSC patients stem from the contributions from two independent lines of research: clinical advances in genetics and phenotyping of TSC (left timeline) and basic research on the TSC-mTOR pathway and mouse models (right timeline). See text for details.
One of the lessons of the early clinical trials of mTOR inhibitors is that the selection of inclusion and exclusion criteria has to be carefully tailored for each outcome measure. In other words, one cannot necessarily look at all outcome measures in one group of subjects. For example, a group of individuals with TSC that has SEGAs may not have the ability to participate in neurocognitive testing batteries because of age or intellectual ability. Therefore, a separate trial needed to be designed to investigate whether mTOR inhibition could improve neurocognition in children with TSC (clinicaltrials.gov: NCT01289912). This placebo-controlled double blind trial of everolimus is currently enrolling children with TSC between the ages of 6 and 21 years, with IQ greater than or equal to 60, who are stable on an anti-seizure medication regimen and have no evidence of SEGAs. The primary endpoint is improvement on neurocognitive tests while autism, seizure frequency, and sleep habits are evaluated as secondary endpoints. This trial highlights one of the biggest challenges in treatment trials in neurodevelopmental disorders: How do we demonstrate that a treatment has had an effect? What are the most sensitive outcome measures that are dynamic within a reasonable time span of treatment? Does improvement in an outcome measure correlate with better function in real life? Significant attention has to be paid to choosing the most sensitive, reliable and quantitative outcome measures appropriate for each group of subjects.
A second lesson from these early trials has been the need for multi-center collaborations. For longitudinal studies in a rare disease such as TSC, traveling long distances for research visits at one center is extremely taxing for families, especially when their children are affected with ASD, epilepsy, sleep, and behavioral disorders, as is often the case in TSC. Therefore, multi-center collaborations are necessary to recruit sufficient subjects and to gather the large amount of data required to test the hypotheses in a reasonable time frame. This is best accomplished in concert with national advocacy organizations — in this case the Tuberous Sclerosis Alliance. Such advocacy organizations have been crucial in supporting basic, translational and clinical studies in TSC and other neurodevelopmental disorders, not only by directly funding research and informing families about study opportunities which is essential for recruitment, but also by bringing together leaders of the field and encouraging collaborations.
Need for biomarkers
An abundance of clinical and basic science evidence suggests that mTOR inhibitors represent a rational candidate for the treatment of neurodevelopmental disabilities in TSC. However, mTOR inhibitors can have side effects, such as immuno-suppression and endocrine dysfunction. Given the prominent role of the mTOR pathway in many normal physiological functions, there is also the theoretical risk of adverse effects on growth and development, especially early in life. Despite these risks, steps can be taken to minimize unnecessary exposure and risk of mTOR inhibitors in the context of drug treatment of ASD in TSC patients. Since not all patients with TSC develop ASD or intellectual disability, an early biomarker that can reliably identify at a young age those children who will develop severe neurodevelopmental deficits would be very helpful. A recent study suggests that treating TSC patients that have an abnormal EEG before onset of infantile spasms with vigabatrin may improve neurological outcome [56••], but the use of EEG as a reliable biomarker of future ASD or even epilepsy has not been rigorously validated. Diffusion tensor imaging (DTI) appears promising as a biomarker of neurological complications and ASD [57•,58••], but needs to be studied in a larger group of children with TSC and preferably in a prospective trial. With a predictive biomarker, the highest risk TSC patients who are most appropriate for a drug treatment (e.g. a clinical trial) could be selected, thus sparing the low-risk TSC patients exposure to a drug with potential risk and likely increasing the efficacy of the treatment for a targeted population. Furthermore, sensitive biomarkers can potentially accelerate treatment trials as quantitative surrogate endpoints. Lastly, mechanistically based biomarkers could help identify subgroups of ‘idiopathic’ ASD patients that can benefit from treatments similar to those effective in homogenous populations like TSC.
Conclusions: other barriers and future directions
While the excitement over our ability to finally offer mechanism-based treatment options for rare genetic diseases is warranted, we should recognize that there are still several obstacles to performing treatment trials in neurodevelopmental disorders. One obvious difficulty is the risk-benefit analysis. For many parents whose children are affected with ASD and other neurodevelopmental disorders, treatment trials offer the hope of their child having more effective communication or enhanced social skills. However, for institutional review boards (IRBs), weighing the risk-benefit ratio of a treatment trial is much more difficult in the area of neurodevelopmental disorders than in oncology. This reluctance to intervene may subside once treatment trials in neurodevelopmental disorders become more common, and if there are therapeutic successes. Second, the number of physician scientists trained in clinical trials for neurodevelopmental disorders is extremely low. As the targets for treatment trials in neuroscience increase (and there is no question they will increase with the general application of next-generation sequencing), the number of physician scientists with this expertise also has to be increased. This will require not only additional mentoring and guidance, but also providing the incentives for young clinicians to choose this path.
Similar to the TSC trials described here, targeted treatment trials have recently been initiated for other known genetic disorders associated with ASD such as fragile X, Rett and Down syndromes (Table 1). There is no doubt that the number of potential treatment targets in neurodevelopmental disorders will increase based on investigations of single-gene disorders; for instance, it was recently discovered that a common anti-cancer drug (topotecan) can turn on the normally dormant paternal Ube3a allele in the brain of the Angelman syndrome mouse model [59••]. Recent evidence from mouse models indicate that even disorders that appear quite similar in terms of cell biology (i.e. regulation of protein synthesis) such as TSC and FXS may have diametrically opposite physiological phenotypes under certain circumstances [31••]. Therefore, a detailed understanding of the circuitry and cellular abnormalities associated with each single-gene disorder is crucial to select the most effective treatment. Otherwise, one type of therapy that relieves neurological symptoms in one disease could potentially be harmful in the other. Until we have better biomarkers to predict the treatment response, using these therapies in idiopathic (genetically undefined) autism is similarly risky. Finally, just like in other areas of clinical medicine, we have to be mindful of the fact that not all trials will succeed. Despite the challenges and barriers, carefully designed treatment trials in genetically defined neurodevelopmental disorders are one of the most exciting avenues of research in science and medicine today, and we hope that they will have a positive impact, improving the lives of children and families affected with these disorders in the near future.
Table 1.
Targeted clinical trials for genetic disorders associated with ASD (from clinicaltrials.gov 1/13/12)
| Genetic disorder associated with ASD | Study drug | ASD related outcome measures | Clinicaltrials.gov study ID |
|---|---|---|---|
| Tuberous sclerosis complex (TSC) | Everolimus (RAD001) | ASD, memory, language skills, cognition, behavior, sleep | NCT01289912 |
| Rett syndrome | rhIGF-1 | Behavior, cognition, motor function, language skills | NCT01253317 |
| Fragile X syndrome | (1) Acamprosate | (1) Hyperactivity, language impairment, irritability, social deficits, and cognitive delay | (1) NCT01300923 |
| (2) Arbaclofen (STX209) | (2) Social withdrawal | (2) NCT01325220 | |
| (3) Minocycline Hydrochloride | (3) Language, behavior, cognition | (3) NCT01053156 | |
| (4) AFQ056 and RO4917523 | (4) Learning, cognition, behavior | (4) NCT01253629 and NCT01015430 | |
| Down syndrome | (1) Memantine | (1) Cognition, memory, language | (1) NCT01112683 |
| (2) Rivastigmine | (2) Language, memory, and executive function | (2) NCT01084135 | |
| (3) RG1662 | (3) Cognition, memory | (3) NCT01436955 |
Acknowledgments
I am grateful to Kira Dies for assistance with the figure/table and Drs. Elizabeth Berry-Kravis, Sarah Spence, David Kwiatkowski and Steven Roberds for critical reading of the manuscript. I would like to thank all members of the TSC communities for many helpful discussions. Owing to limited space I have not quoted all the literature in this field, and I apologize to those whose articles are not referenced. The clinical trial (NCT01289912; PIs Sahin, Franz, De Vries) is funded by Novartis, Autism Speaks and Tuberous Sclerosis Alliance. I have served as a consultant and site-PI for Novartis. Research in my laboratory related to this manuscript was funded by the NIH R01 NS058956, Tuberous Sclerosis Alliance, Autism Speaks, John Merck Fund, Nancy Lurie Marks Family Foundation, Children's Hospital Boston Translational Research Program and the Manton Family Foundation.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Ganz M. The costs of autism. In: Moldin SO, Rubenstein JLR, editors. Understanding Autism: From Basic Neuroscience to Treatment. Taylor & Francis; 2006. pp. 475–502. [Google Scholar]
- 2.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 3.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 4.Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23:183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Courchesne E, Redcay E, Morgan JT, Kennedy DP. Autism at the beginning: microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev Psychopathol. 2005;17:577–597. doi: 10.1017/S0954579405050285. [DOI] [PubMed] [Google Scholar]
- 6•.Freitag CM, Staal W, Klauck SM, Duketis E, Waltes R. Genetics of autistic disorders: review and clinical implications. Eur Child Adolesc Psychiatry. 2010;19:169–178. doi: 10.1007/s00787-009-0076-x. An up-to-date review of the molecular genetics of ASD, including linkage, genome-wide association and copy number variation studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2011;156:255–274. doi: 10.1002/ajmg.b.31159. An extensive and critical review of the more than 30 twin studies performed in ASD and the heritability of autism. [DOI] [PubMed] [Google Scholar]
- 8•.Shen Y, Dies K, Holm I, Bridgemohan C, Sobeih M, Caronna E, Miller K, Frazier J, Silverstein I, Picker J, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125(4):e727–e735. doi: 10.1542/peds.2009-1684. peds.2009-1684v2001. This study demonstrates the utility of chromosomal microarray in detecting clinically significant genetic chances in ASD patients. It makes a strong case for including chromosomal microarray in the initial diagnostic evaluation of individuals with ASD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaaf CP, Zoghbi HY. Solving the autism puzzle a few pieces at a time. Neuron. 2011;70:806–808. doi: 10.1016/j.neuron.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 10.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–668. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- 11.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 12.Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. 2008;23:520–525. doi: 10.1177/0883073807309788. [DOI] [PubMed] [Google Scholar]
- 13.DiMario FJ., Jr Brain abnormalities in tuberous sclerosis complex. J Child Neurol. 2004;19:650–657. doi: 10.1177/08830738040190090401. [DOI] [PubMed] [Google Scholar]
- 14.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005:R251–R258. doi: 10.1093/hmg/ddi260. 4 Spec No. 2. [DOI] [PubMed] [Google Scholar]
- 15.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 18.Tworetzky W, McElhinney DB, Margossian R, Moon-Grady AJ, Sallee D, Goldmuntz E, van der Velde ME, Silverman NH, Allan LD. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;92:487–489. doi: 10.1016/s0002-9149(03)00677-5. [DOI] [PubMed] [Google Scholar]
- 19.Datta AN, Hahn CD, Sahin M. Clinical presentation and diagnosis of tuberous sclerosis complex in infancy. J Child Neurol. 2008;23:268–273. doi: 10.1177/0883073807309250. [DOI] [PubMed] [Google Scholar]
- 20.Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YH, Werner H, Puschel AW. Rheb and mTOR regulate neuronal polarity through Rap1B. J Biol Chem. 2008;283:33784–33792. doi: 10.1074/jbc.M802431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita T, Sobue K. Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem. 2009;284:27734–27745. doi: 10.1074/jbc.M109.008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23•.Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci. 2010;13:163–172. doi: 10.1038/nn.2477. The authors demonstrate that TSC2-mTOR signaling acts downstream of the axon guidance molecules, EphA receptors, to regulate axon guidance in the CNS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 28.Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Barbaro MF, Baraban SC. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci Lett. 2006;401:35–39. doi: 10.1016/j.neulet.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 30••.Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci. 2011;31:8862–8869. doi: 10.1523/JNEUROSCI.1617-11.2011. This is the first report demonstrating that protein synthesis dependent hippocampal mGluR-mediated long-term depression (LTD) is abolished by loss of Tsc1, whereas a protein synthesis-independent form of NMDA receptor-dependent LTD is preserved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31••.Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. This report argues that mutations in FXS and TSC regulate mGluR-mediated LTD in opposite directions. More importantly, knocking out the Fmr1 gene from the Tsc2 mutant mice prevents the learning disability in these mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci. 2006;23:686–692. doi: 10.1111/j.1460-9568.2006.04594.x. [DOI] [PubMed] [Google Scholar]
- 33.Minshew NJ, Williams DL. The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch Neurol. 2007;64:945–950. doi: 10.1001/archneur.64.7.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- 37.Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, Yamada KA, Wozniak DF, Holtzman DM, Gutmann DH, et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2007;28:184–196. doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J, Shrikhande G, Xu J, McKay RM, Burns DK, Johnson JE, Parada LF. Tsc1 mutant neural stem/progenitor cells exhibit migration deficits and give rise to subependymal lesions in the lateral ventricle. Genes Dev. 2011;25:1595–1600. doi: 10.1101/gad.16750211. This is the first demonstration of subependymal nodule-like lesions in a TSC mouse model, suggesting that SENs can arise from post-natal neural stem/progenitor cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G, Lamb R. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002;159:217–224. doi: 10.1083/jcb.jcb.200206108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, Nellist M, Yeung RS, Halley DJ, Nicosia SV, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277:35364–35370. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 41.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 42.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 43.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 44.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 45••.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. The authors demonstrate reduction in SEGA size and seizure frequency in TSC patients treated with an mTOR inhibitor. [DOI] [PubMed] [Google Scholar]
- 46.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Dabora SL, Franz DN, Ashwal S, Sagalowsky A, DiMario FJ, Jr, Miles D, Cutler D, Krueger D, Uppot RN, Rabenou R, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF– D levels decrease. PLoS One. 2011;6:e23379. doi: 10.1371/journal.pone.0023379. This is a multicenter phase II, open label, single arm trial of rapamycin for the treatment of renal angiomyolipomas in adults. Rapamycin treatment induced regression of kidney angiomyolipomas, but angiomyolipomas regrew when rapamycin was discontinued. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Davies DM, de Vries PJ, Johnson SR, McCartney DL, Cox JA, Serra AL, Watson PC, Howe CJ, Doyle T, Pointon K, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17:4071–4081. doi: 10.1158/1078-0432.CCR-11-0445. This is another multicenter phase II, open label, single arm trial of rapamycin for the treatment of renal angiomyolipomas in adults. In this study, 41 out of 48 angiomyolipomas were smaller after treatment, but there was no change in pulmonary function, a secondary outcome measure. [DOI] [PubMed] [Google Scholar]
- 49•.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–1606. doi: 10.1056/NEJMoa1100391. The authors report the results of a multicenter phase III, randomized, placebo-controlled trial of rapamycin for the treatment of lymphangioleiomyomatosis in adults (MILES trial). The results indicate that rapamycin treatment may be useful in selected patients with LAM. However, after discontinuation of rapamycin, the decline in lung function will likely resume. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Tiberio D, Franz DN, Phillips JR. Regression of a cardiac rhabdomyoma in a patient receiving everolimus. Pediatrics. 2011;127:e1335–e1337. doi: 10.1542/peds.2010-2910. This case report describes a 7-year-old boy with a SEGA and a cardiac rhabdomyoma. After everolimus treatment, previously unchanged ventricular rhabdomyoma regressed rapidly in size. [DOI] [PubMed] [Google Scholar]
- 51.Hofbauer GF, Marcollo-Pini A, Corsenca A, Kistler AD, French LE, Wuthrich RP, Serra AL. The mTOR inhibitor rapamycin significantly improves facial angiofibroma lesions in a patient with tuberous sclerosis. Br J Dermatol. 2008;159:473–475. doi: 10.1111/j.1365-2133.2008.08677.x. [DOI] [PubMed] [Google Scholar]
- 52.Haemel AK, O'Brian AL, Teng JM. Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol. 2010;146:715–718. doi: 10.1001/archdermatol.2010.125. [DOI] [PubMed] [Google Scholar]
- 53.Mutizwa MM, Berk DR, Anadkat MJ. Treatment of facial angiofibromas with topical application of oral rapamycin solution (1 mg mL−1) in two patients with tuberous sclerosis. Br J Dermatol. 2011;165:922–923. doi: 10.1111/j.1365-2133.2011.10476.x. [DOI] [PubMed] [Google Scholar]
- 54.Kaufman McNamara E, Curtis AR, Fleischer AB., Jr Successful treatment of angiofibromata of tuberous sclerosis complex with rapamycin. J Dermatol Treat. 2012;23:46–48. doi: 10.3109/09546634.2010.489598. [DOI] [PubMed] [Google Scholar]
- 55.DeKlotz CM, Ogram AE, Singh S, Dronavalli S, MacGregor JL. Dramatic improvement of facial angiofibromas in tuberous sclerosis with topical rapamycin: optimizing a treatment protocol. Arch Dermatol. 2011;147:1116–1117. doi: 10.1001/archdermatol.2011.254. [DOI] [PubMed] [Google Scholar]
- 56••.Jozwiak S, Kotulska K, Domanska-Pakiela D, Lojszczyk B, Syczewska M, Chmielewski D, Dunin-Wasowicz D, Kmiec T, Szymkiewicz-Dangel J, Kornacka M, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15:424–431. doi: 10.1016/j.ejpn.2011.03.010. This open-label trial of vigabatrin in 14 infants with TSC suggests that ‘preventative’ anti-epileptic treatment before clinical seizure onset can improve neurodevelopmental outcomes. [DOI] [PubMed] [Google Scholar]
- 57•.Widjaja E, Simao G, Mahmoodabadi SZ, Ochi A, Snead OC, Rutka J, Otsubo H. Diffusion tensor imaging identifies changes in normal-appearing white matter within the epileptogenic zone in tuberous sclerosis complex. Epilepsy Res. 2010;89:246–253. doi: 10.1016/j.eplepsyres.2010.01.008. Using diffusion tensor imaging in 12 children with TSC, the authors demonstrate that the white matter in the epileptogenic zones had occult abnormalities. This could represent extension of dysplastic cerebral or the effect of ictal activity. [DOI] [PubMed] [Google Scholar]
- 58••.Peters JM, Sahin M, Vogel-Farley VK, Jeste SS, Gregas MC, Prabhu SP, Scherrer B, Warfield SK. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol. 2012;19:17–25. doi: 10.1016/j.acra.2011.08.016. This is the largest study to date (n=40) of diffusion tensor imaging in children with TSC and indicates that this type of imaging can help differentiate TSC patients with ASD from those without ASD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59••.Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW, Jr, Lee HM, Chen X, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185–189. doi: 10.1038/nature10726. Using a high-throughput screen, the investigators identify small molecules that alter the expression of intact paternal UBE3A allele in Angelman syndrome mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]