

Abstract
Norovirus (NoV) is the most common agent of nonbacterial epidemic gastroenteritis and is estimated to cause 21 million cases of the disease in the United States annually. The antigen enzyme-linked immunosorbent assays (ELISAs) currently available for NoV diagnosis detect only certain strains and are approved for use in the United States only in epidemics where NoV is suspected. There is a clear need for simpler, more rapid, and more reliable diagnostic tools for the detection of NoV. In this study, phage display technology was used to screen a library of phage displaying random 12-mer peptides for those that bind to Norwalk virus virus-like particles (NV VLPs). Three phage clones displaying unique peptides were identified, and both the peptide-displaying phages and the peptides were confirmed to bind specifically to NV VLPs. The peptide displayed on phage clone NV-N-R5-1 was determined to bind to the protruding domain of the VP1 capsid protein. This phage also bound to NV VLPs seeded into NoV-negative stool with a limit of detection of 1.56 ng NV VLP. This value was comparable to monoclonal antibody (MAb) 3912, which is currently used in commercially available assays. Furthermore, the NV-N-R5-1 phage exhibited high specificity by detecting NV only in previously characterized NV-positive stool samples in contrast to no detection in NV-negative stool samples. These data demonstrate that the further development of NV-N-R5-1 phage as a diagnostic reagent is possible and might offer several distinct advantages over antibodies, such as decreases in the time and cost of production and ease of isolating phage against other epidemic strains currently circulating as well as those that are emerging.
INTRODUCTION
Human norovirus (NoV) causes an estimated 21 million cases of gastroenteritis in the United States each year (1). NoVs are members of the family Caliciviridae and contain a single-stranded positive-sense RNA genome. NoVs are classified into six genogroups (GI to GVI) and further subdivided into genotypes based on the capsid sequence (2, 3). Most human NoVs fall within the GI and GII genogroups. Norwalk virus (NV), genotype GI.1, was the first NoV isolated and is considered the prototype virus of the genus; nevertheless, genotype GII.4 NoVs are currently the most frequently detected in humans (4). NoV infection is generally self-limiting in healthy adults, requiring only rehydration and supportive therapy; however, the illness can be severe and even fatal in the elderly and young children (4, 5). Furthermore, the infection can be chronic in immunocompromised patients, lasting years and mimicking the symptoms of transplant rejection (6, 7).
NoV's low infectious dose, resistance to many common sterilization procedures, and ease of transmission make epidemic outbreaks common and difficult to control, particularly in semiclosed environments such as schools, nursing homes, hospitals, cruise ships, and military settings (4, 8). NoV has been shown to be the cause of the majority of nonbacterial gastroenteritis epidemics in the United States, resulting in a huge economic burden, particularly in health care settings, when the cost of lost revenue from closures to new admissions, staff absences, and cleaning expenses are considered (1, 9–11). In order to manage severe and chronic NoV infections and effectively control epidemic outbreaks, rapid diagnosis is crucial. In fact, it has been observed that outbreaks were contained an average of 6 days sooner (7.9 versus 15.4 days) when a diagnosis was made within 3 days, rather than 4 or more days, after the first case (12). Only one immunoassay, the RIDASCREEN Norovirus enzyme-linked immunosorbent assay (ELISA) (3rd generation), is currently approved for NoV diagnosis in the United States, but it is currently approved for use only in outbreak settings because it lacks sensitivity (13–16).
Phage display is a powerful technique for rapidly screening large-scale libraries for novel substrates (17). Phage display employs random sequence peptides as fusions to the gene III protein, which is a solvent-accessible capsid protein present in five copies on one end of the phage coat (18). This allows biopanning, in which the phages displaying peptide are allowed to interact with the chosen target, noninteracting phages are washed away, and specifically bound phages are eluted from the target by low pH. Several rounds of biopanning enrich for peptide sequences that interact with the target (19). The advantage of phage display relies on the presence of the genetic material for these peptides within the phage chromosome, linking the genotype and phenotype for easy determination of the sequence of the binding peptide (20). This method has been used to isolate peptide-displaying phages that are useful diagnostic substrates for other bacterial and viral pathogens such as Mycobacterium avium and, more recently, hepatitis B virus (21–23).
In this study, we utilized phage display to screen for peptides that bind to Norwalk virus virus-like particles (NV VLPs). We isolated one peptide-displaying phage that interacted with NV VLPs specifically and with high sensitivity, and we showed that it was useful in detecting NoVs in clinical stool samples. This phage was used for virus detection in an ELISA and has the potential to be further developed, either in the context of the phage or as free peptide, into more sensitive and rapid assay formats.
MATERIALS AND METHODS
VLP production.
NV VLPs were expressed and purified as reported previously (24). Briefly, the VP1 and VP2 capsid proteins were expressed from a baculovirus vector in Sf9 insect cells and purified using a cesium chloride gradient. Proper VLP structure was confirmed by electron microscopy, and the VLPs were stored at 4°C. The NV protruding (P) domain was expressed and purified as described previously (25).
Phage biopanning.
Five micrograms of NV VLP suspended in 100 μl of phosphate-buffered saline (PBS) was immobilized in immunotubes (Nalgene) overnight at 4°C for rounds 1 to 3 of biopanning. Five hundred nanograms of NV VLP suspended in 100 μl of PBS buffer was immobilized in immunotubes (Nalgene) overnight at 4°C for rounds 4 to 5. For all rounds, the coated VLPs were washed twice with 1 ml of PBS, and the tubes were blocked with 2% nonfat dry milk in PBS and shaken gently at room temperature for 2 h. The tubes were washed three more times with PBS. The Ph.D-12 phage display library (New England BioLabs) was diluted to 1011 PFU/ml in PBS with 1% nonfat dry milk and incubated in VLP-coated tubes for 2 h at room temperature with gentle shaking. Subsequent rounds of biopanning were performed with the same amount of amplified phages from the previous biopanning round. Tubes were washed 10 to 30 times (with an increase in the number of washes in each subsequent round) with 1 ml PBS-T (1× PBS, 0.05% Tween 20). Phages that interacted with the VLP target were eluted with 1 ml of elution buffer (0.2 M glycine [pH 2.2], 1 mg/ml bovine serum albumin [BSA]) and neutralized after 10 min with 150 μl of 1 M Tris (pH 9.2).
For phage titration, eluted phages were diluted 10- to 1,000-fold in PBS, and 10 μl of diluted phage was incubated with 200 μl of mid-log-phase Escherichia coli ER2738 cells at room temperature for 10 min before being spread in top agar on LB plates containing 0.2 mM isopropyl-β-d-thiogalactoside (IPTG) and 0.1 mM 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). The plates were incubated overnight at 37°C, and the blue plaques were counted. White plaques, which represent environmental M13-like phage, were also counted. As long as the white plaques represented <5% of the total plaques, the eluted phage population was carried forward to the next round of biopanning.
The eluted phages were amplified by inoculating 20 ml of mid-log-phase E. coli ER2738 cells with 250 μl of the eluted phage. Infected cells were grown with shaking at 37°C for 3 to 5 h, followed by centrifugation at 3,000 × g for 10 min to remove the cells. The supernatant was transferred to a fresh tube, and 5 ml of 20% polyethylene glycol (PEG) 8000, 2.5 M NaCl was added and mixed well. The phages were allowed to precipitate for at least 3 h and pelleted by centrifugation at 3,000 × g for 30 min. Phage pellets were suspended in 1 ml of PBS. Centrifugation at 13,000 × g for 10 min removed cellular debris, and the titers of the phages at dilutions of 108 to 1010 were determined using the methods described above.
DNA sequencing of phage clones.
The solution containing eluted phages was used to infect E. coli, and individual phage plaques were obtained as described for phage titer determinations. For phage amplification, 10 to 15 individual blue phage plaques were picked and used to inoculate 1 ml of mid-log-phase E. coli ER2738 cells in a deep-well 96-well plate. Infected cultures were grown at 37°C with shaking for 3 to 5 h. The cells were removed by centrifugation at 3,000 × g for 15 min, and the supernatant containing the phages was transferred to a fresh 96-well plate.
PCR amplification targeted the region containing the library sequence and was performed with 5 μl of phage supernatant as the template. PCR products were then sequenced to determine the nucleotide sequence and thereby determine the sequence of the displayed peptide (Lone Star Labs, Houston, TX). Peptide sequences were analyzed, and those that demonstrated significant enrichment were chosen for further study. The chosen peptide sequences were compared to identify motifs that were common among them.
ELISA.
Initial ELISAs were performed by directly coating 96-well polystyrene plates with 0 to 5 μg of NV VLP overnight. The wells were blocked with 2% milk in PBS for 2 h at room temperature. After washing 3 times with PBS, 5 × 1010 phage in 1% milk-PBS were added and incubated at room temperature with gentle rocking for 2 h. Plates were washed 10 times with PBS-T. Horseradish peroxidase (HRP)-conjugated M13 antibody (Pharmacia Biotech) was diluted 1:5,000 in 1% milk-PBS, and 100 μl was added to each well and incubated at room temperature for 45 min. After washing with PBS-T, HRP substrate, 3,3′,5′-tetramethylbenzidine (TMB) (Kirkegaard and Perry Laboratories [KPL]), was added to each well. Development was allowed to proceed for 10 to 20 min and stopped with 1 M H3PO4 (KPL). Optical density was measured at 450 nm on a Tecan Infinite M200 Pro plate reader.
Capture ELISAs were performed by adsorbing rabbit polyclonal NV antiserum diluted 1:5,000 in the wells of a 96-well polystyrene plate overnight (26). After blocking the wells with 10% milk for 1 h at room temperature, 0 to 100 ng of VLP was diluted in PBS and incubated in antibody-coated wells for 2 h at room temperature for capture. The wells were washed 3 times with PBS-T and 1 × 1010 PFU/ml NV-N-R5-1 phage, a 1:2,500 dilution of the polyclonal antibody or a 1:500 dilution of monoclonal antibody (MAb) 3912 was prepared in 2% milk, and 100 μl was added to each well (27, 28). The plate was incubated at room temperature for 2 h and then washed 5 times with PBS-T. An HRP-conjugated anti-rabbit secondary antibody, anti-mouse secondary antibody, or M13 phage antibody was used for detection as described above. A negative control either lacking primary antibody or with nonpeptide-displaying M13 phage yielded background absorbance, and these values were subtracted from each well. To determine the limit of detection for the NV-N-R5-1 phage and antibodies, each sample was analyzed in triplicate and the cutoff for positive signal was determined by calculating three standard deviations above the [VLP] = 0 background.
Stool samples were collected from persons infected with NV, a GI.1 virus, as described previously (29) and stored at −80°C. Samples were weighed and suspended in PBS to create a 10 to 20% (wt/vol) solution. The solution was vortexed thoroughly and then clarified by centrifugation at 8,000 × g for 10 min. Stool samples for ELISAs were used at 10% or diluted to stated concentrations in PBS. The samples had been characterized previously by reverse transcriptase (RT)-PCR and were further determined to be positive or negative for NV by antigen ELISAs (29).
SPOT synthesis.
Peptide sequences were synthesized on cellulose membranes by automated SPOT synthesis on a MultiPep RS (Intavis, Bergisch Gladbach, Germany) as described previously (30). After synthesis, the membranes were soaked in methanol for 10 min, followed by two 10-min washes in PBS before being blocked with Superblock blocking buffer (Thermo Scientific) overnight at 4°C with gentle rocking. The following day, the membrane was washed three times for 10 min each with SPOT wash buffer (PBS-T, 1% BSA). NV VLP was diluted to a concentration of 1 μg/ml in SPOT wash buffer and incubated with the membrane for 2 h with gentle shaking at room temperature. After being washed three times with SPOT wash buffer, NV MAb 3912 (31) was diluted 1:1,000 in SPOT wash buffer and incubated with the membrane for 1 h with gentle shaking at room temperature. The membrane was washed three more times with SPOT wash buffer, and then the secondary antibody was diluted 1:5,000 and applied to the membrane at room temperature for 45 min. SPOTs were developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) for 1 min and then exposed to autoradiography film (Denville Scientific).
RESULTS
Screening a random 12-mer peptide phage library for Norwalk-binding peptides.
A commercially available M13 phage display library containing random-sequence 12-mer peptides fused to gene III protein was used for biopanning with immobilized NV VLPs as the target. Five rounds of biopanning against immobilized NV VLPs were performed in two independent library biopanning experiments. After rounds 3 to 5, 10 to 15 individual phage clones from each experiment were sequenced at the 5′ end of gene III encoding the library peptides. Poor-quality sequences were removed, resulting in 8 to 12 sequences per round. Three phage clones encoding sequences that occurred most commonly were chosen to move forward to study VLP-binding characteristics (Table 1). For this purpose, each of the three chosen phage clones was used to infect E. coli to amplify the phage stocks for binding experiments. All three of the chosen sequences contain an SW motif, and two sequences (phage clones NV-O-R5-3 and NV-O-R5-6) contain an extended motif, YRSW.
Table 1.
Amino acid sequences of phage clones identified by biopanning
| Expt no. | Amino acid sequence in biopanning round: |
Phage clone | ||
|---|---|---|---|---|
| 3 | 4 | 5 | ||
| 1 | LDYRSWSPYATS (3×)a,b | LDYRSWAPYATS (5×)b | LDYRSWAPYATS (4×)b | NV-O-R5-3 |
| AGELSPNRSAFL | IQYRSWIPFSYP (2×)b | IQYRSWIPFSYPb | NV-O-R5-6 | |
| QIPPRPPLLTTL | FRSYESPNFRPP (2×) | LSIRSYTSPQWQ (2×) | —c | |
| HNVTWAALMANV | YRSFDPWYPPVH | YRSFDPWYPPVH (2×) | — | |
| SYNTLTQIAKIR | LTQQRSWSPYMP | — | ||
| NSTNPHESRPTS | THQNRQTADIPS | — | ||
| 2 | LPSWYLAYQKII (9×)b | LPSWYLAYQKII (10×)b | LPSWYLAYQKII (11×)b | NV-N-R5-1 |
| ISWADWTQRWRW (2×) | SHVSKLVYQSQS | — | ||
| ALPTFGVISPFS | — | |||
The frequencies of sequences that appeared multiple times are indicated in parentheses.
The chosen sequences contained either the SW motif or the extended YRSW motif.
—, the absence of a chosen clone.
The binding of each phage clone to immobilized NV VLPs was tested by phage ELISAs. For these experiments, 0 to 5 μg of NV VLP or bovine serum albumin (BSA), as a negative control, was adsorbed onto a 96-well plate, and 5 × 1010 PFU of phage was added to each well. Binding was detected with an HRP-conjugated M13 phage antibody (Fig. 1). The results demonstrated that all three phage clones were able to bind to NV VLPs. The NV-O-R5-3 and NV-O-R5-6 phage clones demonstrated dose-dependent binding at the high VLP concentrations used in this assay, while binding of the NV-N-R5-1 clone (Table 1) appeared saturated at all of the concentrations. At the lower VLP concentrations used in subsequent assays, a dose response was seen for NV-N-R5-1 binding (see Fig. 4).
Fig 1.
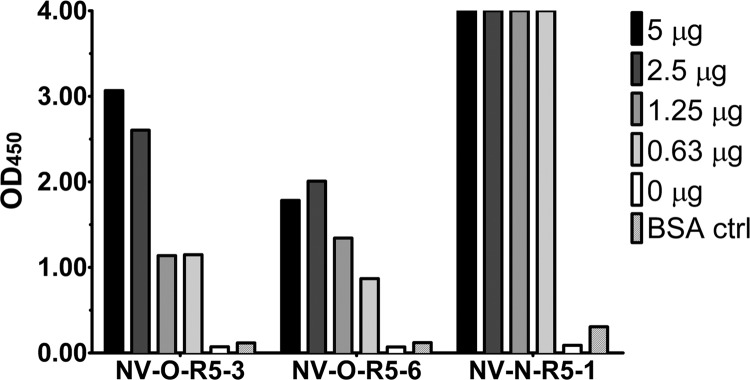
Phage binding to NV VLPs. NV VLPs (5, 2.5, 1.25, and 0.63 μg) or 5 μg of unrelated protein BSA was immobilized, and the binding of each phage, indicated on the x axis, was detected by M13 phage antibody. The signal at optical density at 450 nm (OD450) is indicated on the y axis.
Fig 4.

Phage binding to captured NV VLPs. NV VLPs (0 to 100 ng) were captured by immobilized NV polyclonal antibody. Binding of each phage was detected by HRP-conjugated M13 phage antibody, followed by determination of the OD450 (y axis).
The ability of the peptide encoded in each phage clone to bind NV VLPs in the absence of the phage was tested using a peptide array. SPOT synthesis was performed to create a peptide array that included each of the candidate peptide sequences, as well as point or deletion mutants that were hypothesized to disrupt binding (30). In addition, the array contained T7 and Strep-Tag II peptide sequences, which are unrelated to the phage-encoded peptides, and thereby serve as controls for nonspecific binding. NV VLPs were incubated with the peptide array, and binding was detected with MAb 3912, which recognizes NV VLPs (Fig. 2). The data indicated that NV VLPs bind the phage peptides and do not bind the negative-control peptides. The NV-N-R5-1 mutant peptide showed a loss of VLP binding. However, the mutation of arginine at position 4 of the NV-O-R5-6 peptide showed no significant decrease in binding, indicating that this mutation does not decrease binding below the threshold of detection for this assay and is not critical for binding in the context of the entire peptide sequence. Interestingly, the same mutation in the NV-O-R5-3 peptide led to a complete loss of binding, indicating that this residue is much more important in relation to the overall affinity of this peptide. These results demonstrate that the peptides alone, in the absence of the phage context, interact specifically with NV VLPs.
Fig 2.

NV VLP binding to SPOT peptide array. The NV VLP-binding peptide sequences, mutant peptides, and unrelated peptides indicated were synthesized on a cellulose membrane by SPOT synthesis (30). NV VLPs were incubated with the membrane, and binding was detected with NV VLP MAb 3912.
Phage binding is specific to the protruding domain of VLPs.
The major capsid protein of NoV, VP1, is composed of an inner shell domain and an outer protruding (P) domain, which is further subdivided into the P1 subdomain and the outermost, surface-exposed, P2 subdomain (32). However, we previously observed that VLPs adsorbed to a polystyrene surface can have altered structures due to protein unfolding that exposes different epitopes and affects the binding of antibodies (N. J. Ajami, S. E. Crawford, F. H. Neill, T. D. Parker, T. N. Tanaka, R. Kitamoto, R. Czako, B. Kuo, R. L. Atmar, and M. K. Estes, unpublished data). Phage-displaying peptides that bind to the P domain were desired, as they were hypothesized to be the most likely to bind to native virus. To test for possible P domain binding, purified NV P domain or BSA as a negative control was adsorbed into the wells of a 96-well plate, and each phage was tested for binding as was done previously with NV VLPs (Fig. 3). The results revealed that only one phage clone, NV-N-R5-1, was able to bind specifically to the purified NV P domain. None of the phage demonstrated binding to the shell domain of NV (data not shown). Because the peptide sequences from all three phage clones share a motif, it is likely that they are binding to the same epitope on NV VLPs. However, the surrounding sequence likely affects the ability of the phage to bind to the native P domain structure, which would explain why two phages were able to bind to VLPs adsorbed to the plate surface but not to the properly folded P domain. Alternatively, because the NV-O-R5-3 and NV-O-R5-6 phage clones appear to have a lower affinity for VLPs, the smaller amount of P domain present in the P domain ELISA in comparison to the VLP ELISA might simply be below the limit of detection for these two phage clones.
Fig 3.
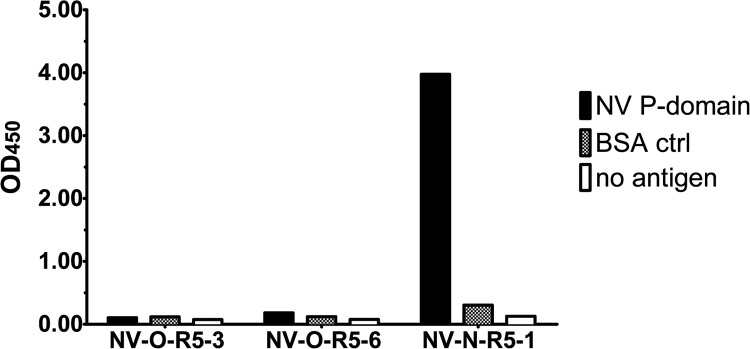
Phage binding to the NV P domain. Five micrograms of NV P domain, unrelated protein BSA, or no protein was immobilized in microtiter wells. Binding of each phage, indicated on the x axis, was detected with HRP-conjugated M13 phage antibody, followed by determination of the OD450 (y axis).
Because the NV-N-R5-1 clone was the only phage to demonstrate binding to purified P domain by ELISA, it was hypothesized that the NV-N-R5-1 clone would also be the only phage that displayed binding to antibody-captured VLPs, which maintain their original conformation in contrast to VLPs directly adsorbed to the plate surface. Polyclonal anti-NV VLP antibody was adsorbed into the wells of a 96-well plate, and 0 to 100 ng of NV VLP was incubated for capture in each well. A total of 1 × 109 phage was added, nonbinders were washed away, and retained phage were detected with an HRP-conjugated M13-phage antibody (Fig. 4). As expected, NV-N-R5-1 was the only phage that bound to the antibody-captured VLPs. Therefore, the NV-N-R5-1 clone was chosen to test for feasibility of use in the detection of NV in clinical stool samples.
NV-N-R5-1 sensitivity determined for NV VLPs in stool samples.
As a first step toward applying the NV-N-R5-1 phage as a sensor for virus in clinical samples, the sensitivity of detection of the phage was determined. For this purpose, an ELISA was performed under the same conditions as described for the antibody-captured VLPs, except that NV VLPs were diluted in a 10% clarified solution of an NoV-negative stool sample (Fig. 5). The results indicated that the NV-N-R5-1 phage is able to interact with NV VLPs under these conditions with a sensitivity similar to that of the diagnostic MAb 3912. Notably, the diverse components of the stool did not inhibit the interaction of the phage with NV VLPs. These data were further used to determine the lowest limit of detection of NV-N-R5-1 for NV VLPs in a 10% suspension of the NoV-negative stool. Using a threshold for specific positive signal of three standard deviations over the [VLP] = 0 background, 1.56 ng of NV VLP was the smallest amount of VLPs to show a positive signal with detection by the NV-N-R5-1 phage. The lowest limit of detection for the polyclonal antibody was also 1.56 ng, and the lowest limit of detection for MAb 3912 was 3.13 ng. Therefore, the NV-N-R5-1 phage was determined to be slightly more sensitive for NV than the commercially available diagnostic MAb 3912 and on par with a widely characterized polyclonal antibody.
Fig 5.

NV-N-R5-1 phage binding to NV VLPs in NoV-negative stool suspension. NV VLPs (0 to 50 ng) (x axis) were diluted in 10% NoV-negative stool suspension and captured by immobilized NV polyclonal antibody. NV VLPs were detected by 1 × 109 PFU NV-N-R5-1 phage, a 1:2,500 dilution of NV polyclonal antibody, or a 1:500 dilution of MAb 3912. Binding was detected by M13 antibody or secondary antibody and determination of the OD450 (y axis).
NV-N-R5-1 phage is able to detect norovirus in clinical stool samples.
Based on the comparisons of the sensitivity of the NV-N-R5-1 phage with the MAb 3912 discussed above, it was hypothesized that the phage would be effective at detecting virus from clinical samples. This was tested by using the anti-NV polyclonal antibody to capture virus from NV-positive stool samples and then using the NV-N-R5-1 phage to detect the presence of the virus (Fig. 6). The results showed that the NV-N-R5-1 phage was able to detect NV in all of the NV-positive stool suspensions with signal similar to that of the NV polyclonal antiserum, and no signal was observed in the previously characterized NoV-negative samples or when using the M13 control phage. The NV-O-R5-3 and NV-O-R5-6 phages did not show any detection in the NV-positive stool suspensions (data not shown). These results demonstrate that the NV-N-R5-1 phage is able to interact with native NV in a specific manner and is not inhibited by any of the components of the stool suspension.
Fig 6.

NV-N-R5-1 phage detection of NoV in stool suspensions from infected volunteers. Stool samples were collected from persons with and without NV infection. NV-positive, NoV-negative, or 100 ng of NV VLP in NoV-negative stool or buffer (x axis) was serially diluted to the indicated concentrations and then captured by immobilized NV polyclonal antibody. Binding was tested for M13KE control phage, NV polyclonal antibody, or NV-N-R5-1 phage. The color key indicates dilutions of the M13KE control, NV antibody, and NV-N-R5-1 samples.
DISCUSSION
In this study, three phage clones that bind to GI.1 VLPs were identified. Only one of these phage clones, NV-N-R5-1, interacted with antibody-captured VLPs, and it was further shown to detect GI.1 NoV in clinical stool samples. Phage ELISAs revealed that the peptide displayed on the NV-N-R5-1 phage binds to the P domain of VP1, and further experiments are in progress to determine the specific binding epitope. Using the NV-N-R5-1 phage, we were able to detect a minimum of 1.56 ng of NV VLP when seeded into an NoV-negative stool sample. This limit of detection is similar to that observed with MAb 3912 for NV, which is used in commercially available diagnostic tests for NoV detection. In addition, it is possible that the measured sensitivity of the phage is underestimated because the current ELISA method requires that we capture the VLPs with an antibody for further detection. Therefore, a limitation might be the binding affinity of the capture antibody to the VLP. Although further work and testing are needed before NV-N-R5-1 is ready for commercial diagnostic use, the work presented here demonstrates that the use of phage as a diagnostic reagent is possible. Furthermore, there are many advantages of phages over antibodies that make the continued development of this phage as a diagnostic reagent attractive.
First, the phage system has great potential for engineering increased binding affinity, and therefore increased NoV detection sensitivity, by utilizing cycles of mutagenesis and biopanning enrichment to find the most favorable peptide sequence. The random peptide phage library that was screened has a diversity of 109 unique sequences of 12-mer peptides, which represents only a small fraction of the 3.8 × 1021 possible 12-mer sequences. Therefore, it is likely that only a few of the residues within the NV-N-R5-1 peptide are contributing to binding and that the rest of the sequence can be optimized for increased affinity. Mutagenesis can also be used to screen for peptides with increased solubility that maintain or increase binding affinity. The NV-N-R5-1 peptide is insoluble when it is not expressed in the context of the phage or a peptide array, which poses a limitation on creating new assays that use the free peptide as a reagent.
Second, the time and cost of production of phage preparations are significantly more favorable than those for antibody production. For example, 1 × 1013 PFU of phage can be obtained from 20 ml of infected E. coli ER2738 culture and purified from the supernatant via a quick and simple PEG precipitation, which provides enough phage for 1,000 diagnostic tests in the current format. This entire amplification and purification protocol can be completed in about 10 h for nominal cost and can easily be scaled up for commercial production.
Third, isolation of phage clones that bind to new NoV variants is simpler and faster than production of new antibodies via biopanning of phage libraries. NoV is a highly diverse group of viruses with new pandemic variants emerging every 2 to 4 years (4). Most of the variations between strains are present in the protruding domain of the major capsid protein, which is the most accessible region for the binding of diagnostic substrates. Therefore, it is likely that new or modified virus recognition reagents that are specific for emerging strains will need to be developed every few years. New phage clones can be isolated and validated from the same original library much more rapidly than antibodies can be raised and with much less financial investment.
The possibility of using phage as a substrate for diagnostic assays has been demonstrated in this work. However, the peptide that is displayed by this phage also represents a potential starting point for diagnostic assays that are simpler and more rapid than ELISA formats. The binding affinity (Kd) of individual peptides for NV VLPs was unable to be determined in this work, partially due to difficulties in solubilizing the free peptide. It is possible that the binding affinity of the individual peptide for NV VLPs is currently too low for the free monomeric peptide to be a useful reagent. On the phage particle, the peptide is displayed in five copies on one end, which can potentially increase binding avidity through multipoint attachment. The large phage particle also holds the peptides apart from one another and therefore in a position to interact with the target. We are currently working to increase the solubility and affinity of this peptide to facilitate its development as a reagent for the direct detection of virus. This will likely require either an increase in affinity of the monomeric peptide or the development of a multivalent peptide reagent such as a dendrimer.
In summary, we have developed a phage ELISA method that is able to detect GI.1 NoV in clinical stool samples with sensitivity equivalent to that of the available monoclonal antibody 3912. This demonstrates the utility of a phage particle displaying a peptide as a reagent for the detection of norovirus. Furthermore, the work demonstrates the power of phage display as a tool for identifying novel diagnostic reagents for the detection of viral pathogens.
ACKNOWLEDGMENTS
This research was funded by Public Health Service grants NIH P01 AI057788 and NIH P30DK56338, Agriculture and Food Research Initiative Competitive grant 2011-68003-30395 from the USDA National Institute of Food and Agriculture, the John S. Dunn Research Foundation, and a Pharmaceutical Sciences Training Program fellowship, Keck Center of the Gulf Coast Consortia, National Institute of General Medical Sciences (NIGMS), grant T32GM089657.
Footnotes
Published ahead of print 3 April 2013
REFERENCES
- 1. Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS. 2006. Norovirus classification and proposed strain nomenclature. Virology 346:312–323 [DOI] [PubMed] [Google Scholar]
- 3. Kroneman A, Vennema H, Deforche K, v d Avoort H, Penaranda S, Oberste MS, Vinje J, Koopmans M. 2011. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51:121–125 [DOI] [PubMed] [Google Scholar]
- 4. Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N. Engl. J. Med. 361:1776–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trivedi TK, DeSalvo T, Lee L, Palumbo A, Moll M, Curns A, Hall AJ, Patel M, Parashar UD, Lopman BA. 2012. Hospitalizations and mortality associated with norovirus outbreaks in nursing homes, 2009-2010. JAMA 308:1668–1675 [DOI] [PubMed] [Google Scholar]
- 6. Kaufmann SS, Chatterjee NK, Fuschino ME, Morse DL, Morotti RA, Magid MS, Gondolesi GE, Florman SS, Fishbein TM. 2005. Characteristics of human calicivirus enteritis in intestinal transplant recipients. J. Pediatr. Gastroenterol. Nutr. 40:328–333 [DOI] [PubMed] [Google Scholar]
- 7. Bok K, Green KY. 2012. Norovirus gastroenteritis in immunocompromised patients. N. Engl. J. Med. 367:2126–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hutson AM, Atmar RL, Estes MK. 2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol. 12:279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fankhauser RL, Noel JS, Monroe SS, Ando T, Glass RI. 1998. Molecular epidemiology of “Norwalk-like viruses” in outbreaks of gastroenteritis in the United States. J. Infect. Dis. 178:1571–1578 [DOI] [PubMed] [Google Scholar]
- 10. Hoffmann S, Batz MB, Morris JG., Jr 2012. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J. Food Prot. 75:1292–1302 [DOI] [PubMed] [Google Scholar]
- 11. Johnston CP, Qiu H, Ticehurst JR, Dickson C, Rosenbaum P, Lawson P, Stokes AB, Lowenstein CJ, Kaminsky M, Cosgrove SE, Green KY, Perl TM. 2007. Outbreak management and implications of a nosocomial norovirus outbreak. Clin. Infect. Dis. 45:534–540 [DOI] [PubMed] [Google Scholar]
- 12. Lopman BA, Reacher MH, Vipond IB, Hill D, Perry C, Halladay T, Brown DW, Edmunds WJ, Sarangi J. 2004. Epidemiology and cost of nosocomial gastroenteritis, Avon, England, 2002-2003. Emerg. Infect. Dis. 10:1827–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parker TD, Kitamoto N, Tanaka T, Hutson AM, Estes MK. 2005. Identification of genogroup I and genogroup II broadly reactive epitopes on the norovirus capsid. J. Virol. 79:7402–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Atmar RL, Estes MK. 2001. Diagnosis of noncultivatable gastroenteritis viruses, the human caliciviruses. Clin. Microbiol. Rev. 14:15–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ambert-Balay K, Pothier P. 2013. Evaluation of 4 immunochromatographic tests for rapid detection of norovirus in faecal samples. J. Clin. Virol. 56:194–198 [DOI] [PubMed] [Google Scholar]
- 16. Kirby A, Gurgel RQ, Dove W, Vieira SC, Cunliffe NA, Cuevas LE. 2010. An evaluation of the RIDASCREEN and IDEIA enzyme immunoassays and the RIDAQUICK immunochromatographic test for the detection of norovirus in faecal specimens. J. Clin. Virol. 49:254–257 [DOI] [PubMed] [Google Scholar]
- 17. Sidhu SS, Fairbrother WJ, Deshayes K. 2003. Exploring protein-protein interactions with phage display. Chembiochem 4:14–25 [DOI] [PubMed] [Google Scholar]
- 18. Samoylova TI, Norris MD, Samoylov AM, Cochran AM, Wolfe KG, Petrenko VA, Cox NR. 2012. Infective and inactivated filamentous phage as carriers for immunogenic peptides. J. Virol. Methods 183:63–68 [DOI] [PubMed] [Google Scholar]
- 19. Rakonjac J, Bennett NJ, Spagnuolo J, Gagic D, Russel M. 2011. Filamentous bacteriophage: biology, phage display and nanotechnology applications. Curr. Issues Mol. Biol. 13:51–76 [PubMed] [Google Scholar]
- 20. Paschke M. 2006. Phage display systems and their applications. Appl. Microbiol. Biotechnol. 70:2–11 [DOI] [PubMed] [Google Scholar]
- 21. Monjezi R, Tan SW, Tey BT, Sieo CC, Tan WS. 2013. Detection of hepatitis B virus core antigen by phage display mediated TaqMan real-time immuno-PCR. J. Virol. Methods 187:121–126 [DOI] [PubMed] [Google Scholar]
- 22. Schofield DA, Sharp NJ, Westwater C. 2012. Phage-based platforms for the clinical detection of human bacterial pathogens. Bacteriophage 2:105–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stratmann J, Strommenger B, Stevenson K, Gerlach GF. 2002. Development of a peptide-mediated capture PCR for detection of Mycobacterium avium subsp. paratuberculosis in milk. J. Clin. Microbiol. 40:4244–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hale AD, Crawford SE, Ciarlet M, Green J, Gallimore C, Brown DW, Jiang X, Estes MK. 1999. Expression and self-assembly of Grimsby virus: antigenic distinction from Norwalk and Mexico viruses. Clin. Diagn. Lab. Immunol. 6:142–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi JM, Hutson AM, Estes MK, Prasad BV. 2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. U. S. A. 105:9175–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gilpatrick SG, Schwab KJ, Estes MK, Atmar RL. 2000. Development of an immunomagnetic capture reverse transcription-PCR assay for the detection of Norwalk virus. J. Virol. Methods 90:69–78 [DOI] [PubMed] [Google Scholar]
- 27. Jiang X, Wang M, Graham DY, Estes MK. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66:6527–6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hardy ME, Tanaka TN, Kitamoto N, White LJ, Ball JM, Jiang X, Estes MK. 1996. Antigenic mapping of the recombinant Norwalk virus capsid protein using monoclonal antibodies. Virology 217:252–261 [DOI] [PubMed] [Google Scholar]
- 29. Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Graham DY. 2008. Norwalk virus shedding after experimental human infection. Emerg. Infect. Dis. 14:1553–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frank R, Overwin H. 1996. SPOT synthesis. Epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods Mol. Biol. 66:149–169 [DOI] [PubMed] [Google Scholar]
- 31. Hale AD, Tanaka TN, Kitamoto N, Ciarlet M, Jiang X, Takeda N, Brown DW, Estes MK. 2000. Identification of an epitope common to genogroup 1 “Norwalk-like viruses.” J. Clin. Microbiol. 38:1656–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290 [DOI] [PubMed] [Google Scholar]