

Abstract
Central nervous system (CNS) antibiotic distribution was described mainly from cerebrospinal fluid data, and only few data exist on brain extracellular fluid concentrations. The aim of this study was to describe brain distribution of cefotaxime (2 g/8 h) by microdialysis in patients with acute brain injury who were treated for a lung infection. Microdialysis probes were inserted into healthy brain tissue of five critical care patients. Plasma and unbound brain concentrations were determined at steady state by high-performance liquid chromatography. In vivo recoveries were determined individually using retrodialysis by drug. Noncompartmental and compartmental pharmacokinetic analyses were performed. Unbound cefotaxime brain concentrations were much lower than corresponding plasma concentrations, with a mean cefotaxime unbound brain-to-plasma area under the curve ratio equal to 26.1 ± 12.1%. This result was in accordance with the brain input-to-brain output clearances ratio (CLin,brain/CLout,brain). Unbound brain concentrations were then simulated at two dosing regimens (4 g every 6 h or 8 h), and the time over the MICs (T>MIC) was estimated for breakpoints of susceptible and resistant Streptococcus pneumoniae strains. T>MIC was higher than 90% of the dosing interval for both dosing regimens for susceptible strains and only for 4 g every 6 h for resistant ones. In conclusion, brain distribution of cefotaxime was well described by microdialysis in patients and was limited.
INTRODUCTION
Cefotaxime is a third-generation extended-spectrum cephalosporin and is recommended as an empirical therapy for bacterial meningitis, ventriculitis, and cerebral abscesses associated with metronidazole (1, 2). One of the limits of this urgent treatment is its central nervous system (CNS) penetration. The mechanism of penetration of microbial pathogens in the CNS remains unclear in bacterial meningitis (3), and it has been shown that some bacteria usually implicated in such infection could have the ability to bind and cross the vascular endothelium of the blood-brain barrier (BBB) (4). The exact location of bacteria in meningitis is still unclear. Few studies, limited to investigations in cerebrospinal fluid (CSF), were performed (5–8), and they found a low central distribution of cefotaxime (5, 6). In humans, the easiest way to study antibiotic penetration into CNS is to measure the concentration in CSF from lumbar puncture or external ventricular drainage. CSF concentrations are usually considered a good surrogate for brain target site concentrations (9). However, qualitative differences, such as anatomy, enzymatic activity, or bulk flow, exist between the BBB and blood-CSF barrier (BCSFB) (9), which could result in differences of drug distribution between CSF and brain extracellular fluid (ECF).
Intracerebral microdialysis is one of the in vivo techniques allowing brain ECF sampling to study ECF distribution of exogenous compounds, such as antibiotics. In humans, feasibility issues of brain microdialysis restrict its use, and this technique concerns only patients who require surgery for brain tumor resection (10) or cerebral metabolism monitoring in neurointensive care units (11). The main advantage in intracerebral microdialysis is to measure continuously brain unbound concentrations as a function of time; the comparison of these brain concentrations with unbound plasma concentrations may provide information on drug transport across the BBB (12).
The main goal of this study was to explore cerebral ECF distribution of cefotaxime in patients with acute brain injury by comparing unbound concentrations in brain and plasma.
MATERIALS AND METHODS
Patients.
This study was performed in the neurointensive care unit at University Hospital of Poitiers (France) in accordance with the Declaration of Helsinki (Edinburgh, Scotland; October 2000) and was approved by the local ethics committee (CPP OUEST III, protocol no. 2008-003311-12). Written informed consent was obtained from a legal representative of the five patients enrolled. Patients (1 woman, 4 men), 39 to 67 years old, were brain injured, sedated with midazolam and fentanyl, and mechanically ventilated. The demographic characteristics are detailed in Table 1. All received cefotaxime (Panpharma, Fougères, France) for the clinical management of a lung infection at a dosing regimen of 2 g three times per day. The exclusion criteria were a renal failure (calculated creatinine clearance of <5 ml · min−1) and cefotaxime contraindication. Routine monitoring for acute brain injury included brain-specific monitoring of intracranial pressure (ICP) (Microsensor ICP monitoring system; Codman & Shurtleff, Inc., Raynham, MA), measurement of the partial pressure of oxygen in brain tissue (PbO2) (Licox; Integra Neurosciences, Lyon, France), and cerebral microdialysis (CMA-70 [20 kDa]; CMA, Stockholm, Sweden) for determination of metabolism parameter concentrations.
Table 1.
Demographic patient characteristics
| Characteristic | Value by patient no.: |
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Age (yrs) | 64 | 43 | 56 | 39 | 67 |
| Ht (cm) | 172 | 164 | 180 | 175 | 175 |
| Wt (kg) | 85 | 58 | 85 | 95 | 100 |
| Creatinine clearance (ml · min−1) | 166 | 193 | 146 | 187 | 143 |
| Serum albumin (g · liter−1) | 28.9 | 28.0 | 30.0 | 28.4 | 24.4 |
| Serum total proteins (g · liter−1) | 52.0 | 51.5 | 60.0 | 52.8 | 52.0 |
| Diagnosisa | TBI | TBI | SAH | TBI | SAH |
| No. of cefotaxime doses before study | 14 | 9 | 9 | 5 | 9 |
| Corticotherapy (3 × 1 mg · kg−1 · d−1) | No | Yes | Yes | No | No |
TBI, trauma brain injury; SAH, subarachnoid hemorrhage.
Microdialysis probe implantation.
Upon admission in the unit (day 0), patients were equipped with microdialysis probe, ICP, and PbO2 catheters for clinical indications. All were inserted through a single triple lumen bolt (Licox; Integra Neuroscience, Lyon, France) in healthy brain tissue, confirmed by a routine computed tomography scan (CT scan) performed on day 2. After insertion, the microdialysis probe was perfused with CNS perfusion fluid (CMA, Stockholm, Sweden) using a microdialysis pump (CMA-106; CMA, Stockholm, Sweden) at a flow rate of 0.3 μl · min−1.
Drug administration and samplings.
Cefotaxime brain pharmacokinetic study was conducted at the steady state between days 3 and 5, after 5 to 14 cefotaxime administrations. After collection of baseline dialysates and blood samples, 2 g of cefotaxime was infused over 0.5 h. Brain dialysates were collected over a maximum period of 9 h at 30-min intervals during the first 3 h and at 1-h intervals for the rest of experiment. Seven to nine blood samples were collected on heparinized Vacutainers over 8 to 10 h, maximum. Blood samples were centrifuged at 2,000 × g for 15 min at 4°C, and plasma was collected to determine total cefotaxime concentrations. Two blood aliquots for each patient were used to determine unbound plasma concentrations of cefotaxime. The first blood sample was chosen at early time (between 0.25 h and 0.75 h after the beginning of infusion), and the second at the end of pharmacokinetic experiment (between 6 and 10 h). Blood was centrifuged, and plasma was then ultrafiltrated (Centrifree; Millipore Corporation, Billerica, MA) at 2,500 × g for 15 min at 4°C. Directly after collection, dialysates, ultrafiltrate (UF), and plasma samples were kept at −80°C until analysis.
Recovery calculations.
For each patient, in vivo probe recovery was determined using retrodialysis by drug over 2.5 h at the end of the experiment, as previously described (12, 13). The next cefotaxime injection was delayed to perform the recovery estimation. Briefly, the microdialysis probe was perfused with 20 μg · ml−1 solution of cefotaxime in CNS perfusion fluid, and after a 1-h equilibration period, three 30-min interval dialysates were collected. Cefotaxime concentrations were determined in the perfusate (Cin) and in dialysates (Cout). The in vivo relative recovery by loss was calculated for each dialysate collected, and the mean value was used to correct dialysate concentrations for pharmacokinetic calculations (13). In the present study, because the cefotaxime residual unbound concentration in the brain was always estimated as less than 1% of cefotaxime concentration administered in probe (Cin), this concentration was not taken into consideration to estimate recovery like it could be previously performed (14).
Cefotaxime assay.
Cefotaxime concentrations were determined by high-performance liquid chromatography with UV detection. The chromatographic system consisted of a Xterra C18 column (150- by 3.8-mm internal diameter [i.d.]; France), a Hitachi pump L-2130 (VWR, Fontenay sous bois, France), and a Hitachi L-2200 autosampler (VWR, Fontenay sous bois, France) connected to a UV detector (Schimadzu SPD 10A; Marne la Vallée, France) at 235 nm. Data were recorded and analyzed on an EZChrom integrator (VWR, Fontenay sous bois, France). The mobile-phase consisted of a solution of KH2PO4 0.01 M mixed with acetonitrile (86:14 [vol/vol]) at a flow rate of 0.4 ml · min−1. Brain dialysates and UF samples were injected directly after dilution with an internal standard solution of dimetridazole (0.5 ng · ml−1). A seven-point calibration standard curve with concentrations between 0.2 and 40 μg · ml−1 was performed. The dialysates and UF cefotaxime intra- and interday variability were, respectively, characterized at four (0.2, 0.5, 5, and 40 μg · ml−1) and three (0.5, 5, and 30 μg · ml−1) levels of concentrations, respectively, and were always lower than 15%. Plasma samples (100 μl) were treated by the addition of 200 μl of acetonitrile containing internal standard (2.5 μg · ml−1) for deproteinization. A seven-point calibration standard curve was prepared with concentrations between 1 and 80 μg · ml−1. The plasma intra- and interday variability were, respectively, characterized at four (1, 2, 10, and 80 μg · ml−1) and three (2, 10, and 60 μg · ml−1) levels of concentrations and were always lower than 20%.
Pharmacokinetic analysis.
Ratios of cefotaxime concentrations in ultrafiltrate on corresponding total plasma concentrations allow for the obtainment of two individual unbound fraction values (fu) of cefotaxime for each patient. A mean was then determined and was used to convert total concentrations into unbound concentrations. Pharmacokinetic parameter values were estimated from unbound plasma and ECF brain unbound concentrations by individual noncompartmental analysis (NCA) and compartmental analysis (Phoenix WinNonlin 6.2; Pharsight).
Noncompartmental analysis.
The areas under the plasma and brain ECF unbound concentration-time curves from dosing time to dosing interval (τ) (AUC0→τ) were calculated using the linear trapezoidal rule. The elimination rate constant kel and corresponding half-lives (t1/2) were determined by least-squares fit of data points (log concentration time) in the terminal phase of the decline. Steady-state unbound body clearance of cefotaxime (CLss,u) and volume of distribution (Vss,u) were calculated according to standard procedures.
Compartmental analysis parameters.
The compartmental analysis was performed in two steps as previously described (12). Briefly, unbound plasma drug concentration-time data were first analyzed using a two-compartment model. Plasma parameters estimated were then fixed to fit brain data. The brain was represented as a compartment characterized by a volume (Vb) which was fixed as a constant corresponding to 1% of the volume of distribution (15). This brain compartment was also characterized by brain input clearance (CLin,brain) and brain output clearance (CLout,brain) (Fig. 1). However, due to the small size of the brain, unbound plasma drug concentrations govern brain unbound drug concentrations but not the reverse. Therefore, drug transport into and out of the brain can be modeled with elimination from the brain, which does not influence plasma drug concentrations (15).
Fig 1.
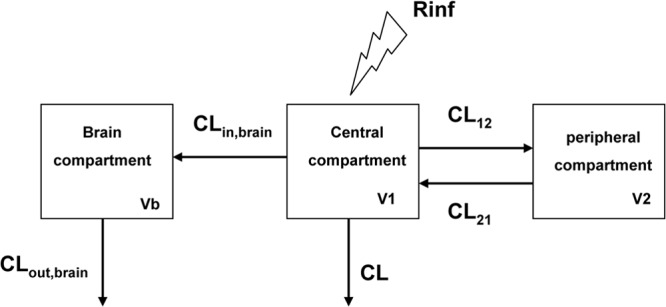
Pharmacokinetic model. V1, V2, and Vb correspond to respective volumes of central, peripheral, and brain compartments. CL12 and CL21 represent distribution clearances between central and peripheral compartments, CLin,brain and CLout,brain, the brain input and output clearances. Rinf is the rate of infusion (2 g over 30 min).
Cefotaxime dosing regimen simulations.
Mean cefotaxime unbound concentrations in brain were simulated after multiple administrations at two dosing regimens, 4 g every 6 h (16 g per day) and 4 g every 8 h (12 g per day), considering that the drug was infused at a constant rate over 30 min using Berkeley Madonna software, version 8.3.18. (University of California, Berkeley, CA). Simulations were performed using mean unbound plasma pharmacokinetic parameters and mean brain clearances (CLin,brain and CLout,brain) obtained from the five patients. Simulated unbound brain ECF concentrations were then compared with MIC values equal to 0.5 and 2 μg · ml−1, corresponding to susceptible and resistant MIC breakpoints for Streptococcus pneumoniae (16). The time during which cefotaxime unbound concentrations in brain exceeded the MIC (T>MIC) was determined for each dosing regimen and MIC. T>MIC was expressed as a percentage of the dosing interval.
Statistical analysis.
Results are expressed as means ± standard deviations (SD). Half-life and mean maximum unbound concentration values in plasma and brain were compared by a no-parametric Wilcoxon test at a significance level of P values of <0.05.
RESULTS
The in vivo probe recoveries estimated were ranging between 37.6 ± 0.1% and 54.8 ± 0.2%.
The estimated mean unbound fraction of cefotaxime in plasma (fu) was ranging between 47.4 ± 3.3% and 68.0 ± 10.3% and did not seem affected by concentrations. Unbound cefotaxime concentration-time curves in the brain were below the corresponding curves in plasma and shifted to the right (tmax,brain = 1.35 ± 0.25 h). Mean maximum unbound concentration in brain (Cmax,ub = 4.5 ± 1.7 μg · ml−1) was about 10-fold and significantly lower than the corresponding value in plasma (Cmax,up = 52.1 ± 13.7 μg · ml−1) (Fig. 2). The mean cefotaxime unbound brain-to-plasma AUC ratio was then equal to 26.1 ± 12.1%. Pharmacokinetic parameters obtained by NCA are presented in Table 2.
Fig 2.

Individual plasma and brain cefotaxime concentrations versus time in five patients after a 30-min intravenous infusion of 2 g of cefotaxime administered every 8 h. ●, experimental unbound concentrations in brain obtained by microdialysis; ▲, experimental unbound concentrations in plasma. Full and dashed lines represent, respectively, estimated concentrations in plasma and in brain by the pharmacokinetic model.
Table 2.
Pharmacokinetic parameters obtained by noncompartmental analysis in patients after a 30-min intravenous infusion of 2 g of cefotaxime administered every 8 ha
| Patient | Vdss,u (liters) | CLss,u (liters/h) | t1/2 plasma (h) | t1/2 brain (h) | Brain ECF-free plasma AUC ratio |
|---|---|---|---|---|---|
| P1 | 51.4 | 29.2 | 2.0 | 2.9 | 0.278 |
| P2 | 40.0 | 27.4 | 2.1 | 3.3 | 0.119 |
| P3 | 55.0 | 33.7 | 1.7 | 0.7 | 0.154 |
| P4 | 88.6 | 68.2 | 1.5 | 1.9 | 0.383 |
| P5 | 42.5 | 36.1 | 1.6 | 2.0 | 0.369 |
| Mean | 55.5 | 38.9 | 1.8 | 2.2 | 0.261 |
| SD | 20.9 | 19.2 | 0.3 | 1.2 | 0.121 |
Individual and mean (±SD) results are presented.
The pharmacokinetic model provided adequate data fitting both in plasma and in brain (Fig. 2). Estimations of CLin,brain and CLout,brain were good, with precision on parameters (CV%) always lower than 20%. For all patients, estimated CLin,brain was always lower than CLout,brain, and a mean corresponding ratio of CLin,brain to CLout,brain equal to 33.1 ± 15.8 (from 13.4 to 52.0) was consistent with the mean brain-to-plasma AUC ratio of 26.1 ± 12.1% (11.9 to 38.3%) estimated by noncompartmental analysis.
DISCUSSION
In the present study, cefotaxime unbound clearance corrected by fu was almost the same as previous values in healthy volunteers (240 ml · min−1 or 14.4 liters · h−1) (17), but patients in this study had a well-preserved renal function (Table 1 and 2). Unbound volume of distribution at steady state was relatively high (55.5 ± 20.9 liters), probably due to an increased extracellular water volume traditionally observed in critical care patients. The mean cefotaxime fu of 59.4% in the present study was in accordance with previous in vitro results in which a mean cefotaxime fu of 63% was found (17).
In vivo probe recovery determination is necessary for pharmacokinetic studies. In the present study, it was evaluated in each patient by the in vivo retrodialysis-by-drug method to adequately estimate cefotaxime brain ECF concentrations (12). Probe recoveries were ranging between 37.6 ± 0.1% and 54.8 ± 0.2%; in practice, a 20% in vivo recovery is usually as satisfactory for precise correction (18). Probe recoveries in this study were higher than those estimated in our previous investigation on meropenem brain distribution in two critical care patients (19% ± 7% and 29% ± 7%), even though larger cutoff probes (CMA 71, 100 kDa), were used with the same flow rate (12).
Brain distribution of antibiotics using microdialysis has been reported only in rare occasions with various molecules, including vancomycin, rifampin, fosfomycin, doripenem, and meropenem (12, 19–22). In vivo recovery was estimated in only two occasions, either by no net flux (12) or by retrodialysis by drug (20). In each of these studies, free brain-to-plasma AUC ratios were estimated lower than unity, attesting to the limited brain distribution of these antibiotics. However, without precise probe recovery estimation, these should be interpreted very carefully.
The present study is the first one to investigate cefotaxime extracellular brain concentrations by microdialysis. Previous studies in human have explored only cefotaxime distribution in cerebrospinal fluid (CSF) (5–8). Among them, two in adults (5, 6) and one in children (7) reported CSF-to-plasma AUC ratios lower than 20%, indicating poor penetration of cefotaxime in CSF as reported now in brain (26.1 ± 12.1% in the present study). It is well established that physicochemical properties lead partly to compound distribution in the CNS and that a low molecular size, an appropriate lipophilicity, and a large amount of nonionized form facilitate compound penetration in brain (23). Cefotaxime exhibits a relatively low molecular mass (425.5 kDa), an intermediate protein binding, and a moderate lipophilicity, and it is a weak acid with a pKa at 3.4, meaning that at pH 7.4 most of cefotaxime is ionized, which should restrict its penetration in CNS. But limited cefotaxime CNS penetration should be explained mostly by active transport systems located on barriers between blood and CNS, like the blood-brain barrier (BBB) and the blood-CSF barrier (BCSFB). Some β-lactam antibiotics were shown to be transported in vitro or in vivo by different transporters, like organic anion transporter 3 (OAT-3), peptide transporter 2 (PEPT-2), and multidrug-resistant associated protein 4 (MRP4) (24–26). Among these transporters, MRP4 is present at the luminal side of both brain capillary endothelial cells and epithelial cells of choroid plexus (blood sides) (9). Cephalosporins were shown to have a high in vitro affinity for MRP4 transporters, with ceftriaxone exhibiting one of the highest affinities, but cefotaxime was not tested (24). OAT-3 is both expressed in the brush border membrane of choroid plexus (9, 27) and at the brain level of the BBB (9, 27, 28) and is involved, with coactivation of transporters localized at the blood level, in drug efflux from CNS to blood (25). PEPT-2 is present at the apical (luminal) side of the choroidal epithelium (29), but its presence was not described in endothelial cells of the BBB (30). In the present study, the cefotaxime active transport out of brain ECF was responsible for higher brain efflux clearances (CLout,brain) than influx clearances (CLin,brain) estimated in all patients. In fact, if only passive diffusion processes were involved in cefotaxime brain distribution, CLin,brain and CLout,brain should be equal (13). Higher CLout,brain than CLin,brain values explain the lower cefotaxime unbound brain ECF than unbound plasma concentrations observed in the present study, with a mean brain-to-plasma AUC ratio equal to 26.1 ± 12.1% (13).
Cefotaxime, like other β-lactams, is a time-dependent agent, meaning that efficacy is correlated with the time during which concentrations are over the MIC (T>MIC). Cefotaxime in the present study was administered at a usual dose to treat lung infection due to mechanical ventilation in critical care patients (2 g every 8 h). However, traditional cefotaxime dose regimens for treatment of bacterial meningitis are 4 g every 6 h or 4 g every 8 h for adults (31), and the major issue in the treatment of bacterial meningitis is the risk of antibiotherapy failure. Consequently, simulations of mean brain ECF concentrations were performed from mean patient pharmacokinetic parameters using cefotaxime doses used in meningitis treatment. The simulated cefotaxime brain concentrations in the present study were consistent with those previously observed in a critical care patient treated by cefotaxime at the meningitis dose (4 g three times per day) (32). The experimental Cmax observed in this patient (11.4 μg · ml−1) was close to the present simulated value for the same dosing regimen (8.7 μg · ml−1). Considering MIC breakpoints of Streptococcus pneumoniae, T>MIC values were estimated for susceptible strains to be 99.9% and 99.8% of the 4 g/8 h and 4 g/6 h dosing intervals, respectively, and estimated to be 63.7% and 89.8% of the same dosing intervals for resistant strains (Fig. 3). For β-lactams used in meningitis, a time over minimal bactericidal concentration (MBC) at least equal to 50% of the dosing interval (33) and even greater than 90% (34) has been required for a bacterial eradication. Considering that for bactericidal agents like β-lactams the MBC value is usually the same as the MIC value or two times higher (35), the target was reached for susceptible strains whatever the dosing regimens (4 g/6 h and 4 g/8 h), while 4 g every 6 h was required for resistant strains. However, three limitations should be taken into consideration with these simulations. First, it should be assumed that brain distribution is linear in the range of concentrations used. Second, the desacetylcefotaxime metabolite of cefotaxime was not estimated in this study because of difficulties of quantification by microdialysis. Since this metabolite is active, it should be necessary to characterize its brain distribution to evaluate complete treatment efficacy. Third, our patients were all brain injured, but none of them were treated for meningitis. Antibiotic penetration through inflamed meninges is increased compared to noninflamed ones, and it should be expected to get higher cefotaxime concentrations in case of meningitis (36).
Fig 3.
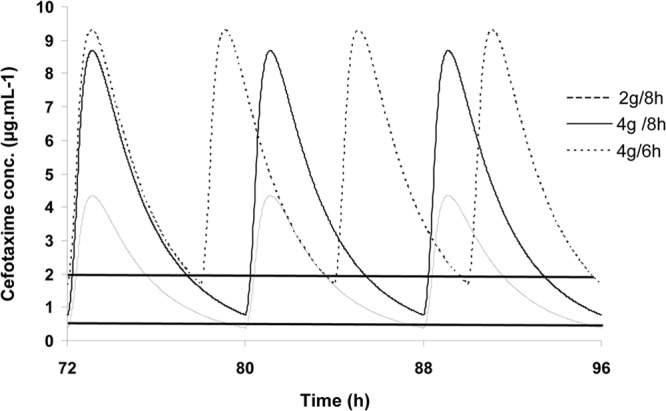
Simulations of mean cefotaxime unbound concentrations in brain, at steady state, after drug infusion at a constant rate over 30 min at different dosing regimens, 2 g every 8 h and 4 g every 6 h or 8 h. The horizontal lines at 0.5 and 2 μg · ml−1 represent, respectively, the MIC of susceptible and resistant strains of Streptococcus pneumoniae.
In conclusion, this study is the first one to quantify brain distribution of cefotaxime in patients by microdialysis. In CSF, cephalosporins are known to have a limited distribution, due mainly to active transporters. This study describes also a limited distribution of cefotaxime in the extracellular fluid.
ACKNOWLEDGMENTS
This work was supported by a public health grant from the regional Programme Hospitalier de Recherche Clinique (PHRC) from the French Ministère de l'Education et de la Recherche.
Footnotes
Published ahead of print 9 April 2013
REFERENCES
- 1. Chaudhuri A, Martinez-Martin P, Kennedy PG, Andrew Seaton R, Portegies P, Bojar M, Steiner I. 2008. EFNS guideline on the management of community-acquired bacterial meningitis: report of an EFNS task force on acute bacterial meningitis in older children and adults. Eur. J. Neurol. 15:649–659 [DOI] [PubMed] [Google Scholar]
- 2. van de Beek D, Drake JM, Tunkel AR. Nosocomial bacterial meningitis. N. Engl. J. Med. 362:146–154 [DOI] [PubMed] [Google Scholar]
- 3. Gerber J, Nau R. 2010. Mechanisms of injury in bacterial meningitis. Curr. Opin. Neurol. 23:312–318 [DOI] [PubMed] [Google Scholar]
- 4. Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala'Aldeen DA, Tuomanen EI. 2009. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest. 119:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nau R, Prange HW, Muth P, Mahr G, Menck S, Kolenda H, Sorgel F. 1993. Passage of cefotaxime and ceftriaxone into cerebrospinal fluid of patients with uninflamed meninges. Antimicrob. Agents Chemother. 37:1518–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Humbert G, Leroy A, Nair SR, Cherubin CE. 1984. Concentrations of cefotaxime and the desacetyl metabolite in serum and CSF of patients with meningitis. J. Antimicrob. Chemother. 13:487–494 [DOI] [PubMed] [Google Scholar]
- 7. Trang JM, Jacobs RF, Kearns GL, Brown AL, Wells TG, Underwood FL, Kluza RB. 1985. Cefotaxime and desacetylcefotaxime pharmacokinetics in infants and children with meningitis. Antimicrob. Agents Chemother. 28:791–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goldwater PN. 2005. Cefotaxime and ceftriaxone cerebrospinal fluid levels during treatment of bacterial meningitis in children. Int. J. Antimicrob. Agents 26:408–411 [DOI] [PubMed] [Google Scholar]
- 9. Westerhout J, Danhof M, De Lange EC. 2011. Preclinical prediction of human brain target site concentrations: considerations in extrapolating to the clinical setting. J. Pharm. Sci. 100:3577–3593 [DOI] [PubMed] [Google Scholar]
- 10. Blakeley JO, Olson J, Grossman SA, He X, Weingart J, Supko JG. 2009. Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J. Neurooncol. 91:51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hlatky R, Valadka AB, Goodman JC, Robertson CS. 2004. Evolution of brain tissue injury after evacuation of acute traumatic subdural hematomas. Neurosurgery 55:1318–1323 [DOI] [PubMed] [Google Scholar]
- 12. Dahyot-Fizelier C, Timofeev I, Marchand S, Hutchinson P, Debaene B, Menon D, Mimoz O, Gupta A, Couet W. 2010. Brain microdialysis study of meropenem in two patients with acute brain injury. Antimicrob. Agents Chemother. 54:3502–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dahyot C, Marchand S, Bodin M, Debeane B, Mimoz O, Couet W. 2008. Application of basic pharmacokinetic concepts to analysis of microdialysis data: illustration with imipenem muscle distribution. Clin. Pharmacokinet. 47:181–189 [DOI] [PubMed] [Google Scholar]
- 14. Karjagin J, Lefeuvre S, Oselin K, Kipper K, Marchand S, Tikkerberi A, Starkopf J, Couet W, Sawchuk RJ. 2008. Pharmacokinetics of meropenem determined by microdialysis in the peritoneal fluid of patients with severe peritonitis associated with septic shock. Clin. Pharmacol. Ther. 83:452–459 [DOI] [PubMed] [Google Scholar]
- 15. Hammarlund-Udenaes M, Paalzow LK, de Lange EC. 1997. Drug equilibration across the blood-brain barrier—pharmacokinetic considerations based on the microdialysis method. Pharm. Res. 14:128–134 [DOI] [PubMed] [Google Scholar]
- 16. EUCAST 2012. Breakpoint tables and interpretation of MICs and zone diameters. Version 2.0. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/EUCAST_breakpoints_v_2.0_120101.pdf
- 17. Esmieu F, Guibert J, Rosenkilde HC, Ho I, Le Go A. 1980. Pharmacokinetics of cefotaxime in normal human volunteers. J. Antimicrob. Chemother. 6(Suppl A):83–92 [DOI] [PubMed] [Google Scholar]
- 18. Bouw MR, Hammarlund-Udenaes M. 1998. Methodological aspects of the use of a calibrator in in vivo microdialysis-further development of the retrodialysis method. Pharm. Res. 15:1673–1679 [DOI] [PubMed] [Google Scholar]
- 19. Caricato A, Pennisi M, Mancino A, Vigna G, Sandroni C, Arcangeli A, Antonelli M. 2006. Levels of vancomycin in the cerebral interstitial fluid after severe head injury. Intens. Care Med. 32:325–328 [DOI] [PubMed] [Google Scholar]
- 20. Mindermann T, Zimmerli W, Gratzl O. 1998. Rifampin concentrations in various compartments of the human brain: a novel method for determining drug levels in the cerebral extracellular space. Antimicrob. Agents Chemother. 42:2626–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brunner M, Reinprecht A, Illievich U, Spiss CK, Dittrich P, van Houte M, Muller M. 2002. Penetration of fosfomycin into the parenchyma of human brain: a case study in three patients. Br. J. Clin. Pharmacol. 54:548–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poeppl W, Zeitlinger M, Donath O, Wurm G, Muller M, Botha F, Illievich UM, Burgmann H. 2012. Penetration of doripenem in human brain: an observational microdialysis study in patients with acute brain injury. Int. J. Antimicrob. Agents 39:343–345 [DOI] [PubMed] [Google Scholar]
- 23. Nau R, Sorgel F, Eiffert H. 2010. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 23:858–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akanuma S-I, Uchida Y, Ohtsuki S, Kamiie J-i, Tachikawa M, Terasaki T, Hosoya K-I. 2011. Molecular-weight-dependent, anionic-substrate-preferential transport of Î2-lactam antibiotics via multidrug resistance-associated protein 4. Drug Metab. Pharmacokinetics 26:602–611 [DOI] [PubMed] [Google Scholar]
- 25. Spector R. 2010. Nature and consequences of mammalian brain and CSF efflux transporters: four decades of progress. J. Neurochem. 112:13–23 [DOI] [PubMed] [Google Scholar]
- 26. Shen H, Ocheltree SM, Hu Y, Keep RF, Smith DE. 2007. Impact of genetic knockout of PEPT2 on cefadroxil pharmacokinetics, renal tubular reabsorption, and brain penetration in mice. Drug Metab. Dispos. 35:1209–1216 [DOI] [PubMed] [Google Scholar]
- 27. Kusuhara H, Sugiyama Y. 2005. Active efflux across the blood-brain barrier: role of the solute carrier family. NeuroRx 2:73–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loscher W, Potschka H. 2005. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog. Neurobiol. 76:22–76 [DOI] [PubMed] [Google Scholar]
- 29. Shen H, Keep RF, Hu Y, Smith DE. 2005. PEPT2 (Slc15a2)-mediated unidirectional transport of cefadroxil from cerebrospinal fluid into choroid plexus. J. Pharmacol. Exp. Ther. 315:1101–1108 [DOI] [PubMed] [Google Scholar]
- 30. Shen H, Smith DE, Keep RF, Brosius FC., III 2004. Immunolocalization of the proton-coupled oligopeptide transporter PEPT2 in developing rat brain. Mol. Pharm. 1:248–256 [DOI] [PubMed] [Google Scholar]
- 31. Société de pathologie infectieuse de langue française 2009. 17th consensus conference. Consensus conference on bacterial meningitis. Short text. Med. Malad. Infect. 39:175–186 [DOI] [PubMed] [Google Scholar]
- 32. Frasca D, Dahyot-Fizelier C, Couet W, Debaene B, Mimoz O, Marchand S. 2012. Brain microdialysis distribution study of cefotaxime in a patient with traumatic brain injury. Br. J. Anaesth. 109:830–831 [DOI] [PubMed] [Google Scholar]
- 33. Andes DR, Craig WA. 1999. Pharmacokinetics and pharmacodynamics of antibiotics in meningitis. Infect. Dis. Clin. North Am. 13:595–618 [DOI] [PubMed] [Google Scholar]
- 34. Lutsar I, Friedland IR. 2000. Pharmacokinetics and pharmacodynamics of cephalosporins in cerebrospinal fluid. Clin. Pharmacokinet. 39:335–343 [DOI] [PubMed] [Google Scholar]
- 35. Turnidge JD. 1998. The pharmacodynamics of beta-lactams. Clin. Infect. Dis. 27:10–22 [DOI] [PubMed] [Google Scholar]
- 36. Rodriguez-Cerrato V, McCoig CC, Michelow IC, Ghaffar F, Jafri HS, Hardy RD, Patel C, Olsen K, McCracken GH., Jr 2001. Pharmacodynamics and bactericidal activity of moxifloxacin in experimental Escherichia coli meningitis. Antimicrob. Agents Chemother. 45:3092–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]