

Abstract
Despite recent progress in the development of direct-acting antiviral agents against hepatitis C virus (HCV), more effective therapies are still urgently needed. We and others previously identified three phenothiazine compounds as potent HCV entry inhibitors. In this study, we show that phenothiazines inhibit HCV entry at the step of virus-host cell fusion, by intercalating into cholesterol-rich domains of the target membrane and increasing membrane fluidity. Perturbation of the alignment/packing of cholesterol in lipid membranes likely increases the energy barrier needed for virus-host fusion. A screening assay based on the ability of molecules to selectively increase the fluidity of cholesterol-rich membranes was subsequently developed. One compound that emerged from the library screen, topotecan, is able to very potently inhibit the fusion of liposomes with cell culture-derived HCV (HCVcc). These results yield new insights into HCV infection and provide a platform for the identification of new HCV inhibitors.
INTRODUCTION
Hepatitis C virus (HCV) infects at least 130 million people worldwide and is the major cause of chronic liver disease. Infected patients are at risk of developing fibrosis, cirrhosis, and liver cancer (1–3). Although HCV was identified in 1989, advances in treatment have been augmented since the development of cell culture-grown HCV (HCVcc) in 2005 (4–6). No vaccine is available, and the current treatment for HCV infection involves a weekly injection of pegylated alpha interferon and a twice-daily weight-based dose of ribavirin for 24 to 48 weeks. This standard of care is plagued by a long duration, limited efficacy, and serious side effects (7). Although the recent addition of new direct-acting antivirals (DAAs) targeting HCV NS3-4A protease—telaprevir and boceprevir—to the anti-HCV therapeutic arsenal have improved the cure rates, they must be used in combination with interferon, as HCV has a remarkable ability to overcome a single DAA. Telaprevir and boceprevir only work in patients infected with genotype 1 HCV and are both not very effective in patients who did not respond to pegylated interferon-ribavirin therapy (8). In addition, both telaprevir and boceprevir appear to worsen the already problematic side effects of the standard therapy, such as rashes and anemia (9). Currently approved DAAs and most molecules in the pipeline are protease inhibitors, nucleoside inhibitors, nonnucleoside inhibitors, and NS5A inhibitors (10). A major obstacle in combating HCV is the low fidelity of the viral replication machinery, enabling the virus to quickly develop resistance (11). To date, ITX-5061 is the only inhibitor of HCV entry that has entered clinical testing. ITX-5061 blocks a postbinding step in the viral entry process by directly interacting with the entry factor scavenger receptor B1 (SR-B1) (12). New DAAs targeting entry steps critical to viral infection with additive potency when combined with existing DAAs and exhibiting low cytotoxicity are highly desirable.
HCV is an enveloped, positive-sense RNA virus belonging to the Flaviviridae family. The 9.6-kb viral genome encodes a single large polyprotein that is processed by viral and cellular proteases to produce the virion structural proteins (core and glycoproteins E1 and E2), P7, and nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). HCV infection involves multiple steps. Viruses first attach to target cells via glycosaminoglycans and low-density lipoprotein (LDL) receptors. After recruitment to the membrane, HCV binds sequentially to entry factors involving SR-B1, the tetraspanin CD81, the Niemann-Pick C1-like 1 (NPC1L1) cholesterol (Cho) uptake receptor, and proteins of tight junctions, i.e., CLDN1 and OCLD (13). HCV then enters cells at the tight junction via clathrin-mediated endocytosis and fuses with the host membrane in the late endosome. Progress in defining the molecular mechanism of HCV entry raises the opportunity to exploit new viral and host targets for therapeutic intervention. Entry inhibitors have the potential to limit the expansion of the infected cell reservoir, prevent reinfection after liver transplantation, and complement the many protease and polymerase inhibitors currently under development. Although the discovery of drugs targeting the entry stage is still in its infancy, antibodies against SR-B1 (14), CD81 (15), and CLDN1 (16), as well as a number of small-molecule inhibitors, have recently been developed and are able to effectively block HCV entry (17–24).
Phenothiazines are a group of nitrogen- and sulfur-containing tricyclic compounds that were first synthesized by Bernthsen in 1883. Phenothiazines with dialkylaminoalkyl groups and small groups substituted at positions 10 and 2, respectively, were found to interact with the dopamine receptors and have exhibited valuable activities, such as neuroleptic, antiemetic, antihistaminic, antipruritic, analgesic, and anthelmintic activities (25). To date, more than 100 phenothiazines have been used in clinics to treat psychotic disorders, and over 5,000 phenothiazine derivatives have been synthesized. Other receptors that can be modulated by phenothiazines include histamine H1, adrenergic α1 and α2, muscarinic (cholinergic), and serotonergic receptors (25). In addition to neurotransmitter receptors, phenothiazines have also been reported to bind to calmodulin and block its calcium signal-transduction activity, inhibit clathrin-coated pit formation, and activate rynodine receptors (26). Antiviral and antimicrobial activities have also been described for phenothiazines and related compounds (27).
Our lab and others recently identified three phenothiazines—fluphenazine, trifluoperazine, and prochlorperazine—as potent HCV entry inhibitors (28, 29). In this work, we wanted to understand the antiviral mode of action of this family of compounds, which presumably inhibit HCV entry through a common mechanism of action. This information will assist in future endeavors to identify new and more potent inhibitors of HCV entry. We found that phenothiazines inhibit the virus-cell fusion step of the HCV life cycle by intercalating into the host cholesterol-rich membrane. In the presence of phenothiazines, cholesterol-rich membranes become more permeable to water molecules, leading to increased membrane fluidity. We subsequently developed a high-throughput screening assay. We screened a library of 2,752 compounds and identified a molecule, topotecan, that dose-dependently inhibits HCVcc-liposome fusion. This study suggests that alteration of target cholesterol-rich membrane fluidity may be a novel mode for suppressing HCV entry and should facilitate the identification of new HCV inhibitors with unique modes of action.
MATERIALS AND METHODS
Cells, plasmids, compounds, and reagents.
Huh-7.5 cells and plasmids encoding HIV Gag-Pol (30) and the envelope proteins of HCV H77/J6 (30) and vesicular stomatitis virus (VSV) were kindly provided by Charles Rice (Rockefeller University, NY). HEK 293T cells were purchased from Invitrogen (Carlsbad, CA). Trifluoperazine, prochlorperazine, mesoridazine, promazine, triflupromazine, and cis-flupentixol were purchased from Sigma-Aldrich (St. Louis, MO). Chlorpromazine and thioridazine were from MP Biomedicals (Solon, OH). Fluphenazine and bafilomycin were from Alfa Aesar (Ward Hill, MA) and Axxora (San Diego, CA), respectively. All phenothiazine compounds were dissolved in dimethyl sulfoxide (DMSO) to a 10 mM stock concentration. Bafilomycin was dissolved in DMSO to a 250 μM stock concentration. Laurdan and Prodan fluorescent probes were purchased from Anaspec (Fremont, CA). Laurdan and Prodan were dissolved in methanol and DMSO to final concentrations of 0.5 mM and 10 mM, respectively. The human anti-CD81 JS81 monoclonal antibody (MAb) was obtained from BD Biosciences (San Jose, CA). CellTiter-Glo luminescent cell viability assay kits and BioLux Gaussia luciferase assay kits were purchased from Promega (Madison, WI) and New England BioLabs (Ipswich, MA), respectively. The growth medium for all cell culture work was Dulbecco's modified Eagle's medium (DMEM) containing 4,500 mg/liter glucose, 4.0 mM l-glutamine, and 110 mg/liter sodium pyruvate (Thermo Scientific HyClone, Logan, UT) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 1× nonessential amino acids (Thermo Scientific HyClone, Logan, UT). Dulbecco's phosphate-buffered saline (DPBS) was purchased from Thermo Scientific HyClone (Logan, UT). Octadecyl rhodamine B chloride (R18) was purchased from Invitrogen (St. Aubin, France), and all other lipids (99% pure) were from Avanti Polar Lipids (Alabaster, AL).
Production of HCVcc and pseudotyped lentiviruses.
The production and titer determination of Jc1 Gluc HCVcc (31) in Huh-7.5 cells were performed as previously described (28). Jc1 Gluc HCV contains the Gaussia luciferase (Gluc) reporter gene between the HCV genes encoding the p7 and NS2 proteins. Pseudotyped lentiviruses were produced by cotransfecting 293T cells with plasmids encoding HIV Gag-Pol (30), a provirus (pTRIP-Gluc) (28), and the appropriate envelope protein, using TransIT reagent (Mirus, Madison, WI) following the manufacturer's protocol. The supernatants containing the pseudoparticles were collected, pooled, and filtered (0.45 μm pore size) at 48 h posttransfection and then stored at 4°C for up to 1 week or at −80°C for long-term storage. For production of lentiviruses pseudotyped with the envelope proteins from HCV genotype 1a H77 (H77 HCVpp), HCV genotype 2b J6 (J6 HCVpp), and vesicular stomatitis virus (VSV-Gpp), plasmids H77 E1E2 pcDNA3, J6 E1E2 pcDNA3, and pVSVG, respectively, were used (30). A control pseudotyped lentivirus lacking any envelope protein (Env−pp) was generated using the same protocol, except that the envelope protein-encoding plasmid was replaced with empty vector (pcDNA3).
HCVcc infection assays.
To determine whether phenothiazines are virucidal, Jc1 Gluc HCVcc (6.4 × 105 50% tissue culture infective doses [TCID50]/ml) was incubated with phenothiazines (50 or 5 μM), PD 404,182 (150 μM), or DMSO (0.5%) for 1 h at 37°C, and the virus-compound mixtures were diluted 100-fold in growth medium and used to infect Huh-7.5 cells seeded 4 to 6 h earlier in 96-well plates at 3.2 × 104 cells/well. For controls, virus and drugs were diluted 100-fold separately and mixed before infecting Huh-7.5 cells. Cells were thoroughly washed at 14 to 16 h post-virus inoculation to remove residual drug and virus. Supernatant Gluc activity was measured at 48 h postinfection. The percentages of virus entry and spread were determined relative to those of the DMSO control.
To determine whether phenothiazines act on host cells, Huh-7.5 cells were infected with Jc1 Gluc HCVcc at various times after drug removal. Briefly, Huh-7.5 cells were seeded in 48-well plates at 4 × 104 cells/well. After attachment, these cells were treated with phenothiazines (5 μM), PD 404,182 (150 μM), bafilomycin (10 nM), or DMSO (0.5%) for 2 h at 37°C. For sets 1 to 3, these cells were washed thoroughly to remove residual drugs and then inoculated with Jc1 Gluc HCVcc (multiplicity of infection [MOI] = 1) or VSV-Gpp (100-fold dilution) at 0, 4, or 24 h post-drug removal at 37°C. At 15 minutes post-virus inoculation, these cells were thoroughly washed to remove any remaining viruses and returned to the 37°C and 5% CO2 incubator. For set 4, cells were infected with the same viruses, but in the presence of the drug, and were continuously incubated in drug-containing medium after the infection period. Supernatant Gluc activity was measured at 72 h postinfection, normalized to viable cell levels, and used as an indication of viral infection.
To determine the anti-HCV activities of phenothiazines (Table 1) and topotecan (see Fig. 6C), Huh-7.5 cells (1.6 × 104 cells/well) seeded 4 to 6 h earlier were infected with Jc1 Gluc HCVcc (MOI = 0.01) in the presence of increasing concentrations of the compounds. Supernatant Gluc activity was measured at 48 to 72 h postinfection and normalized to the DMSO (0.02 to 0.5%) treatment control. The cell viability was measured using the CellTiter-Glo assay (Promega, Madison, WI) to gauge the compound toxicity. The 50% inhibitor concentration (IC50), 50% effective concentration (EC50), and 50% cytotoxic concentration (CC50) were calculated using the sigmoidal fit function in OriginLab (OriginLab, Northampton, MA).
Table 1.
aIC50 and CC50 values were calculated using a Gluc reporter HCVcc infection assay and represent averages ± standard deviations for two independent experiments.
Fig 6.

Characterization of topotecan. (A) Topotecan preferentially increases the fluidity of cholesterol-rich membranes (POPC/Cho/SM) compared to cholesterol-free membranes (POPC). Liposomes composed of pure POPC or POPC with 32 mol% cholesterol (Cho) and 14 mol% sphingomyelin (SM) were incubated with Laurdan (5 μM) or Prodan (15 μM) for 15 min at room temperature prior to the addition of topotecan (0.1, 0.5, 1, or 2 μM) or DMSO (1%). The mixture was incubated at 37°C for another 30 min, and the fluorescence shifts were determined. delta GP, GPtopotecan − GPDMSO. The values and error bars represent means and standard deviations, respectively, for 2 independent experiments. (B) Topotecan dose-dependently inhibits HCVcc-liposome fusion in vitro. HCVcc (∼1.6 × 105 particles) was mixed with R18 dye-labeled liposomes in the presence of increasing concentrations of topotecan. Fusion between HCVcc and liposomes was triggered by the addition of HCl to lower the solution pH. Dequenching of R18 dye was monitored by determining the increase in fluorescence. The x axis corresponds to the duration of the process. (C) Topotecan dose-dependently inhibits HCVcc infection in cell culture. The chemical structure of topotecan is shown in the inset. Huh-7.5 cells were infected with Jc1 Gluc HCVcc (MOI = 0.01) in the absence or presence of increasing concentrations of topotecan. Infectivity (black bars) was quantified by measuring the supernatant Gluc activity at 72 h postinfection and then normalized to the DMSO (0.02%) control level. Drug cytotoxicity in the absence of HCV infection (solid circles) was determined by the CellTiter-Glo assay. The values and error bars represent means and standard deviations, respectively, for 2 independent experiments.
Synchronized HCVcc infection assay.
To determine the step of entry inhibited by phenothiazines, we carried out a synchronized infection assay (Fig. 1B). Huh-7.5 cells were seeded in 48-well plates at 4 × 104 cells/well. The next day, virus-cell attachment was initiated by incubating the cells with Jc1 Gluc HCVcc (MOI = 1) at 4°C for 1.5 h. Unbound viruses were removed by thorough washing with complete growth medium, and then infection of bound viruses was initiated by moving the cell culture plates to a 37°C and 5% CO2 incubator. Fluphenazine (5 μM), bafilomycin (10 nM), JS81 (2 μg/ml), or DMSO (0.05%) was added to the cells at different time points after the temperature shift. Cells were washed thoroughly with complete growth medium at 5 h post-drug addition. Supernatant Gluc activity was measured at 48 h postinfection, using a BioLux Gaussia luciferase assay kit (New England BioLabs, Ipswich, MA), and was used as an indication of viral infection.
Fig 1.

Phenothiazines inhibit HCV fusion. (A) Schematic of synchronized HCVcc infection assay. Huh-7.5 cells were incubated with Jc1 Gluc HCVcc (MOI = 1) for 1.5 h at 4°C, washed extensively, and shifted to 37°C. Fluphenazine (5 μM), bafilomycin (10 nM), JS81 (2 μg/ml), or DMSO (0.05%) was added at different times after the temperature shift and removed at 5 h post-compound addition. Supernatant Gluc activity was measured at 48 h postinfection, normalized to the DMSO control level, and used as an indication of infection efficiency. (B) Fluphenazine inhibits HCVcc entry at a step similar to that of bafilomycin inhibition and later than JS81 inhibition. Fluphenazine (5 μM), JS81 (2 μg/ml), or bafilomycin (10 nM) was added to the appropriate wells at different times post-temperature shift. Representative data from at least 5 independent experiments are presented. The error bars represent standard deviations for triplicate samples. (C) In vitro liposome fusion assay confirms that fluphenazine, trifluoperazine, and promazine dose-dependently inhibit HCV fusion. Concentrated H77 HCVpp (∼2 × 105 particles) was mixed with R18 dye-labeled liposomes and increasing concentrations of the phenothiazines. Fusion between HCVpp and liposomes was triggered by the addition of HCl to lower the solution pH. Dequenching of the R18 dye, corresponding to fusion between HCVpp and liposomes, was monitored by determining the increase in fluorescence. The x axis corresponds to the duration of the fusion process. Curves representative of at least 4 independent experiments are presented.
HCVpp/cc-liposome fusion assay.
To evaluate the ability of compounds to inhibit HCV fusion in vitro, we carried out an HCV-liposome fusion assay. Fusion between HCVpp and liposomes was assayed as described elsewhere (32). H77 HCVpp collected from the cell supernatant was purified and concentrated 100-fold by use of ultracentrifugation devices, to a titer of ∼107 IU/ml. One microliter of liposomes (25 μM final lipid concentration) composed of phosphatidylcholine (PC), cholesterol, and R18 (65:30:5 mol%) was added to a 37°C thermostated cuvette containing 20 μl concentrated H77 HCVpp (∼2 × 105 viral particles) in phosphate-buffered saline (PBS) at pH 7.2. Fluphenazine, trifluoperazine, or promazine dissolved in DMSO was added to the mixture at a final concentration of 2.5, 5, 10, or 20 μM. After thermic equilibration, fusion was initiated by adding 2 μl diluted HCl (final pH, 5.0). Lipid mixing was measured as the dequenching of R18 (excitation, 560 nm; emission, 590 nm), resulting in an increase of the fluorescence signal. Recordings were performed with a Tecan Infinite-1000 spectrofluorometer. Maximal R18 dequenching was measured after addition of Triton X-100 to the cuvette at a 0.1% final concentration. Fusion between JFH-1 HCVcc and liposomes was determined similarly. Freshly prepared JFH1 HCVcc, concentrated 100-fold through a 20% sucrose cushion, was used in the fusion experiment (33).
For the modified HCVpp-liposome fusion experiment, 20 μl concentrated H77 HCVpp or liposomes (working suspension at a 1:20 dilution from stock) was preincubated with fluphenazine, trifluoperazine, or promazine at a 5 μM final concentration for 3 min at 37°C. This mixture was then diluted 10-fold with PBS to reach a 0.5 μM final drug concentration, and 1 μl untreated liposomes or 20 μl HCVpp was added, respectively. Fusion was determined in the same way as that described above.
The study of the effect of topotecan on HCVcc, membrane fusion was performed as described previously (33). Briefly, JFH-1 HCVcc (∼1.6 × 105 particles), suspended in 150 mM NaCl, 10 mM Tricine-NaOH, pH 7.4, was added to a cuvette containing R18-labeled PC-cholesterol liposomes (15 μM final lipid concentration) in the absence or presence of increasing concentrations of topotecan. After temperature equilibration, fusion was initiated by HCl addition to the cuvette, and kinetics were recorded using a dual-channel PicoFluor handheld fluorometer (Turner Biosystems, Sunnyvale, CA) operated under the “rhodamine” channel (excitation and emission wavelengths of 540 ± 20 and >570 nm, respectively). Maximal R18 dequenching was measured after the addition of 0.1% Triton X-100 (final concentration) to the cuvette.
Fluorescence spectroscopy.
To determine whether phenothiazines affect membrane fluidity, we calculated the generalized polarization (GP) of the fluorescent dyes Laurdan and Prodan incorporated into the liposomes in the absence or presence of phenothiazines. POPC (11-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), sphingomyelin (SM), and cholesterol (Cho) (Avanti Polar Lipids, Inc., Alabaster, AL) dissolved in chloroform at 10 mg/ml were mixed at a molar ratio of 1:0:0 (100% POPC), 2.3:0:1 (70% POPC plus 30% Cho), or 3.9:1:2.3 (54% POPC plus 14% SM plus 32% Cho). To remove solvent, the lipids were first dried under a stream of nitrogen and then lyophilized. The lyophilized lipid mixtures were resuspended in DPBS at a 400 μM final lipid concentration, sonicated at room temperature in a water bath sonicator for 10 min, and extruded repeatedly through a 100-nm polycarbonate membrane filter (Avanti Polar Lipids, Inc.) to obtain uniformly sized liposomes. Extruded liposomes were stored at 4°C for up to a week. For GP determination, liposomes (200 μM final concentration) were first incubated with Laurdan (5 μM final concentration) or Prodan (15 μM final concentration) for 15 min at room temperature in the dark. Increasing concentrations of the compounds were added to the mixture, and 100 μl/well of liposome-drug mixture was transferred to a white 384-well plate. The plate was incubated at 23°C for 30 min in the dark, and fluorescence spectra were collected in a Gemini EM spectrofluorometer (Molecular Devices, San Francisco, CA) with an excitation wavelength of 310 to 350 nm for both dyes and the emission spectra recorded at 440 and 480 nm for Prodan and 440 and 490 nm for Laurdan. GP was calculated according to the following equation (34): GP = (IB − IR)/(IB + IR), where IB and IR are the fluorescence intensities at the blue and red edges of the emission spectrum, respectively. Data were corrected for the background signal measured with liposomes deprived of a probe. After the fluorescence spectra were measured, the plate was returned to a 37°C incubator for another 30 min, after which the spectra were measured again.
Screen for inhibitors.
A high-throughput screening assay was developed based on the ability of a compound to selectively increase the GP of cholesterol-rich liposomes in comparison to the DMSO control. The library screening was conducted at The National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease (NSRB). Liposomes composed of POPC alone or POPC, Cho, and SM at a 3.9:1:2.3 molar ratio (200 μM final concentration) were incubated with Prodan (15 μM final concentration) for 15 min at room temperature in the dark. Eighty microliters of the mixture was dispensed into each well of white 384-well plates by use of a Matrix WellMate liquid dispenser (Matrix, Hudson, NH). One hundred nanoliters of the drug library (with concentrations ranging from 2 to 5 mg/ml) was then added to each well by use of a Seiko D-TRAN XM3106-31 PN 4-axis Cartesian robot pin transferor (Caliper Life Science, Waltham, MA). The plates were incubated in the dark for 30 min at room temperature, and the fluorescence spectra were collected in an SGM 610 FlexStation III spectrofluorometer (Molecular Devices, San Francisco, CA) with an excitation wavelength of 310 nm and the emission spectra recorded at 440 and 480 nm. We screened the Biomol4 (Enzo Life Sciences, Plymouth Meeting, PA) and Chembridge 3 libraries for molecules whose GP differed from that of the 0.25% DMSO control by a value of >0.05 (positive hit). Although we screened only 2,752 compounds in this study, this assay is amenable to high-throughput screening.
Replication inhibition and qRT-PCR.
To determine the effect of topotecan on viral replication, we quantified the amounts of HCV RNA in the appropriate cells (see Fig. 7A). Huh-7.5 cells were electroporated with Jc1 Gluc HCV genomic RNA according to a previously described protocol (28) and seeded into 24-well plates (1.4 × 105 cells/well). The desired compounds were added to the medium at 6 h postelectroporation. The medium was replaced with fresh, compound-containing medium at 48 h postelectroporation, and the supernatant Gluc activity was determined using a BioLux Gaussia luciferase assay kit (New England BioLabs, Ipswich, MA) at 72 h postelectroporation. After removing all the supernatant, these cells were washed once with Dulbecco's phosphate-buffered saline (Thermo Scientific HyClone, Logan, UT) and underwent one freeze-thaw cycle at −80°C before RNA extraction using an EZNA Total RNA kit (Omega Bio-Tek, Norcross, GA). The amount of HCV RNA was quantified via TaqMan quantitative reverse transcription-PCR (qRT-PCR) (qScript One-Step Fast kit; Quanta Biosciences, Gaithersburg, MD), using previously described primers (35).
Fig 7.
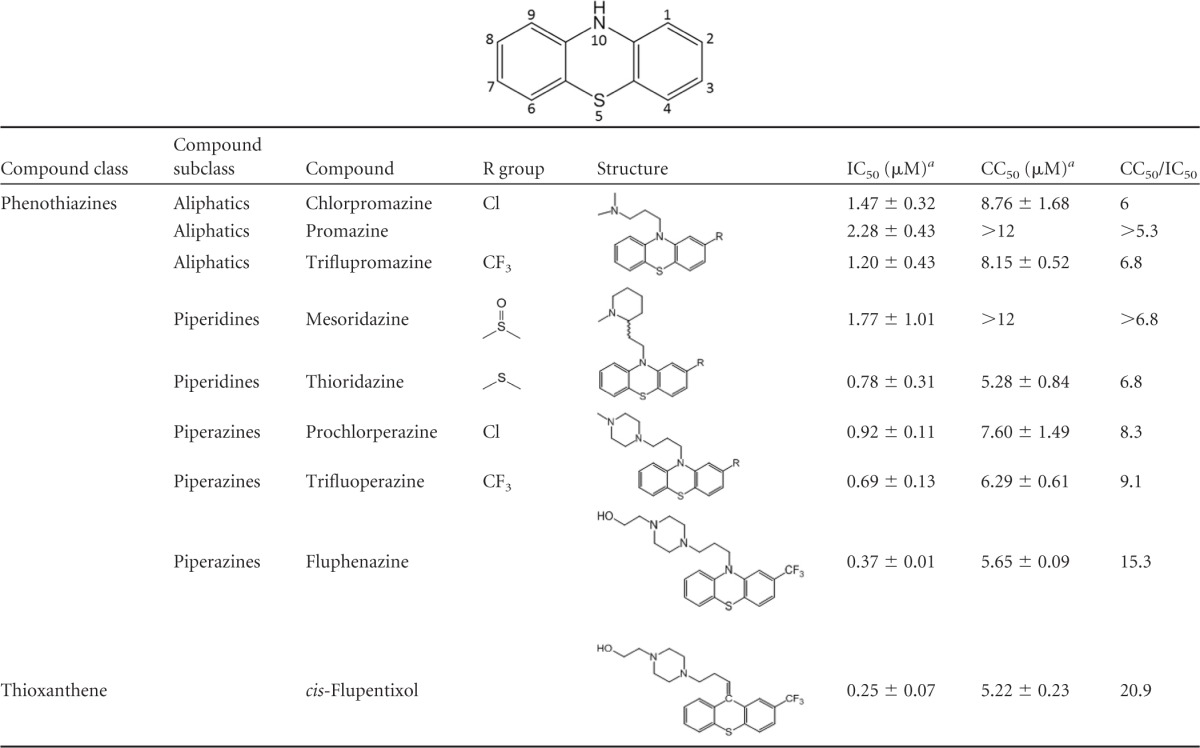
Topotecan primarily inhibits HCV replication. (A) Effect of topotecan on HCVcc replication. Huh-7.5 cells were electroporated with Jc1 Gluc HCV genomic RNA and treated with topotecan, DMSO (0.125%), or 2′CMA at 6 h postelectroporation. The amount of Gluc reporter in the supernatant and the intracellular HCV RNA level were quantified at 72 h postelectroporation. 2′CMA, 2′-C-methyladenosine. (B) Topotecan does not inhibit H77 HCVpp entry. Huh-7.5 cells were transduced with 5-fold-diluted H77 HCVpp or Env−pp in the presence of topotecan, JS81 (2 μg/ml), or DMSO (0.02%). The supernatant Gluc reporter activity was measured at 48 h post-compound removal, normalized to signals from Env−pp, and used as an indication of the level of infection. Values represent the means for 2 independent experiments, and error bars represent the standard deviations.
Virus entry inhibition assay.
To determine the effect of topotecan on HCV entry, uptake of HCVpp in the presence of the compound was evaluated. Huh-7.5 cells were seeded in poly-l-lysine-treated 96-well flat-bottom tissue culture plates at 1.8 × 104 cells/well and incubated at 37°C and 5% CO2 for 5 to 6 h to allow cell attachment. Compounds were added to the appropriate wells 1 h before transduction. HCVpp and Env−pp were added to the cells at a 1:5 dilution. The next day, these cells were washed 4 times with 100 μl/well of complete growth medium to remove unbound pseudoparticles and preexisting Gluc reporter, and 100 μl of fresh medium was added to each well. Supernatant Gluc activity was quantified at 48 h postwashing by use of a BioLux Gaussia luciferase assay kit (New England BioLabs, Ipswich, MA). The presented HCVpp entry data represent differences in supernatant Gluc activities between HCVpp- and Env−pp-transduced cells at the specified compound concentrations.
Statistical analysis.
Statistical significance between different samples was evaluated using Student's t test. A P value of <0.01 was considered statistically significant. All analysis was done using Microsoft Excel.
RESULTS
Identification of additional phenothiazine-like HCV inhibitors.
Since phenothiazines and like compounds have been used extensively in humans, we first explored the anti-HCV activities of 6 additional FDA-approved phenothiazine and similar compounds by using a Gluc HCVcc assay (Table 1). All tested compounds exhibited anti-HCV activity at submicromolar to micromolar concentrations and were able to specifically inhibit HCV entry (see Fig. S1 in the supplemental material). The most potent compound, cis-flupentixol, exhibited an IC50 of 0.25 μM and a therapeutic index (CC50/IC50) of 20. Comparing the anti-HCV potencies of different phenothiazines, we noticed that molecules with a piperazine ring at position 10 appeared to be slightly more potent than those with a tertiary amine (prochlorperazine > chlorpromazine; trifluoperazine and fluphenazine > triflupromazine), and the presence of a propanol group on the piperazine ring (fluphenazine) further increased the anti-HCV potency. A trifluoromethyl group at position 2 of the phenothiazine nucleus also enhanced the overall anti-HCV activity (trifluoperazine > prochlorperazine; triflupromazine > chlorpromazine).
Phenothiazines inhibit HCV fusion.
HCV entry involves three main steps: (i) attachment of virions to the cell surface, (ii) movement of virus particles from the cell surface to the tight junction through interaction with different receptors, and (iii) entry into the host cell through clathrin-mediated endocytosis and fusion of the viral membrane with the endosome upon acidification (36). To elucidate the anti-HCV mechanism of action of phenothiazines, we first determined the entry step inhibited by phenothiazines in a synchronized infection experiment (Fig. 1A). Fluphenazine retained maximum entry inhibition when it was added after the temperature shift to 37°C, indicating that this compound inhibits a postattachment step of HCV entry. Similar profiles were obtained for all other phenothiazine inhibitors in a similar assay using HCVpp (see Fig. S2 in the supplemental material). The inhibitory activity of fluphenazine was lost at a later time than that of a CD81 antibody (JS81) and at a step similar to that of inhibition by bafilomycin, an H+-ATPase inhibitor that blocks the fusion of HCV by suppressing endosome acidification, indicating that fluphenazine and, possibly, other phenothiazines inhibit HCV entry at a step later than CD81 binding, likely during fusion (Fig. 1B).
To determine the effects of phenothiazines on HCV fusion directly, we carried out an in vitro HCVpp-liposome fusion experiment. For this experiment, we chose fluphenazine and trifluoperazine, both phenothiazines with a piperazine substitution at position 10, as well as promazine, which has an aliphatic substitution at this position. All three phenothiazines dose-dependently inhibited HCVpp-liposome fusion in vitro (Fig. 1C), with fluphenazine exhibiting the strongest fusion inhibition, followed by trifluoperazine and promazine, consistent with their anti-HCV potencies determined via Gluc HCVcc assay (Table 1). This activity was not due to unspecific molecular quenching of R18 fluorescence (data not shown) and was therefore fully attributable to fusion inhibition. These results confirm the fusion-inhibitory activity of these three phenothiazines. The ability of phenothiazines with both piperazine and aliphatic substitutions at position 10 to inhibit HCV fusion suggests that fusion inhibition is independent of the substitution at position 10 and is likely a feature shared by other phenothiazines. In addition, since proteins are absent from the liposome, these data also indicate that a cellular protein(s)/receptor(s) is not required for phenothiazine-mediated HCV fusion inhibition.
Phenothiazines inhibit HCV fusion by acting on the host cell.
We sought to elucidate whether phenothiazines act on cells or the virus. To determine whether phenothiazines are virucidal, Jc1 Gluc HCVcc was mixed with fluphenazine, prochlorperazine, or trifluoperazine and then diluted 100-fold (pretreatment), or each component was first individually diluted and then mixed (control) (Fig. 2A). The infectivities of HCVcc samples pretreated with phenothiazines were similar to that observed with the control (nonpretreated) (Fig. 2B). The positive control, the virucidal compound PD 404,182, reduced HCV infectivity >90% during the same period (37). These results indicate that phenothiazines do not inhibit HCV entry by inactivating the virus directly.
Fig 2.
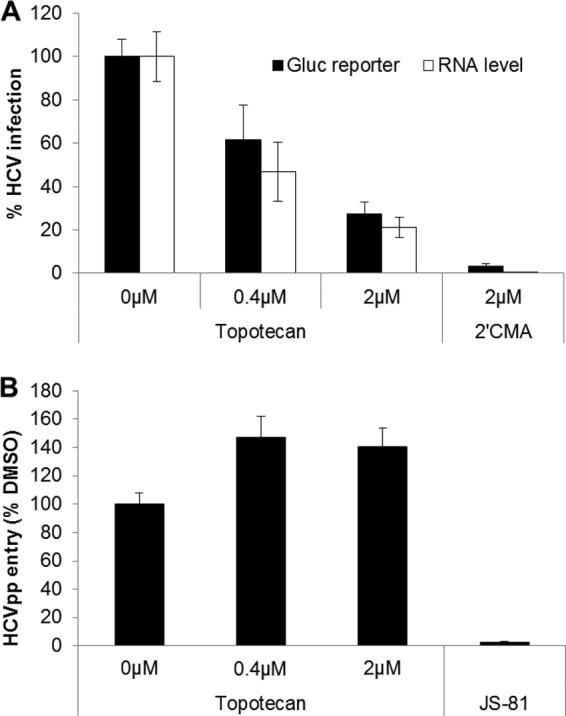
Phenothiazines do not act directly on HCV. (A) Schematics of experiments to determine a compound's effect on virions. In the pretreatment group, Jc1 Gluc HCVcc (6.4 × 105 TCID50/ml) was incubated with the appropriate compound at 37°C for 1 h and then diluted 100-fold and used to infect Huh-7.5 cells. In the control group, the same amounts of HCVcc and drug were incubated separately at 37°C for 1 h, diluted 100-fold, and then mixed and used to infect Huh-7.5 cells. The final titers of HCVcc and the concentrations of drug were identical between the pretreatment and control groups. (B) Viruses pretreated with phenothiazines retain similar infectivity to that of the control. Supernatant Gluc activity was measured at 48 h postinfection and normalized to the DMSO control (0.5%) level. Fluph, fluphenazine; Triflu, trifluoperazine; Proch, prochlorperazine; PD, PD 404,182; Bafilo, bafilomycin. The values and error bars represent means and standard deviations, respectively, for at least 2 independent experiments. Statistical significance was determined by Student's t test (*, P < 0.01).
To evaluate whether phenothiazines interact with the host cell, Huh-7.5 cells were treated with a phenothiazine at 37°C for 2 h and extensively washed to remove residual drug prior to infection with Jc1 Gluc HCVcc or VSV-Gpp for 15 min at 0, 4, or 24 h post-drug removal (Fig. 3A). Cells pretreated with phenothiazines became significantly more resistant to infection by HCVcc but not VSV-Gpp, suggesting that phenothiazines inhibit the entry of HCV by acting on the host cell (Fig. 3B). Cells treated with the control compound bafilomycin were similarly resistant to both HCVcc and VSV-Gpp infections. It is worth noting that although some phenothiazines are able to inhibit clathrin-coated pit formation and endosome acidification, this effect is likely not at play in the observed phenothiazine-mediated HCV inhibition phenomenon, because inhibition of VSV-Gpp, which also enters cells through clathrin-mediated endocytosis (38), was not observed at the same concentration of phenothiazines (Fig. 3B). Collectively, these results suggest that phenothiazines inhibit HCV entry by acting on a nonprotein host cell component, likely the lipid membrane.
Fig 3.
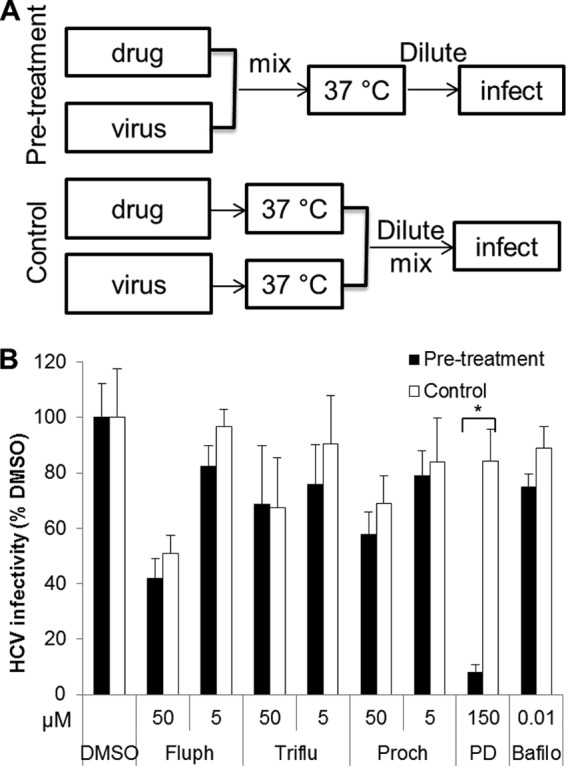
Phenothiazines inhibit HCV by acting on host cells. (A) Schematic of experiment to determine whether phenothiazines inhibit HCVcc entry by acting on host cells. Huh-7.5 cells were treated with the specified compounds at 37°C for 2 h, extensively washed at 0 h, and then infected with Jc1 Gluc HCVcc (MOI of ∼1) or VSV-Gpp (diluted 500-fold) for 15 min at 37°C at 0, 4, or 24 h post-compound removal (sets 1 to 3). For set 4, the cells were infected with the same viruses in the presence of the drugs. (B) Cells pretreated with phenothiazines became resistant to HCVcc but not VSV-Gpp infection, indicating that phenothiazines selectively inhibit HCV entry. The supernatant Gluc activity was measured at 72 h post-drug removal, normalized first to the cell viability and then to the cell viability-normalized DMSO control (0.5%). The values and error bars represent means and standard deviations, respectively, for at least 2 independent experiments. Fluph, 5 μM fluphenazine; Triflu, 5 μM trifluoperazine; Proch, 5 μM prochlorperazine; Bafi, 10 nM bafilomycin. Statistical significance was determined by Student's t test (*, P < 0.01).
To confirm that phenothiazines inhibit HCV by interacting with the target lipid membrane, we designed a modified liposome-HCV fusion experiment (Fig. 4A) in which liposomes or virus was pretreated with the appropriate compound prior to dilution of the mixture and addition of the other component. In set 1, cholesterol-rich liposomes were preincubated with phenothiazines (5 μM). This mixture was diluted 10-fold to lower the drug concentration and then mixed with HCVpp. In set 2, HCVpp was preincubated with phenothiazines (5 μM), and the mixture was diluted 10-fold prior to addition of liposomes. In both sets, the final concentration of phenothiazine in the mixture was 0.5 μM, a concentration that is expected to have little to no effect on liposome-HCVpp fusion. In set 3, liposomes, phenothiazines (final concentration, 5 μM), and HCVpp were all mixed together at the same time. Finally, in set 4, liposomes and HCVpp were mixed in the absence of phenothiazines. We reasoned that if phenothiazines inhibit HCV-membrane fusion by interacting with the liposomal membrane, set 1 (liposomes pretreated with phenothiazine) would exhibit a similar extent of fusion inhibition to that of set 3 (all components mixed together), despite the much lower phenothiazine concentration at the time of fusion in set 1, while set 2 (HCVpp pretreated with compound) would exhibit minimal fusion inhibition. On the other hand, if phenothiazines interact with the virus directly, set 2 would exhibit a similar extent of fusion inhibition to that of set 3. Stronger fusion inhibition was observed in sets 1 and 3 than in set 2 (Fig. 4B), providing further evidence that phenothiazines inhibit HCV fusion by acting on the target liposome/cell membrane. Set 1 exhibited slightly stronger fusion inhibition than set 3, despite the 10-fold lower concentration in set 1 at the time of fusion. This may have been due to the presence of additional vesicles (e.g., exosomes) in the concentrated HCVpp samples used in set 3, which could have competed with R18-labeled liposomes for the phenothiazines during membrane fusion.
Fig 4.
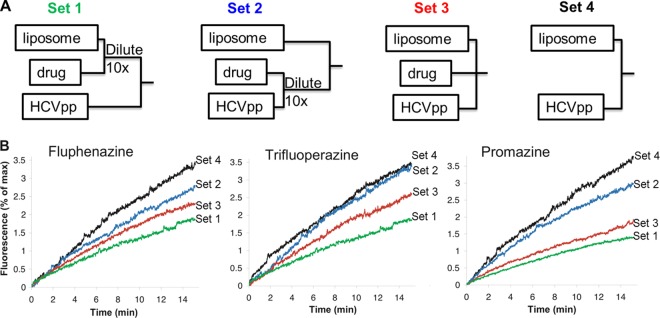
Phenothiazines inhibit HCVpp-liposome fusion by interacting with the target membrane. (A) Modified drug addition protocol. Set 1, a phenothiazine and liposomes were premixed and diluted 10-fold prior to the addition of HCVpp; set 2, HCVpp and a phenothiazine were premixed and diluted 10-fold prior to the addition of liposomes; set 3, liposomes, a phenothiazine, and HCVpp were mixed together; set 4, liposomes and HCVpp were mixed in the absence of phenothiazines. The final concentrations of phenothiazines in sets 1 and 2 were 10% of those in set 3. (B) Fusion between HCVpp and liposomes was initiated by decreasing the pH to 5.0 (time zero) and were recorded as R18 fluorescence dequenching over time. Concentrated H77 HCVpp (∼2 × 105 particles) was used in each assay. Curves representative of at least 4 independent experiments are presented.
Phenothiazines likely inhibit HCV fusion by increasing the target membrane fluidity.
To gain insight into how phenothiazines inhibit HCV-membrane fusion, we studied their effect on lipid membrane fluidity. The lipophilic nature of phenothiazines enables this class of molecules to intercalate into lipid membranes and alter their fluidity (39). However, this effect was never evaluated at low concentrations or in membranes with high cholesterol concentrations. Membrane fluidity was gauged by the GP generated by the fluorescent dyes Laurdan and Prodan. Both Laurdan and Prodan probes are lipophilic dyes able to insert into lipid bilayers and become fluorescent. The GP value is higher for rigid/ordered lipid membranes, as fewer water molecules have access to the probes embedded inside the membrane (40). The Laurdan probe inserts deep in the hydrophobic core of the lipid membrane, while Prodan preferentially partitions to the lipid head groups (41).
We determined the effect of phenothiazines on the GP of liposomes composed of 100% POPC or POPC with 30 mol% cholesterol. Membranes composed of 100% POPC resemble the basal cellular membrane, while those containing additional cholesterol mimic lipid raft-containing membranes (42). Phenothiazines significantly reduced the GP of cholesterol-rich but not cholesterol-free membranes, in a dose-dependent manner (Fig. 5; see Fig. S3 in the supplemental material). These results suggest that phenothiazines specifically reduce the rigidity of cholesterol-rich membranes. Since similar GP reductions were observed for both the Laurdan and Prodan dyes, phenothiazines likely insert deep in the hydrophobic core of the lipid membrane. It is possible that phenothiazines also intercalate into POPC membranes, but since these membranes are naturally very fluidic, the presence of phenothiazines does not appear to further increase the membrane fluidity at the low concentrations used. The ability of phenothiazines to significantly reduce the GP of cholesterol-rich but not cholesterol-free membranes may account for the minimal toxicity at the tested concentrations. We also determined the effect of phenothiazines on liposomes containing both cholesterol and sphingomyelin, a composition that more closely resembles that of lipid rafts (42). Incorporation of sphingomyelin did not have much effect on GP reduction (see Fig. S3), suggesting that cholesterol may be the major effector of phenothiazines. Lipid rafts are believed to be the location of HCV-cell fusion (32). Collectively, our data indicate that an increase of lipid raft membrane fluidity could be a means through which phenothiazine-induced HCV entry inhibition occurs.
Fig 5.

Fluphenazine preferentially increases the fluidity of cholesterol-rich membranes. Liposomes composed of pure POPC or POPC with 30% cholesterol (Cho) were incubated with Laurdan (5 μM) or Prodan (15 μM) for 15 min at room temperature prior to the addition of fluphenazine (5, 10, or 20 μM) or DMSO (1.25%). The mixture was incubated at 37°C for another 30 min, and the fluorescence shifts were determined. delta GP, GPfluphenazine − GPDMSO. The values and error bars represent means and standard deviations, respectively, for 2 independent experiments.
Screening for additional HCV fusion inhibitors.
Based on the above results, we hypothesized that compounds capable of increasing the fluidity of cholesterol-rich membranes will be able to inhibit HCV entry. We developed a screening assay using cholesterol-containing liposomes incorporating the Prodan dye and then screened 2,752 compounds. One compound, topotecan, was found to preferentially increase the fluidity of cholesterol-rich membranes at concentrations comparable to those approved for therapy in humans (43) (Fig. 6A). An in vitro membrane fusion assay confirmed that topotecan inhibits HCVcc-liposome fusion (Fig. 6B). This result underscores the importance of membrane fluidity on HCV entry and validates our membrane fluidity-based screening approach for HCV entry inhibitor discovery. Topotecan dose-dependently inhibited HCVcc infection in cell culture, with an estimated EC50 of 0.2 μM (Fig. 6C) and a CC50 of 88.1 μM (see Fig. S4 in the supplemental material). However, the inhibitory effect of topotecan was due mainly to its inhibition of HCVcc replication rather than entry (Fig. 7) (see Discussion).
DISCUSSION
Insight into HCV entry gained over the last few years has allowed for the discovery and development of inhibitors acting at different stages of viral uptake. Addition of new entry inhibitors to current therapies will increase the resistance barrier, inhibit expansion of the infected pool, and reduce the rate and extent of reinfection after liver transplantation (44). We and others recently identified 3 phenothiazine compounds—trifluoperazine, fluphenazine, and prochlorperazine—as inhibitors of HCV entry (28, 29). Phenothiazines are a large class of chemicals, many of which are currently used in clinics to treat psychotic disorders (25). To determine whether other phenothiazines can also inhibit HCV infection, we tested 6 additional FDA-approved phenothiazines and similar molecules and discovered that all these molecules exhibit anti-HCV activity (Table 1). The most potent inhibitor, cis-flupentixol, exhibited an IC50 of 0.25 μM and a therapeutic index of 20. The presence of a piperazine ring at position 10 enhances but is not required for HCV entry inhibition. The presence of a trifluoromethyl group at position 2 also appears to enhance anti-HCV activity. This information should assist future structure-activity relationship studies to identify more potent phenothiazine-based anti-HCV inhibitors.
We showed that phenothiazines inhibit HCV-cell fusion by specifically interacting with the host/target membrane. Incorporation of phenothiazines into the leaflets of the target membrane increases the water permeability/fluidity of cholesterol-rich membranes (Fig. 5) and reduces the rate of virus-liposome lipid mixing and hemifusion (Fig. 4). The effects of lipid composition on viral infection, particularly the influence of cholesterol and sphingolipids, have been studied widely. Many viruses enter host cells via cholesterol-rich microdomains (lipid rafts), such as West Nile, Ebola, Marburg, herpes simplex, and vaccinia viruses, retroviruses, and alphaviruses (45). In some cases, the cholesterol dependence is due to clustering of viral receptors in the lipid raft, while in other cases it is due to specific interactions between the viral envelope glycoproteins and (a lipid of) the target membrane, as is the case for the fusion protein of alphaviruses and cholesterol (46). Concerning HCV, both phenomena are believed to occur. In vitro cell culture studies have shown that HCV entry is adversely affected by cholesterol depletion (47, 48). The cholesterol absorption receptor NPC1L1 was recently identified as an HCV entry factor (49) which forms cholesterol-enriched microdomains together with flotillins (50). Concerning the tetraspanin HCV receptor CD81, two cholesterol binding sites have been mapped in the three-dimensional model of this molecule (51). It therefore appears that cholesterol might play a role in HCV entry through the local mobilization of receptors at specific membrane regions.
Our in vitro fusion studies showed that the presence of cholesterol significantly enhances the fusion of both HCV envelope protein-pseudotyped lentiviruses and cell culture-produced virions with liposomes, further confirming the important role of cholesterol in HCV-mediated fusion (32, 33, 52). In these receptor-free assays, cholesterol is likely to play a direct role in the fusion process.
Cholesterol is one of the most important lipid species in eukaryotic cells and has several different functions. Two of the primary and essential roles of cholesterol are to decrease permeability and increase the stability of the membrane bilayer (53). Membranes rich in cholesterol have a rough surface due to clustering of cholesterol molecules into small patches (microdomains) (54) and the different membrane thicknesses of cholesterol-rich regions (55, 56). The local inhomogeneity and curvature in the target membrane can influence the early interaction of a fusion protein/peptide with the target membrane and, ultimately, virus fusion (57, 58). The ability of phenothiazines to significantly increase the fluidity of cholesterol-rich membranes indicates that these molecules may interfere with cholesterol clustering and decrease the packing of cholesterol-rich microdomains, leading to reduced local inhomogeneity, which is important for HCV fusion.
A second possibility, which is not completely independent of the first one, is that the incorporation of phenothiazines can affect interaction of the 3β-OH of cholesterol with the HCV envelope protein/fusion peptide. The 3β-OH molecule participates in H-bond interactions with the head groups of various lipids, water in the solvent, and membrane proteins. In addition, 3β-OH can influence the folding of peptides at the water-membrane interface (59). The 3β-OH in cholesterol is required for the fusion of Semliki Forest virus (60, 61) and the optimal fusion of HCV with liposomes (32). Insertion of phenothiazines into the cholesterol-rich membrane may adversely affect the interaction of 3β-OH with HCV envelope proteins, thus inhibiting HCV-cell fusion.
Building upon our observation that HCV entry can be inhibited by increasing target membrane fluidity, we developed a screen using liposomes and the lipophilic dye Prodan. We screened a library of 2,752 compounds and identified a molecule, topotecan, that preferentially increases the fluidity of cholesterol-rich membranes (Fig. 6A). Using a well-established HCVcc-liposome fusion assay (32, 33), we showed that topotecan dose-dependently inhibits HCVcc-membrane fusion in vitro, validating the screen as a tool for discovering inhibitors of virus-cell fusion (Fig. 6B). However, in cell culture assays, the anti-HCV activity of topotecan appears to derive primarily from its inhibition of HCV replication rather than entry, with an estimated EC50 of ∼0.2 μM (Fig. 7). Topotecan (Hycamtin) is a topoisomerase I inhibitor that acts by stabilizing the single-stranded DNA (ssDNA)-topoisomerase I complex and causing DNA cleavage (62). It is currently used in chemotherapy for various cancers. In addition to topoisomerase, topotecan is known to affect many other cellular pathways, including downregulation of the phosphatidylinositol 3-kinase (PI3K)–Akt signaling pathway (63) and disruption of the hypoxia inducible factor 1 (HIF-1) signaling pathway (64). Topotecan is also a substrate of the ABC transporters P-glycoprotein (P-gp/MDR1) and breast cancer resistance protein (BCRP) and is actively cleared by the cell (65). The lack of HCV entry inhibition by topotecan in cell culture assays may be due in part to active cellular extrusion and/or intracellular trafficking of the compound. Nevertheless, the ability of topotecan to inhibit HCV replication may warrant additional clinical studies of this compound.
In conclusion, our studies shed light on the mechanism of action of phenothiazines as inhibitors of HCV entry and showed, for the first time, that alteration of target host cell membrane fluidity can inhibit HCV entry. It is possible that the same mechanism is responsible for the antiviral activities of phenothiazines toward other viruses, such as inhibiting the budding of measles and herpes simplex viruses (27). Based on these insights, we developed a high-throughput screen for modulators of cholesterol-rich membrane fluidity and screened a library of 2,752 compounds. One hit from this screen—topotecan—was found to both increase the fluidity of cholesterol-rich membranes and inhibit the fusion of these membranes with HCV. Targeting an entry step independent of viral proteins may also be an effective way to retard the development of drug resistance and inhibit HCV deletion mutants, which were found to reduce the antiviral effects of interferon therapy for chronic hepatitis C patients (66). This study represents an exciting new paradigm for exploring additional membrane-targeting antivirals.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Charles Rice for providing Jc1 Gluc reporter HCV and Huh-7.5 cells. We thank Su Chiang, Tao Ren, Dave Wrobel, Jennifer Nale, and Douglas Flood at NSRB for assisting with the compound library screening.
This work was supported by the NIH, the Texas Engineering Experimental Station, the ANRS (to E.-I.P.), and NERCE (grant U54 AI057159).
Footnotes
Published ahead of print 25 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02593-12.
REFERENCES
- 1. Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41–52 [DOI] [PubMed] [Google Scholar]
- 2. Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. 2000. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 132:296–305 [DOI] [PubMed] [Google Scholar]
- 3. Poynard T, Yuen MF, Ratziu V, Lai CL. 2003. Viral hepatitis C. Lancet 362:2095–2100 [DOI] [PubMed] [Google Scholar]
- 4. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 5. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fried MW. 2002. Side effects of therapy of hepatitis C and their management. Hepatology 36:S237–S244 [DOI] [PubMed] [Google Scholar]
- 8. Welsch C, Jesudian A, Zeuzem S, Jacobson I. 2012. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut 61(Suppl 1):i36–i46 [DOI] [PubMed] [Google Scholar]
- 9. Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB, American Association for Study of Liver Diseases 2011. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:1433–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Asselah T, Marcellin P. 2012. Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver Int. 32(Suppl 1):88–102 [DOI] [PubMed] [Google Scholar]
- 11. Behrens SE, Tomei L, DeFrancesco R. 1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 15:12–22 [PMC free article] [PubMed] [Google Scholar]
- 12. Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17:589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Samreen B, Khaliq S, Ashfaq UA, Khan M, Afzal N, Shahzad MA, Riaz S, Jahan S. 2012. Hepatitis C virus entry: role of host and viral factors. Infect. Genet. Evol. 12:1699–1709 [DOI] [PubMed] [Google Scholar]
- 14. Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, Vitelli A, Nicosia A. 2007. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J. Virol. 81:8063–8071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meuleman P, Hesselgesser J, Paulson M, Vanwolleghem T, Desombere I, Reiser H, Leroux-Roels G. 2008. Anti-Cd81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 48:1761–1768 [DOI] [PubMed] [Google Scholar]
- 16. Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, Schnober EK, Mee C, Soulier E, Royer C, Lambotin M, Grunert F, Thi VLD, Dreux M, Cosset FL, McKeating JA, Schuster C, Baumert TF. 2010. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 51:1144–1157 [DOI] [PubMed] [Google Scholar]
- 17. Coburn GA, Fisch DN, Moorji SM, de Muys JM, Murga JD, Paul D, Provoncha KP, Rotshteyn Y, Han AQ, Qian D, Maddon PJ, Olson WC. 2012. Novel small-molecule inhibitors of hepatitis C virus entry block viral spread and promote viral clearance in cell culture. PLoS One 7:e35351 doi:10.1371/journal.pone.0035351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baldick CJ, Wichroski MJ, Pendri A, Walsh AW, Fang J, Mazzucco CE, Pokornowski KA, Rose RE, Eggers BJ, Hsu M, Zhai W, Zhai G, Gerritz SW, Poss MA, Meanwell NA, Cockett MI, Tenney DJ. 2010. A novel small molecule inhibitor of hepatitis C virus entry. PLoS Pathog. 6:e1001086 doi:10.1371/journal.ppat.1001086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, Manns MP, Ott M, Wedemeyer H, Meuleman P, Pietschmann T, Steinmann E. 2011. The green tea polyphenol epigallocatechin-3-gallate (EGCG) inhibits hepatitis C virus (HCV) entry. Hepatology 54:1947–1955 [DOI] [PubMed] [Google Scholar]
- 20. Calland N, Albecka A, Belouzard S, Wychowski C, Duverlie G, Descamps V, Hober D, Dubuisson J, Rouille Y, Seron K. 2012. (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 55:720–729 [DOI] [PubMed] [Google Scholar]
- 21. Wagoner J, Negash A, Kane OJ, Martinez LE, Nahmias Y, Bourne N, Owen DM, Grove J, Brimacombe C, McKeating JA, Pecheur EI, Graf TN, Oberlies NH, Lohmann V, Cao F, Tavis JE, Polyak SJ. 2010. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 51:1912–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haid S, Novodomska A, Gentzsch J, Grethe C, Geuenich S, Bankwitz D, Chhatwal P, Jannack B, Hennebelle T, Bailleul F, Keppler OT, Poenisch M, Bartenschlager R, Hernandez C, Lemasson M, Rosenberg AR, Wong-Staal F, Davioud-Charvet E, Pietschmann T. 2012. A plant-derived flavonoid inhibits entry of all HCV genotypes into human hepatocytes. Gastroenterology 143:213–222 [DOI] [PubMed] [Google Scholar]
- 23. Wagoner J, Morishima C, Graf TN, Oberlies NH, Teissier E, Pecheur EI, Tavis JE, Polyak SJ. 2011. Differential in vitro effects of intravenous versus oral formulations of silibinin on the HCV life cycle and inflammation. PLoS One 6:e16464 doi:10.1371/journal.pone.0016464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Teissier E, Zandomeneghi G, Loquet A, Lavillette D, Lavergne JP, Montserret R, Cosset FL, Bockmann A, Meier BH, Penin F, Pecheur EI. 2011. Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS One 6:e15874 doi:10.1371/journal.pone.0015874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pluta K, Morak-Mlodawska B, Jelen M. 2011. Recent progress in biological activities of synthesized phenothiazines. Eur. J. Med. Chem. 46:3179–3189 [DOI] [PubMed] [Google Scholar]
- 26. Takacs D, Cerca P, Martins A, Riedl Z, Hajos G, Molnar J, Viveiros M, Couto I, Amaral L. 2011. Evaluation of forty new phenothiazine derivatives for activity against intrinsic efflux pump systems of reference Escherichia coli, Salmonella Enteritidis, Enterococcus faecalis and Staphylococcus aureus strains. In Vivo 25:719–724 [PubMed] [Google Scholar]
- 27. Amaral L, Viveiros M, Molnar J. 2004. Antimicrobial activity of phenothiazines. In Vivo 18:725–731 [PubMed] [Google Scholar]
- 28. Chockalingam K, Simeon RL, Rice CM, Chen Z. 2010. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc. Natl. Acad. Sci. U. S. A. 107:3764–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gastaminza P, Whitten-Bauer C, Chisari FV. 2010. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. U. S. A. 107:291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 31. Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lavillette D, Bartosch B, Nourrisson D, Verney G, Cosset FL, Penin F, Pecheur EI. 2006. Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J. Biol. Chem. 281:3909–3917 [DOI] [PubMed] [Google Scholar]
- 33. Haid S, Pietschmann T, Pecheur EI. 2009. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J. Biol. Chem. 284:17657–17667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parasassi T, De Stasio G, d'Ubaldo A, Gratton E. 1990. Phase fluctuation in phospholipid membranes revealed by Laurdan fluorescence. Biophys. J. 57:1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takeuchi T, Katsume A, Tanaka T, Abe A, Inoue K, Tsukiyama-Kohara K, Kawaguchi R, Tanaka S, Kohara M. 1999. Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology 116:636–642 [DOI] [PubMed] [Google Scholar]
- 36. Shulla A, Randall G. 2012. Hepatitis C virus-host interactions, replication, and viral assembly. Curr. Opin. Virol. 2:725–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chamoun AM, Chockalingam K, Bobardt M, Simeon R, Chang J, Gallay P, Chen Z. 2012. PD 404,182 is a virocidal small molecule that disrupts hepatitis C virus and human immunodeficiency virus. Antimicrob. Agents Chemother. 56:672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun X, Yau VK, Briggs BJ, Whittaker GR. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 338:53–60 [DOI] [PubMed] [Google Scholar]
- 39. Wesolowska O, Michalak K, Hendrich AB. 2011. Direct visualization of phase separation induced by phenothiazine-type antipsychotic drugs in model lipid membranes. Mol. Membr. Biol. 28:103–114 [DOI] [PubMed] [Google Scholar]
- 40. Fadel O, El Kirat K, Morandat S. 2011. The natural antioxidant rosmarinic acid spontaneously penetrates membranes to inhibit lipid peroxidation in situ. Biochim. Biophys. Acta 1808:2973–2980 [DOI] [PubMed] [Google Scholar]
- 41. Parasassi T, Krasnowska EK, Bagatolli L, Gratton E. 1998. LAURDAN and PRODAN as polarity-sensitive fluorescent membrane probes. J. Fluoresc. 8:365–373 [Google Scholar]
- 42. Pike LJ. 2003. Lipid rafts: bringing order to chaos. J. Lipid Res. 44:655–667 [DOI] [PubMed] [Google Scholar]
- 43. Freeman BB, Iacono LC, Panetta JC, Gajjar A, Stewart CF. 2006. Using plasma topotecan pharmacokinetics to estimate topotecan exposure in cerebrospinal fluid of children with medulloblastoma. Neuro-Oncol. 8:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong-Staal F, Syder AJ, McKelvy JF. 2010. Targeting HCV entry for development of therapeutics. Viruses 2:1718–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Teissier E, Pecheur EI. 2007. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur. Biophys. J. 36:887–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kielian M, Chatterjee PK, Gibbons DL, Lu YE. 2000. Specific roles for lipids in virus fusion and exit. Examples from the alphaviruses. Subcell. Biochem. 34:409–455 [DOI] [PubMed] [Google Scholar]
- 47. Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 81:374–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, Lai MM, Miyamura T, Wakita T, Suzuki T. 2008. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 82:5715–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sainz B, Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18:281–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ge L, Qi W, Wang LJ, Miao HH, Qu YX, Li BL, Song BL. 2011. Flotillins play an essential role in Niemann-Pick C1-like 1-mediated cholesterol uptake. Proc. Natl. Acad. Sci. U. S. A. 108:551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seigneuret M. 2006. Complete predicted three-dimensional structure of the facilitator transmembrane protein and hepatitis C virus receptor CD81: conserved and variable structural domains in the tetraspanin superfamily. Biophys. J. 90:212–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lavillette D, Pecheur EI, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset FL. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 81:8752–8765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rawat SS, Viard M, Gallo SA, Rein A, Blumenthal R, Puri A. 2003. Modulation of entry of enveloped viruses by cholesterol and sphingolipids. Mol. Membr. Biol. 20:243–254 [DOI] [PubMed] [Google Scholar]
- 54. Wennberg CL, van der Spoel D, Hub JS. 2012. Large influence of cholesterol on solute partitioning into lipid membranes. J. Am. Chem. Soc. 134:5351–5361 [DOI] [PubMed] [Google Scholar]
- 55. Spector AA, Yorek MA. 1985. Membrane lipid composition and cellular function. J. Lipid Res. 26:1015–1035 [PubMed] [Google Scholar]
- 56. McIntosh TJ. 1978. The effect of cholesterol on the structure of phosphatidylcholine bilayers. Biochim. Biophys. Acta 513:43–58 [DOI] [PubMed] [Google Scholar]
- 57. Stiasny K, Heinz FX. 2004. Effect of membrane curvature-modifying lipids on membrane fusion by tick-borne encephalitis virus. J. Virol. 78:8536–8542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pecheur EI, Martin I, Bienvenue A, Ruysschaert JM, Hoekstra D. 2000. Protein-induced fusion can be modulated by target membrane lipids through a structural switch at the level of the fusion peptide. J. Biol. Chem. 275:3936–3942 [DOI] [PubMed] [Google Scholar]
- 59. Rog T, Pasenkiewicz-Gierula M, Vattulainen I, Karttunen M. 2007. What happens if cholesterol is made smoother: importance of methyl substituents in cholesterol ring structure on phosphatidylcholine-sterol interaction. Biophys. J. 92:3346–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kielian MC, Helenius A. 1984. Role of cholesterol in fusion of Semliki Forest virus with membranes. J. Virol. 52:281–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marquardt MT, Phalen T, Kielian M. 1993. Cholesterol is required in the exit pathway of Semliki Forest virus. J. Cell Biol. 123:57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lorusso D, Pietragalla A, Mainenti S, Masciullo V, Di Vagno G, Scambia G. 2010. Role of topotecan in gynaecological cancers: current indications and perspectives. Crit. Rev. Oncol. Hematol. 74:163–174 [DOI] [PubMed] [Google Scholar]
- 63. Nakashio A, Fujita N, Tsuruo T. 2002. Topotecan inhibits VEGF- and bFGF-induced vascular endothelial cell migration via downregulation of the PI3K-Akt signaling pathway. Int. J. Cancer 98:36–41 [DOI] [PubMed] [Google Scholar]
- 64. Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. 2005. Identification of a novel small-molecule inhibitor of the hypoxia-inducible factor 1 pathway. Cancer Res. 65:605–612 [PubMed] [Google Scholar]
- 65. Shen J, Carcaboso AM, Hubbard KE, Tagen M, Wynn HG, Panetta JC, Waters CM, Elmeliegy MA, Stewart CF. 2009. Compartment-specific roles of ATP-binding cassette transporters define differential topotecan distribution in brain parenchyma and cerebrospinal fluid. Cancer Res. 69:5885–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kohno T, Tsuge M, Hayes CN, Hatakeyama T, Ohnishi M, Abe H, Miki D, Hiraga N, Imamura M, Takahashi S, Ochi H, Tanaka S, Chayama K. 2012. Identification of novel HCV deletion mutants in chronic hepatitis C patients. Antivir. Ther. 17:1551–1561 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.