

Abstract
Despite β-lactamase inhibitors being available for clinical use for nearly 30 years, a paucity of data exists describing the pharmacokinetic-pharmacodynamic (PK-PD) determinants of efficacy for these agents. Herein, we describe dose fractionation studies designed to determine the exposure measure most predictive of tazobactam efficacy in combination with ceftolozane and the magnitude of this measure necessary for efficacy in a PK-PD in vitro infection model. The challenge organism panel was comprised of an isogenic CTX-M-15-producing Escherichia coli triplet set, genetically engineered to transcribe different levels of blaCTX-M-15. These recombinant strains exhibited ceftolozane MIC values of 4, 16, and 64 μg/ml representing low, moderate, and high levels of CTX-M-15, respectively. Different blaCTX-M-15 transcription levels were confirmed by relative quantitative real-time PCR (qRT-PCR) and β-lactamase hydrolytic assays. The exposure measure associated with efficacy was the percentage of the dosing interval that tazobactam concentrations remained above a threshold (%Time>threshold), regardless of enzyme expression (r2 = 0.938). The threshold concentrations identified were 0.05 μg/ml for low and moderate and 0.25 μg/ml for the high-β-lactamase expression strain constructs. The magnitudes of %Time>threshold for tazobactam associated with net bacterial stasis and a 1- and 2-log10 CFU reduction in bacteria at 24 h were approximately 35, 50, and 70%, respectively. These data provide an initial target tazobactam concentration-time profile and a paradigm to optimize tazobactam dosing when combined with ceftolozane.
INTRODUCTION
β-Lactamase inhibitors (BLIs) have been used clinically for nearly 30 years. Clavulanic acid was the first BLI discovered and, in combination with amoxicillin, was introduced to clinical practice in the mid-1980s (1). Shortly thereafter, clavulanic acid was combined with ticarcillin, an intravenous carboxypenicillin (2, 3). By the mid-1980s and early 1990s, clavulanic acid was joined in clinical practice by the penicillanic acid sulfone BLIs sulbactam and tazobactam (4, 5, 6, 7). Sulbactam was combined with the intravenous aminopenicillin ampicillin, while tazobactam was combined with the intravenous ureidopenicillin piperacillin.
Due to the rapidly rising clinical concern of β-lactamase-producing members of the family Enterobacteriaceae, new BLI/β-lactam combinations are under clinical development. Some represent novel β-lactam and BLI combinations, such as the carbapenem biapenem in combination with RPX-7009 (8, 9). Others are U.S. Food and Drug Administration (FDA) approved β-lactams, such as the antistaphylococcal cephalosporin ceftaroline and the antipseudomonal cephalosporin ceftazidime combined with a novel BLI, avibactam (10, 11). Still others are new β-lactams, such as the antipseudomonal cephalosporin ceftolozane combined with an older U.S. FDA-approved BLI, tazobactam (12).
Despite BLIs being available for nearly 3 decades, a paucity of data exists describing the pharmacokinetic-pharmacodynamic (PK-PD) determinants of efficacy for these agents. Preclinical models, such as PK-PD animal and in vitro infection models are tools that can be utilized to describe the potential efficacy of antimicrobial agents (13). Through these models, one can gain important insight about the time course of antimicrobial effect, from which exposure-response relationships can be constructed. These exposure-response relationships, in turn, can be used to determine the exposure measure predictive of efficacy and the magnitude of this measure necessary for efficacy (13).
The objectives of the PK-PD in vitro infection model studies described herein were 3-fold. The first objective was to identify the exposure measure (e.g., area under the concentration-time curve [AUC], maximal concentration [Cmax], or the percentage of the dosing interval that the drug concentration remains above a threshold concentration [%Time>threshold]) that best predicts tazobactam efficacy in combination with ceftolozane against an isogenic set of three CTX-M-15-producing Escherichia coli strains with different transcription levels of blaCTX-M-15. The second objective was to determine the magnitude of the exposure measure associated with net bacterial stasis and a 1- and 2-log10 CFU reduction in bacteria at 24 h. The third objective was to determine the impact of various β-lactamase transcription levels on the magnitude of the exposure measure associated with efficacy.
MATERIALS AND METHODS
Bacteria, antimicrobial agent, and β-lactamase inhibitor.
Ceftolozane and tazobactam were provided by Cubist Pharmaceuticals (Lexington, MA). Three isogenic E. coli strains, each producing different levels of CTX-M-15, were utilized in these studies. The blaCTX-M-15 gene and different upstream promoter regions were inserted into a promoterless cloning vector. Three distinct upstream promoter regions were designed to provide different transcription levels of blaCTX-M-15, which reflected different amounts of CTX-M-15 production. These recombinant vectors were then transformed into a clinical wild-type ceftolozane-susceptible E. coli strain known to be associated with multilocus sequence typing lineage MLST131. The transcription levels of blaCTX-M-15 in all three isolates were determined by quantitative real-time PCR (qRT-PCR) assays using an endogenous reference gene (rpsL). These levels of expression were compared to the level of expression in the E. coli strain demonstrating the lowest level of enzyme production based on MIC results and hydrolysis assays for β-lactams. The hydrolytic activity of the CTX-M-15 enzyme produced by each recombinant strain was measured by observing the changes in absorbance due to the opening of the β-lactam ring in an Ultrospec 3300 pro UV-visible spectrophotometer (GE Healthcare Biosciences, Pittsburgh, PA) during a 2-min interval. The degree of hydrolytic activity was calculated using the following formula: hydrolytic activity = [(ΔA/minute)/protein concentration] × −1,000 (factor) where ΔA is the change in absorbance and the protein concentration is given in micrograms per milliliter.
Media and in vitro susceptibility studies.
Susceptibility studies, using cation-adjusted Mueller-Hinton (MH) broth (BD Laboratories, Franklin Lakes, NJ) microdilution methods, were conducted in accordance with Clinical and Laboratory Standards Institute guidelines (14). Strain susceptibility to ceftolozane was determined alone and in combination with a fixed tazobactam concentration (4 μg/ml). Susceptibility studies were performed in triplicate over a 2-day period and presented as modes.
PK-PD in vitro model and sample processing.
The one-compartment PK-PD in vitro infection model utilized in these studies has been described previously (15). Briefly, the model consists of a central infection compartment containing growth medium, the challenge isolate, and magnetic stir bars to ensure the homogeneity of the drug(s) within the compartment. The central infection compartment was attached to a stir plate, and the entire unit was placed within a temperature- and humidity-controlled incubator set at 35°C. Drug-free growth medium was pumped into the central infection compartment via a computer-controlled peristaltic pump, while growth medium was simultaneously removed through an exit port and captured in a waste container. The challenge isolate was aseptically inoculated directly into the central infection compartment, and the peristaltic rate of diffusion was set at a flow rate that allowed for the simulation of the concentration-time profile of the drug(s) under study in humans. The test compounds were infused via computer-controlled syringe pumps that allow simulation of the desired half-lives, dosing frequencies, and concentrations. Specimens for CFU determination and drug concentration assay were collected from the central infection compartment using a sterile syringe and needle through a rubber septum at predetermined time points.
In these experiments, initial inocula of 1.0 × 106 CFU/ml of each of the challenge isolates were prepared from a culture grown overnight on subinhibitory concentrations of 1 μg/ml ceftriaxone (Sigma-Aldrich, St. Louis, MO) in MH agar (BD Laboratories). Isolates were taken from the overnight cultures and grown to mid-logarithmic phase in a flask of MH broth set in a shaking water bath at 35°C and 125 rotations per minute. The bacterial concentration within the flask of MH broth was determined by measuring the optical density and comparison to a previously confirmed growth curve for each challenge isolate.
Bacteria were then exposed to changing concentrations of ceftolozane and tazobactam simulating half-lives of 2.5 h for ceftolozane and 1 h for tazobactam in humans. One-milliliter specimens were collected for CFU determination at 0, 2, 4, 6, 8, 12, and 24 h. Each sample was centrifuged, washed, and resuspended with sterile normal saline twice to prevent drug carryover and was then cultured onto Trypticase soy agar enriched with 5% sheep blood. Plated samples were incubated at 35°C for 24 h. One-milliliter specimens for drug assay were collected at 1, 3, 5, 7, 9, 11, 13, and 24 h and then immediately frozen at −80°C until assayed for drug concentration.
Dose range studies.
Dose range studies were conducted using each isogenic E. coli strain to determine the dose-response relationship. In these studies, a range of ceftolozane and tazobactam doses were administered either alone or in combination, giving the dose every 8 h, as this is the dosing interval that is expected to be clinically applied. The ceftolozane dose ranged from 125 to 1,000 mg, while the tazobactam dose ranged from 31.25 to 2,000 mg.
Dose fractionation studies.
The ceftolozane and tazobactam dosing regimens selected for the dose fractionation studies were guided by results of the dose range studies, and those chosen were associated with one-half the maximal effect of the drug combination. The total daily tazobactam exposure, as measured by free-drug area under the concentration-time curve from 0 to 24 h (AUC0–24), was held constant but fractionated into 6-, 8-, 12-, and 24-h dosing intervals. In all tazobactam dose fractionation studies, ceftolozane was administered every 8 h. All dose fractionation studies were performed in duplicate for each of the three isogenic strains.
Analytical method.
All samples were assayed by liquid chromatography followed by tandem mass spectrometry (LC/MS/MS) (Waters, Milford, MA). Standard curves ranged from 0.1 to 500 μg/ml for ceftolozane and from 0.1 to 100 μg/ml for tazobactam. Both standard curves were linear over their respective ranges (r2 = 1.000 and 0.999) for ceftolozane and tazobactam, respectively. The lower limit of quantification was 0.1 μg/ml for both compounds. The intraday coefficient of variation (CV) for ceftolozane was ≤7.2% and ≤3.4% at concentrations of 0.3 μg/ml and 400 μg/ml, respectively; for tazobactam, intraday CVs were ≤8.5% and ≤15.8% at concentrations of 0.3 and 80 μg/ml, respectively. Interday CVs were 8.3% and 3.0% at ceftolozane concentrations of 0.3 and 400 μg/ml, respectively. For tazobactam, interday CVs were 7.0% and 13.0% at 0.3 and 80 μg/ml, respectively.
Pharmacokinetic-pharmacodynamic analysis.
Data from the dose fractionation studies were evaluated using Hill-type models and nonlinear least-squares regression. The data were weighted using the inverse of the estimated measurement variance. Relationships between change in log10 CFU at 24 h and AUC, Cmax, and %Time>threshold were evaluated. %Time>threshold was identified through an iterative process where candidate threshold concentrations of 0.01, 0.05, 0.1, 0.25, 0.5, 0.75, and 1 μg/ml were evaluated. Threshold value discrimination was based on the evaluation of the dispersion of data along the exposure axis and optimization of r2 values for the relationship between change in log10 CFU at 24 h and %Time>threshold.
Nucleotide sequence accession numbers.
The nucleotide sequences of blaCTX-M-15 and upstream promoter regions designed for constructing the plasmid vectors utilized herein have been submitted to the EMBL/GenBank/DDBJ sequence databases and assigned the accession numbers KC355190, KC355191, and KC355192.
RESULTS
In vitro susceptibility testing.
The MIC values for ceftolozane alone were determined to be 4, 16, and 64 μg/ml when tested against the low-, moderate-, and high-level CTX-M-15-producing E. coli strains, respectively. The MIC value for ceftolozane in the presence of 4 μg/ml tazobactam was 0.25 μg/ml for each of the three strains. The hydrolytic activity rates, represented here as the amount (mg) of ceftolozane hydrolyzed per minute per milligram of protein were 36, 120, and 580 for the low-, moderate-, and high-level CTX-M-15 producers, respectively. The relative transcription levels of blaCTX-M-15 detected in the moderate- and high-level CTX-M-15-producing strains were, respectively, 8.3 and 43.8-fold higher than that obtained for the isogenic strain producing the smallest amount of enzyme (Table 1).
Table 1.
Susceptibility testing results and hydrolytic activity rates for ceftolozane alone and in combination with tazobactam at 4 μg/ml against E. coli strains producing different levels of CTX-M-15a
| E. coli strain | MIC (μg/ml) |
Hydrolytic activityc | qRT-PCRd | |
|---|---|---|---|---|
| Ceftolozane alone | Ceftolozane + TAZb (4 μg/ml) | |||
| Control | 0.25 | 0.25 | −3 | ND |
| Low producer | 4 | 0.25 | 36 | 1 |
| Moderate producer | 16 | 0.25 | 120 | 8.3 |
| High producer | 64 | 0.25 | 580 | 43.9 |
The transcription levels of blaCTX-M-15 are also shown.
TAZ, tazobactam.
Hydrolytic activity rates expressed as the amount (mg) of ceftolozane hydrolyzed per minute per milligram of protein.
Expression of blaCTX-M-15 relative to the E. coli strain demonstrating the lowest CTX-M-15 production based upon MIC results and hydrolysis assays for β-lactams. ND, not detected.
Pharmacokinetics.
The targeted ceftolozane and tazobactam pharmacokinetic profiles were simulated well in the PK-PD in vitro model for all dosing regimens, as evidenced by the agreement between observed and targeted concentration-time profiles for each drug. Figure 1 shows the relationship between the observed and targeted concentrations from all dosing regimens studied for ceftolozane and tazobactam. The r2 values for these relationships were 0.972 for ceftolozane and 0.984 for tazobactam.
Fig 1.
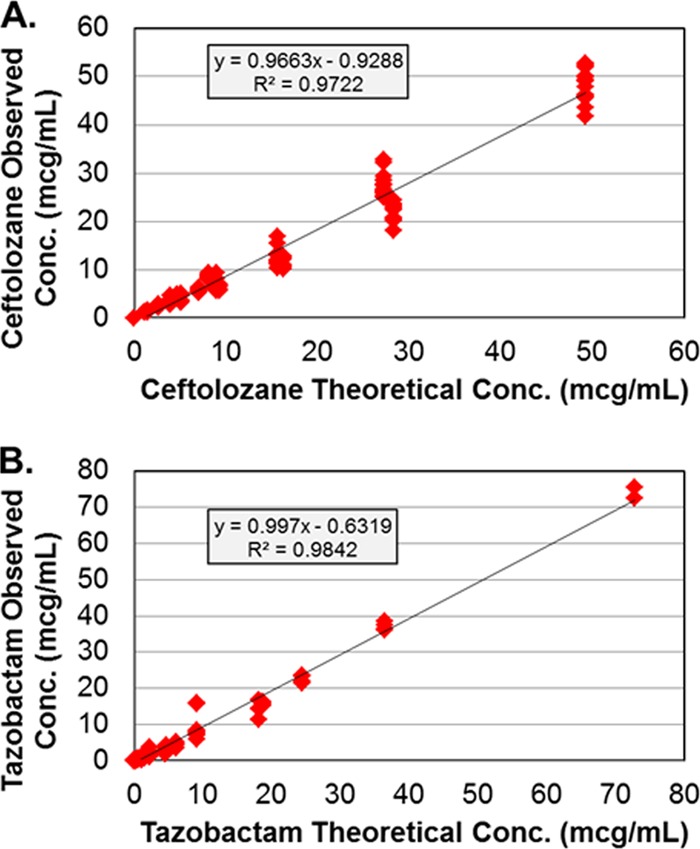
Relationships between the observed and targeted ceftolozane (A) and tazobactam (B) concentrations or regimens studied. The drug concentrations are given in micrograms per milliliter.
Dose range studies.
Bacteria in the no-treatment control arms grew well, reaching a bacterial density of greater than 1.0 × 108 CFU/ml by 12 h in each control arm. The range of tazobactam dosages used in combination with a fixed dosage of ceftolozane performed well and provided a full spectrum of drug effect. Low-tazobactam exposures resulted in CFU/ml values similar to that of the growth control by 24 h, while high-tazobactam exposures resulted in reductions of greater than 2.5 log10 CFU relative to the baseline at 24 h. Intermediate-tazobactam exposures resulted in cell kill of a magnitude that was between that observed in low- and high-exposure study arms (data not shown).
Given the dose range study results, the data were deemed sufficient to identify tazobactam dosing regimens for dose fractionation studies for each of the three isogenic strains. For the dose fractionation study, a total daily dose of 750 mg of tazobactam was chosen for evaluation of the low- and moderate-β-lactamase expression constructs, while a total daily dose of 3,000 mg of tazobactam was chosen for the evaluation of the high-β-lactamase expression construct. The ceftolozane doses given every 8 h were 125, 500, and 1,000 mg for the low-, moderate-, and high-β-lactamase expression constructs, respectively, and were constant regardless of tazobactam fractionation schedule.
Dose fractionation studies.
Averaged time-kill results from the duplicate dose fractionation studies are presented in Fig. 2 for each of the three isogenic strains. Bacteria in the no-treatment control arms grew well, reaching a bacterial density of greater than 1.0 × 108 CFU/ml by 12 h in each control arm. For each isogenic strain in the active-treatment arms, cell kill was similar over the initial 8 h of the experiment. Thereafter, between-tazobactam dosing regimen differences were noted. The 6- and 8-h tazobactam dosing regimens (tazobactam given every 6 h [q6h] or every 8 h [q8h]) performed similarly, regardless of β-lactamase enzyme construct, with almost identical cell kill (greater than 2 log10 CFU reduction) at the 24-h time point. The performances of the 12- and 24-h tazobactam dosing regimens were comparatively worse with lesser magnitudes of cell kill at 24 h, regardless of the β-lactamase enzyme construct.
Fig 2.
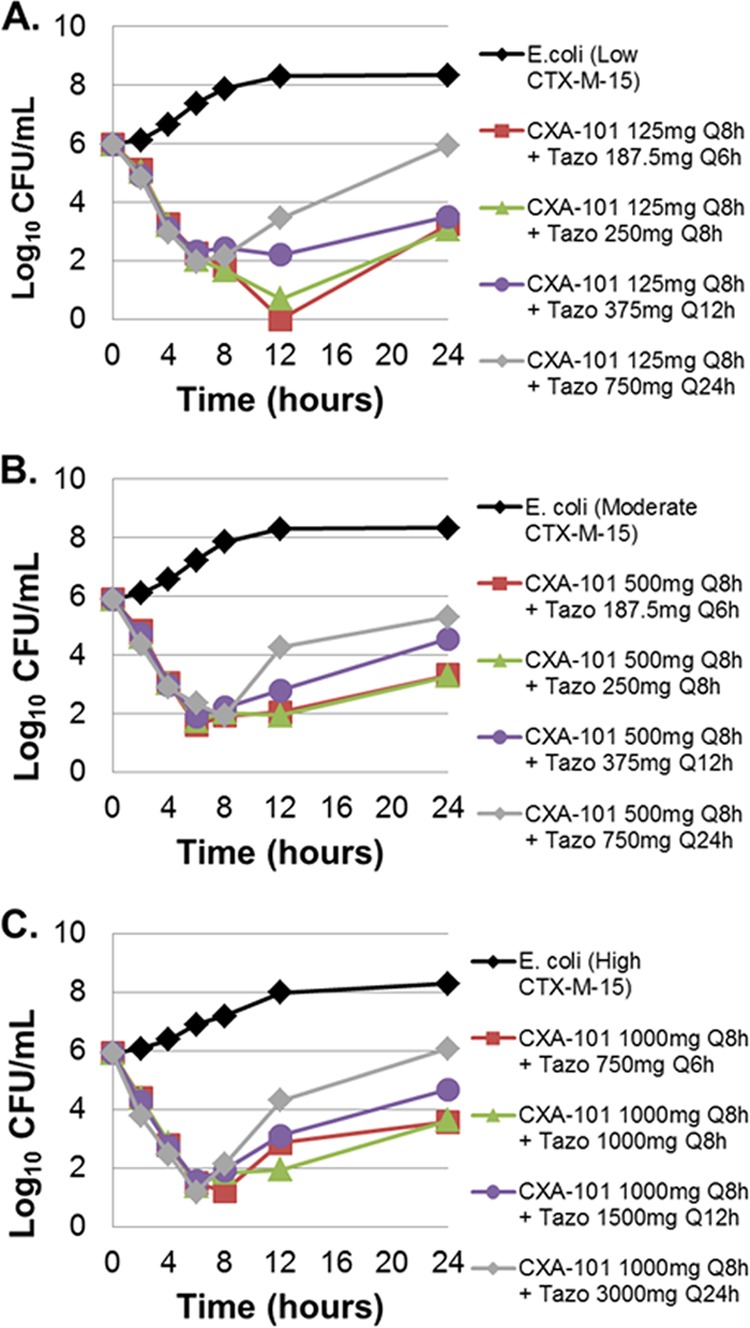
Dose fractionation study results for the low- (A), medium- (B) and high-level (C) CTX-M-15-producing E. coli. The effect of each active regimen is shown relative to the no-treatment controls. CXA-101, ceftolozane; Tazo, tazobactam; Q8h, every 8 h.
Pharmacokinetic-pharmacodynamic analysis.
The relationships between bacterial density and the three tazobactam exposure measures, AUC, Cmax, and %Time>threshold, are presented in Fig. 3. The tazobactam exposure measure that best correlated with efficacy was %Time>threshold, with an r2 value of 0.938. The threshold concentrations were 0.05 μg/ml for the low- and moderate-β-lactamase expression constructs (r2 = 0.975 and 0.972, respectively) and 0.25 μg/ml for the high-β-lactamase expression construct (r2 = 0.914). The parameter estimates (standard errors shown in the parentheses) for the relationship between change in CFU and %Time>threshold shown in Fig. 3 were the basal effect, which corresponds to the response when the exposure is zero (E0) (0.2087), maximum effect attributable to the drug (Emax) 5.8112 (0.9927), Hill's constant 2.0799 (0.8369), and 50% effective exposure (EC50) 39.7123 (7.2324).
Fig 3.
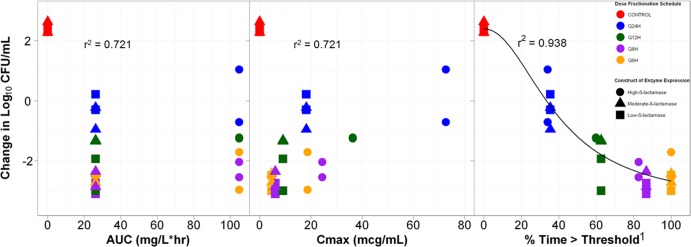
Relationships between three tazobactam exposure measures, AUC, Cmax, and %Time>threshold, and the change in log10 CFU of isogenic CTX-M-15-producing E. coli after 24 h of therapy in a PK-PD in vitro infection model. The color of the symbols represent the different dose fractionation schedules, while the shape of the symbol represents the level of β-lactamase production. Cmax is shown in micrograms per milliliter. 1, the threshold concentration was 0.05 μg/ml for the low- and moderate-β-lactamase genetic constructs and 0.25 μg/ml for the high-β-lactamase genetic construct.
Figure 4 shows the relationships between bacterial density and %Time>threshold for the low-, moderate-, and high-β-lactamase expression constructs. The %Time>thresholds that were necessary for net bacterial stasis and a 1- and 2-log10 CFU reduction in bacteria at 24 h were approximately 35, 50, and 70% of the dosing interval, respectively, regardless of enzyme production.
Fig 4.
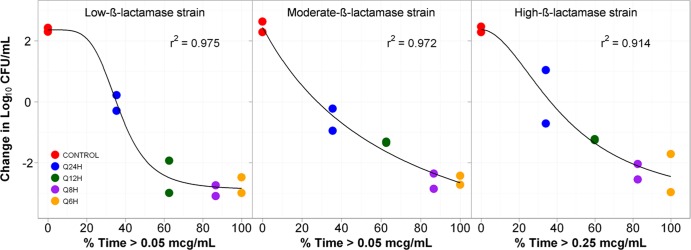
Relationships between tazobactam %Time>threshold and the change in log10 CFU of low-, medium- and high-level CTX-M-15-producing E. coli after 24 h of therapy in a PK-PD in vitro infection model. The threshold concentrations are given in micrograms per milliliter.
DISCUSSION
The objectives of this study were (i) to identify the exposure measure that best predicts tazobactam efficacy in combination with ceftolozane against an isogenic CTX-M-15-producing E. coli triplet set, which differed only in enzyme transcription levels; (ii) to determine the magnitude of the exposure measure required for efficacy; and (iii) to determine the impact of various β-lactamase transcription levels on the magnitude of the exposure measure associated with efficacy.
%Time>threshold was identified as the exposure measure for tazobactam that was most closely associated with efficacy against an isogenic challenge set of CTX-M-15-producing E. coli strains. To our knowledge, this is the first dose fractionation study to conclusively identify the exposure measure, the target threshold concentrations associated with this exposure measure and the magnitude of such an exposure measure predictive of efficacy for a BLI. In this regard, there are two publications in the literature that merit comment, as the conclusions reached by their authors conflict. One studied piperacillin in combination with tazobactam, while the other examined ceftaroline combined with avibactam (16, 17).
In the early 1990s, Strayer and colleagues studied two tazobactam administration schedules in combination with piperacillin, and their results mildly suggested AUC as the exposure measure most closely associated with tazobactam efficacy (16). As an incomplete dose fractionation study design was utilized, the results did not conclusively discriminate which exposure measure best described tazobactam efficacy. Most recently, Louie et al. utilized a dose fractionation study design to examine the PK-PD of ceftaroline combined with avibactam in a hollow-fiber infection model (17). Unfortunately, unlike the current study, the dosing regimens were administered in a manner that resulted in identical Cmax concentrations in all study arms. Thus, the authors were not able to discriminate Cmax from other exposure measures. Nonetheless, the study results did suggest that the duration of time above a threshold concentration, rather than AUC, was likely to be the exposure measure associated with avibactam efficacy. Moreover, unlike the current study, the study by Louie et al. did not identify a target threshold or a target time above that target threshold for avibactam in combination with ceftaroline and did not evaluate the impact of β-lactamase transcription level on the target threshold.
In the current study, the threshold concentration was dependent upon β-lactamase gene transcription levels. The threshold concentration was greater for the high-level CTX-M-15 producer (0.25 μg/ml) than for the low- and medium-level β-lactamase constructs (0.05 μg/ml each). Given that it is unlikely that the affinity of the BLI for the β-lactamase enzyme is lower in the high-level β-lactamase construct, the greater threshold concentration observed is likely due to the difference in the amount of enzyme. Finally, the magnitudes of the %Time>threshold for tazobactam associated with net bacterial stasis and a 1- and 2-log10 CFU reduction in bacteria at 24 h were approximately 35, 50, and 70%, respectively, regardless of enzyme transcription level.
There remain at least five important questions that were not evaluated in our study that deserve comment. First, we do not know the implication, if any, of the presence of additional β-lactamase enzymes, to which ceftolozane is stable to hydrolysis (e.g., an E. coli strain expressing TEM-1, TEM-2, and/or OXA-30), on the tazobactam exposure needed for efficacy. Second, we do not know the implication of the presence of additional β-lactamase enzymes to which ceftolozane is unstable to hydrolysis on the tazobactam exposure needed for efficacy. Third, we do not know the impact of between-strain variability on the %Time>threshold target. Due to the isogenic nature of the strains utilized in our study, the population between-strain variability could not be captured. Fourth, the tazobactam exposure that prevents the amplification of a resistant subpopulation of bacteria remains unknown. Finally, we do not know whether the threshold concentration identified for tazobactam in combination with ceftolozane would change if the BLI was paired with a different β-lactam, such as piperacillin. Each of these questions requires further study.
It is important to note that the challenge organisms selected for these studies, CTX-M-15-producing E. coli, was derived from a clinically relevant linage (i.e., MLST131 lineage). CTX-M-15 and related enzymes have proliferated not only among isolates from hospitalized patients, but also among patients with complicated community-acquired infections (18, 19). In addition, E. coli strains associated with MLST131 belong to a recently emerging lineage of virulent strains which frequently carry blaCTXM-15 (20).
Finally, it should be appreciated that the magnitudes of tazobactam exposure necessary for efficacy identified herein are influenced by the dose of ceftolozane and the shape of the cetolozane exposure. For instance, had we utilized larger ceftolozane doses or administered the partner β-lactam as a continuous infusion, it is likely a lesser tazobactam exposure would have been needed for a given amount of efficacy. Similarly, had much lower doses of the ceftolozane been utilized, larger tazobactam exposures would have been required for a given amount of efficacy.
In conclusion, we successfully utilized a PK-PD in vitro infection model to identify the exposure measure that was best associated with tazobactam efficacy in combination with ceftolozane against an isogenic CTX-M-15-producing E. coli triplet set, which differed only in enzyme expression. Moreover, we were able to identify the magnitude of tazobactam exposure necessary for efficacy. Finally, we demonstrated that β-lactamase expression had little impact on the magnitude of the exposure associated with efficacy. These data provide initial tazobactam target exposure measures associated with efficacy and a paradigm to optimize tazobactam dosing when combined with ceftolozane.
ACKNOWLEDGMENTS
We thank Kim A. Charpentier at the Institute for Clinical Pharmacodynamics (ICPD) (Latham, NY, USA) and Lalitagauri M. Deshpande at JMI Laboratories (North Liberty, IA, USA) for manuscript assistance and technical support.
This study was sponsored by Cubist Inc., Lexington, MA.
The Institute for Clinical Pharmacodynamics (B.V., C.C.B., O.O.O., S.M.B., A.F., and P.G.A.) has received research support from Achaogen, Astellas, AstraZeneca, Basilea Pharmaceutica, Bayer HealthCare, Bristol-Meyers Squibb, Cempra Pharmaceuticals, Cerexa, Cubist Pharmaceuticals, Durata Pharmaceuticals, Fedora Pharmaceuticals, Forest Research Institute, Furiex Pharmaceuticals, GlaxoSmithKline, Meiji Seika Pharma, Nabriva Therapeutics, Nimbus, Pfizer, PolyMedix, Rib-X, Roche Bioscience, Rock Therapeutics, Tetraphase Pharmaceuticals, and The Medicines Company. JMI Laboratories, Inc. (R.E.M., M.C., and R.N.J.) has received research and educational grants in 2009 to 2012 from American Proficiency Institute (API), Anacor, Astellas, AstraZeneca, Bayer, Cempra, Cerexa, Contrafect, Cubist, Daiichi, Dipexium, Enanta, Furiex, GlaxoSmithKline, Johnson & Johnson (Ortho McNeil), LegoChem Biosciences Inc., Meiji Seika Kaisha, Merck, Nabriva, Novartis, Pfizer (Wyeth), Rempex, Rib-X Pharmaceuticals, Seachaid, Shionogi, The Medicines Company, Theravance, and ThermoFisher. Some JMI employees are advisors/consultants for Astellas, Cubist, Pfizer, Cempra, Cerexa-Forest, J&J, and Theravance. The JMI employee authors have no conflicts of interest regarding speaker fees and stock options. L.V.F. and J.N.S. are both employees and stockholders of Cubist Pharmaceuticals.
Footnotes
Published ahead of print 29 April 2013
REFERENCES
- 1. Reading C, Cole M. 1977. Clavulanic acid: a beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 11:852–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. File TM, Jr, Tan TS, Salstrom SJ, Johnson LA, Douglas GF. 1984. Timentin versus piperacillin or moxalactam in the therapy of acute bacterial infections. Antimicrob. Agents Chemother. 26:310–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Todd PA, Benfield P.1990. Amoxicillin/clavulanic acid. An update of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 39:264–307 [DOI] [PubMed] [Google Scholar]
- 4. Davies BI, Maesen FP, van Noord JA. 1984. Clinical, bacteriological and pharmacokinetic results from an open trial of sultamicillin in patients with acute exacerbations of chronic bronchitis. J. Antimicrob. Chemother. 13:161–170 [DOI] [PubMed] [Google Scholar]
- 5. Jackson D, Cockburn A, Cooper DL, Langley PF, Tasker TC, White DJ. 1985. Clinical pharmacology and safety evaluation on timentin. Am. J. Med. 79:44–55 [DOI] [PubMed] [Google Scholar]
- 6. Gould IM, Ansari A, Harvey G, Douglas JG, Smith CC, Reid TM. 1991. Piperacillin/tazobactam in the treatment of serious acute soft tissue infection. Drugs Exp. Clin. Res. 17:187–190 [PubMed] [Google Scholar]
- 7. Fuchs PC, Barry AL, Jones RN. 1985. In vitro activity and disk susceptibility of timentin: current status. Am. J. Med. 79:25–32 [DOI] [PubMed] [Google Scholar]
- 8. Castanheira A, Becker HK, Rhomberg PR, Jones RN. 2012. Pre-clinical evaluation of a carbapenem/β-lactamase inhibitor combination (RPX2003/RPX7009) tested against serine-carbapenemase-producing pathogens, abstr F-856. Abstr. 52nd Annu. Intersci. Conf. Antimicrob. Agents Chemother American Society for Microbiology, Washington, DC [Google Scholar]
- 9. Rafailidis PI, Ioannidou EN, Falagas ME. 2007. Ampicillin/sulbactam: current status in severe bacterial infections. Drugs 67:1829–1849 [DOI] [PubMed] [Google Scholar]
- 10. Castanheira M, Sader HS, Farrell DJ, Mendes RE, Jones RN. 2012. Activity of ceftaroline-avibactam tested against Gram-negative organism populations, including strains expressing one or more β-lactamases and methicillin-resistant Staphylococcus aureus carrying various staphylococcal cassette chromosome mec types. Antimicrob. Agents Chemother. 56:4779–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bryson HM, Brogden RN. 1994. Piperacillin/taxobactam: a review of its antibacterial activity, pharmacokinetic properties and therapeutic potential. Drugs 47:506–535 [DOI] [PubMed] [Google Scholar]
- 12. Chandorkar G, Huntington JA, Gotfried MH, Rodvold KA, Umeh O. 2012. Intrapulmonary penetration of ceftolozane/tazobactam and piperacillin/tazobactam in healthy adult subjects. J. Antimicrob. Chemother. 67:2463–2469 [DOI] [PubMed] [Google Scholar]
- 13. Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, Drusano GL. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin. Infect. Dis. 44:79–86 [DOI] [PubMed] [Google Scholar]
- 14. Clinical and Laboratory Standards Institute 2012. Performance standards for antimicrobial susceptibility testing. Standard M100-S22. 22nd supplement Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 15. Garrison MW, Vance-Bryan K, Larson TA, Toscano JP, Rotschafer JC. 1990. Assessment of effects of protein binding on daptomycin and vancomycin killing of Staphylococcus aureus by using an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 34:1925–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strayer AH, Gilbert DH, Pivarnik P, Medeiros AA, Zinner SH, Dudley MN. 1994. Pharmacodynamics of piperacillin alone and in combination with tazobactam against piperacillin-resistant and -susceptible organisms in an in vitro model of infection. Antimicrob. Agents Chemother. 38:2351–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, Vanscoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob. Agents Chemother. 56:258–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castanheira M, Mendes RE, Rhomberg PR, Jones RN. 2008. Rapid emergence of blaCTX-M among Enterobacteriaceae in U.S. medical centers: molecular evaluation from the MYSTIC Program (2007). Microb. Drug Resist. 14:211–216 [DOI] [PubMed] [Google Scholar]
- 19. Peirano G, Pitout JD. 2010. Molecular epidemiology of Escherichia coli producing CTX-M β-lactamases: the worldwide emergence of clone ST131 O25:H4. Int. J. Antimicrob. Agents 35:316–321 [DOI] [PubMed] [Google Scholar]
- 20. Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. 2010. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. coli infections in the United States. Clin. Infect. Dis. 51:286–294 [DOI] [PubMed] [Google Scholar]