

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) and Streptococcus pyogenes (group A streptococcus [GrAS]) cause serious and sometimes fatal human diseases. They are among the many Gram-positive pathogens for which resistance to leading antibiotics has emerged. As a result, alternative therapies need to be developed to combat these pathogens. We have identified a novel bacteriophage lysin (PlySs2), derived from a Streptococcus suis phage, with broad lytic activity against MRSA, vancomycin-intermediate S. aureus (VISA), Streptococcus suis, Listeria, Staphylococcus simulans, Staphylococcus epidermidis, Streptococcus equi, Streptococcus agalactiae (group B streptococcus [GBS]), S. pyogenes, Streptococcus sanguinis, group G streptococci (GGS), group E streptococci (GES), and Streptococcus pneumoniae. PlySs2 has an N-terminal cysteine-histidine aminopeptidase (CHAP) catalytic domain and a C-terminal SH3b binding domain. It is stable at 50°C for 30 min, 37°C for >24 h, 4°C for 15 days, and −80°C for >7 months; it maintained full activity after 10 freeze-thaw cycles. PlySs2 at 128 μg/ml in vitro reduced MRSA and S. pyogenes growth by 5 logs and 3 logs within 1 h, respectively, and exhibited a MIC of 16 μg/ml for MRSA. A single, 2-mg dose of PlySs2 protected 92% (22/24) of the mice in a bacteremia model of mixed MRSA and S. pyogenes infection. Serially increasing exposure of MRSA and S. pyogenes to PlySs2 or mupirocin resulted in no observed resistance to PlySs2 and resistance to mupirocin. To date, no other lysin has shown such notable broad lytic activity, stability, and efficacy against multiple, leading, human bacterial pathogens; as such, PlySs2 has all the characteristics to be an effective therapeutic.
INTRODUCTION
Gram-positive pathogens such as Streptococcus pyogenes (group A streptococci [GrAS]), Staphylococcus aureus, Streptococcus agalactiae (group B streptococci [GBS]), and Listeria monocytogenes are responsible for millions of serious and sometimes fatal infections worldwide. Additionally, resistance to conventional antibiotics has been on the rise, resulting in increased infection rates, morbidity, mortality, and treatment costs. Consequently, new therapeutic methods need to be developed to reduce the antibiotic pressure on these pathogens.
S. pyogenes annually infects over 750 million people (1), resulting in 25% mortality among the ∼650,000 cases that progress to severe infection (1). This pathogen is responsible for a broad range of infections, such as pharyngitis, impetigo, scarlet fever, erysipelas, cellulitis, toxic shock syndrome, and necrotizing fasciitis; it can lead to serious sequelae, such as rheumatic fever and acute glomerulonephritis (2–4). S. aureus is capable of producing severe, secondary infections in immunocompromised individuals, as well as causing disease in otherwise-healthy people. Besides skin and soft tissue infections (SSTIs), S. aureus can cause sepsis, pneumonia, necrotizing fasciitis, pyomyositis, endocarditis, toxic shock syndrome, and scalded skin syndrome (5, 6). Unfortunately, many S. aureus strains, such as methicillin-resistant S. aureus (MRSA) and (less often) vancomycin-resistant S. aureus (VRSA), have acquired resistance to one or more antibiotics used as standard therapy. MRSAs account for more than 50% of hospital isolates causing pneumonia and septicemia (7), particularly in intensive care units, resulting in 30 to 40% mortality (8, 9). While health care-associated MRSA strains usually infect susceptible patients, community-associated MRSA (CA-MRSA) strains can infect healthy individuals (10–13). CA-MRSA strains are often more virulent and are capable of causing more severe diseases (14, 15).
A novel antimicrobial strategy to control resistant bacterial pathogens involves the use of lytic enzymes (also known as endolysins, or lysins) whose genes are carried by bacteriophages (or phages) (reviewed in references 16 and 17). At the end of phage replication and assembly inside a host bacterium, the progeny phage must escape. To accomplish this, phage produce lysins—peptidoglycan hydrolases that degrade the bacterial cell wall—that result in hypotonic lysis of the bacterium and release of phage progeny. When applied exogenously, these enzymes are likewise able to access the peptidoglycan layer in the Gram-positive cell envelope (due to its lack of an outer membrane) and produce the same lytic effect. While no lysin has yet been FDA approved, these enzymes could be used to treat antibiotic-resistant bacteria. Unlike antibiotics, an important feature of phage lysins is their rapid, lethal effect on bacteria (18–20). Lysins are notable for the potencies and specificities they demonstrate, generally toward the species that the phage carrying the lysin gene infects or closely related organisms (17–19, 21, 22). As such, they presumably exert a less dramatic effect on the normal flora than conventional antibiotics.
Several lysins have been developed against MRSA (21, 23, 24) and S. pyogenes (25); to date, however, no lysin has shown high lytic activity against multiple species of different bacterial pathogens. While developing a lysin with activity against the zoonotic pathogen Streptococcus suis, we discovered an enzyme (PlySs2) with activity against a wide range of Gram-positive pathogens and in vivo efficacy against MRSA and S. pyogenes. In this report, we describe the initial characterization of the first broadly acting lysin that could be used against multiple Gram-positive pathogens.
MATERIALS AND METHODS
Bacterial strains.
All strains were stored at −80°C (see Table S1 in the supplemental material). Staphylococcus, Streptococcus, Listeria, Enterococcus, Pseudomonas, and Bacillus strains were cultivated in brain heart infusion (BHI) broth, unless the medium was replaced with Mueller-Hinton (MH) medium for MIC determinations, as described below. Lactobacillus strains were cultivated in de Man, Rogosa, and Sharpe (MRS) broth (Sigma). Escherichia coli was grown in Luria-Bertani (LB) broth. All media were acquired from Becton, Dickinson, and Company (Sparks, MD), unless otherwise stated. Bacteria were propagated at 37°C and shaken at 200 rpm, if necessary.
Genomic sequence analysis and cloning of PlySs2.
The sequenced genomes of 8 S. suis isolates in GenBank were manually inspected for the presence of integrated prophage regions. If a prophage was suspected, the theoretical translations of each open reading frame (ORF) in that region were subjected to BLASTP and Pfam analyses to locate potential lysin-encoding genes.
A candidate lysin gene (PlySs2 from S. suis strain 89/1591) was PCR cloned from genomic DNA with the following primers: AATGCTAGCCTGATACACAGTTAGAGACC (forward) and CCTAAGCTTCTTTTCACAAATCATAATCCCCAG (reverse). The underlined nucleotides represent engineered restriction sites (NheI and HindIII), which were cut with the corresponding enzymes (NEB, Ipswich, MA) to clone PlySs2 into the pBAD24 expression plasmid (pBAD24_PlySs2) carrying genes for ampicillin selection and arabinose induction. The pBAD24_PlySs2 vector was transformed into E. coli TOP10 cells (Invitrogen).
Recombinant expression and purification of PlySs2.
The aforementioned clone was grown as a patch on LB agar supplemented with 0.2% arabinose, permeabilized by a 10-min exposure to chloroform vapor, and overlaid with soft agar containing heat-killed S. suis bacteria. A streptococcal clearing zone around the E. coli patch confirmed active recombinant expression of PlySs2 (26).
For PlySs2 purification, the above clone was propagated in LB broth (37°C, 220 rpm aeration) with 100 μg/ml ampicillin. Recombinant expression was induced at an optical density at 600 nm (OD600) of ∼0.8 by addition of arabinose (0.2%, final concentration). Following overnight incubation, the cells were pelleted and resuspended in 15 mM phosphate buffer (PB; pH 8.0; buffer A) supplemented with protease inhibitor cocktail tablets (Roche). Cells were lysed with an EmulsiFlex C-5 homogenizer. After debris removal via ultracentrifugation (35,000 × g, 1 h), the supernatant was adjusted to pH 7.4 with the addition of ∼4 volumes of buffer A.
The sample was passed through a HiTrap fast flow DEAE anion-exchange column (General Electric), and the flowthrough (which contained the desired PlySs2) was subjected to ammonium sulfate precipitation at 225 g/liter (40% saturation). The precipitated protein was resolubilized in 40 ml of 15 mM PB, pH 6.7 (buffer B) for every liter of initial E. coli culture. This solution was dialyzed extensively against buffer B. Finally, the dialysate was passed through a HiTrap fast flow carboxymethyl (CM) cation-exchange column (General Electric). The CM column was washed in buffer B plus 17 mM NaCl, which resulted in gradual, pure elution of PlySs2.
The presence of PlySs2 was confirmed based on lytic activity (clearing zones on agar plates containing embedded, autoclaved Pseudomonas aeruginosa [26]) and verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis with Coomassie stain. All fractions containing PlySs2 were pooled and stored at −80°C. For in vivo tests, purified PlySs2 was dialyzed (into 15 mM NaCl, 5 mM PB; pH 7.4), frozen to −80°C, lyophilized overnight, and resuspended in approximately 1/10 of the initial volume before lyophilization. A bicinchoninic acid (BCA) assay (Sigma) was used to determine the protein concentration.
PlySs2 specificity.
The OD600 of log-phase bacterial cultures was adjusted with buffer A to ∼1.0 in 96-well microtiter plates (Falcon). PlySs2, at 32 μg/ml, or buffer B control vehicle was added to each sample well. In each run, S. suis 7997 was included as a positive control. Spectrophotometric readings (OD600) of each well were taken by using a Spectramax Plus 384 apparatus (Molecular Devices) every minute over 60 min at room temperature. Lysin activity was gauged by the degree of turbidity reduction (based on the OD600) following enzyme addition.
Bactericidal assay.
Log-phase bacteria were resuspended in buffer A to an OD600 of 0.1 (0.5 McFarland; ∼108 CFU/ml), and aliquots were added to wells of a polypropylene microtiter plate (Costar). Actual inoculum titers for each experiment were derived from plating serial dilutions of each inoculum. For each organism, buffer B control vehicle or PlySs2 was added at 128 μg/ml to wells in triplicate. Plates were sealed and incubated at 37°C with agitation every 5 min for 1 h. After incubation, cells were serially diluted in 10-fold increments and plated on BHI agar. Death was calculated as follows: −log[(number of cells surviving under test condition)/(number of cells surviving under control condition)].
MIC analysis.
The protocol described by Wiegand et al. (27) was followed to determine MICs, with adjustments as detailed below. Briefly, a final suspension of ∼5 × 105 cells/ml in MHB (or BHI for S. pyogenes) plus sterile-filtered lysin or control vehicle was distributed within a 96-well microtiter plate in triplicate (27). Cells were challenged with 0.5 to 1,024 μg/ml PlySs2 in triplicate. MICs were determined by detection of cell pellet formation in the bottom of rounded wells of polystyrene plates; they were corroborated colorimetrically with alamarBlue vital dye (Invitrogen), following the manufacturer's protocol.
In vitro resistance studies.
According to an established protocol for the in vitro development of mupirocin resistance (28, 29), S. aureus CA-MRSA MW2, S. aureus MSSA 8325, and S. pyogenes MGAS 5005 were grown in the presence of PlySs2 in liquid culture. Initially, bacterial cells at 5 × 108 CFU/ml were grown overnight in the presence of 1/32× the MIC of PlySs2 against each strain (37°C; BHI broth for S. pyogenes with cap secured during gentle shaking; MHB for S. aureus with 220 rpm aeration). The cells were pelleted and subdivided into two aliquots.
One aliquot was diluted 10-fold into fresh MHB medium with double the concentration of PlySs2; a portion of the other was spread onto the surface of MHA containing the PlySs2 MIC for that species. The MIC of 4 resultant colonies was recalculated to determine if a resistant clone had emerged (defined as a 4-fold increase in the MIC). The above procedure was repeated over an 8-day period, and the concentration of PlySs2 in the liquid culture was serially doubled from 1/32× to 4× the original MIC. This process was also conducted with mupirocin for each MRSA strain, to serve as an antibiotic resistance positive control.
In vitro characterization of PlySs2.
The optimal biochemical conditions for PlySs2 enzymatic activity against log-phase pathogenic S. suis 7997 were screened using the same spectrophotometric analysis for evaluating PlySs2 specificity, as described above. The pH dependence of the enzyme was first addressed using two buffer sets with overlapping pH ranges: citrate/phosphate (pH 4.6 to 8.0) and bis-Tris propane (pH 7.0 to 9.7). Concentrations of NaCl, EDTA, and dithiothreitol (DTT) were also varied.
PlySs2 stability.
The thermal stability of PlySs2 was studied by preexposing the enzyme to temperature conditions for defined scales of time: various high temperatures for 30 min, 37°C for hours, 4°C for days, and −80°C for months. The activity of each aliquot against S. suis 7997 was determined spectrophotometrically as described above. PlySs2 activity was also tested after consecutive freeze-thaw cycles between −80°C and room temperature.
In vivo murine model.
The Rockefeller University's Institutional Animal Care and Use Committee approved all in vivo protocols. A systemic infection model described by Daniel et al. (21) was used to test the in vivo efficacy of PlySs2 against multiple Gram-positive bacteria. Briefly, 4- to 5-week-old female FVB/NJ mice (weight range, 15 to 20 g) were obtained from The Jackson Laboratory (Bar Harbor, ME). After a period of acclimation, mice were injected intraperitoneally (i.p.) with 0.5 ml of mid-log-phase (OD600, 0.5) bacteria diluted with 5% hog gastric mucin (Sigma) in saline. Bacterial suspensions contained ∼5 × 105 CFU/ml of MW2, ∼1 × 107 of S. pyogenes MGAS 5005, or a simultaneous combination of both bacteria at the above concentrations for the mixed infection experiments. Actual bacterial inoculation titers were calculated by serial dilution and plating to Columbia blood agar plates for each experiment.
Mice became bacteremic within 1 to 3 h and contained MRSA and/or S. pyogenes within multiple organs, including spleen, liver, kidney, and heart/blood (reference 21 and unpublished observations). Three hours postinfection, the animals were divided into 4 to 5 treatment groups per infection type and were administered i.p. 0.5 ml of either 20 mM phosphate buffer, 2 mg/ml of the streptococcal-specific lysin PlyC (25), 2 mg/ml of the staphylococcal-specific lysin ClyS (21), 2 to 4 mg/ml PlySs2, or a combination of 2 mg/ml PlyC and 2 mg/ml ClyS. A PlySs2 stock of 4 mg/ml was used for S. pyogenes-infected mice to increase survival over an initial 70% survival rate with treatment of 2 mg/ml mg of PlySs2 (data not shown). While this dosage was possible with PlySs2, it was above our obtainable PlyC or ClyS stock concentrations at the time.
The survival rate for each experimental group was monitored every 12 h for the first 24 h and then every 24 h for up to 10 days postinfection. The data were statistically analyzed by using Kaplan-Meier survival curves with standard errors, 95% confidence intervals, and significance levels (log rank/Mantel-Cox test) calculated using the Prism computer program (GraphPad Software, La Jolla, CA).
RESULTS
Identification of PlySs2.
PlySs2 was identified in a prophage region of a serotype 2 strain of S. suis 89/1591 (the ORF was originally annotated in GenBank as SH3 type 5 domain protein; ZP_03625529 [S. Lucas, A. Copeland, A. Lapidus, et al., unpublished data]). The putative lysin sequence corresponding to PlySs2 has the greatest homology among S. suis sequences to a surface antigen, but this sequence has only 35% identity over 53% coverage, with an E value of <10−7. On S. suis overlay plates, clearing zones formed around an E. coli strain transformed with an expression plasmid for PlySs2 (pBAD24_PlySs2), confirming the successful cloning and soluble expression of the lysin (26). Computational sequence alignment indicated that PlySs2 encodes a predicted N-terminal CHAP catalytic domain (cysteine-histidine amidohydrolase/peptidase; PF05257) and a C-terminal SH3 type 5 binding domain (PF08460) (see Fig. S1 in the supplemental material). CHAP domains are catalytically diverse and can possess either alanine-amidase activity (18) or cross-bridge endopeptidase activity (21). Based on its primary sequence, the PlySs2 CHAP domain is divergent from other database CHAP domains (all pairwise E values were <10−15), including those in characterized streptococcal (25) and staphylococcal (21) phage lysins (data not shown, but see Fig. S2 in the supplemental material).
Purification and yield of PlySs2.
With a predicted pI of 9.01, PlySs2 flowed directly through a DEAE column at pH 7.4 (see Fig. S3, lane 4, in the supplemental material), and (following an ammonium sulfate-precipitation step) eluted cleanly both in the shoulder of the flowthrough peak of a CM column and in the 17 mM NaCl wash (see Materials and Methods for details). The preparation yielded ∼60 mg of protein per liter of E. coli culture, with >99% purity (see Fig. S3, lane 6). All experiments were performed with this preparation. Concentrating PlySs2 to 20 mg/ml had no deleterious effect on solubility or activity.
Broad lytic activity.
Purified PlySs2 was tested against a wide range of bacterial species and strains to determine the range of lytic activity. Starting at an OD600 of ∼1.0, all tested strains of S. aureus, including strains resistant to methicillin, vancomycin, daptomycin, mupirocin, and lysostaphin, were reduced to an OD600 ratio ≤0.3 after lysis by PlySs2 over 30 min of exposure (Fig. 1). Readings were also taken after 60 min (see Fig. S4 in the supplemental material). The OD600 ratios of other staphylococci, including Staphylococcus simulans and Staphylococcus epidermidis, were reduced to ∼0.2.
Fig 1.

PlySs2 displayed activity against various species. Multiple strains of staphylococci (including MRSA, MSSA, and VISA), streptococci, enterococci, Listeria, bacilli, and lactobacilli were tested for susceptibility to PlySs2 activity. Escherichia and Pseudomonas were tested as Gram-negative controls. Log-phase cultures were exposed to 32 μg/ml PlySs2 for 30 min in PB (for 60-min readings) (see also Fig. S4 in the supplemental material). The final OD600 of the treated samples was divided by the final OD600 of the untreated samples to generate the normalized values. Complete lysis registered a ratio of ∼0.02. ST, serotype.
With streptococci, PlySs2 lysed most tested M serotypes of S. pyogenes, including M1, M3, M4, M6, M18, M49, and an M-negative variant, as well as unencapsulated and highly encapsulated strains, decreasing their OD600 ratio to ≤0.4. PlySs2 also exhibited strong lytic activity against S. suis, Streptococcus equi zooepidemicus, Streptococcus equi, S. agalactiae type II (encapsulated), and S. agalactiae 090R. The pathogenic Streptococcus sanguinis and group G and E streptococci were moderately sensitive to PlySs2. For Streptococcus mutans, group C streptococci, Streptococcus oralis, Streptococcus rattus, and Streptococcus sobrinus, the OD600 ratio was only reduced to between 0.7 and 0.9. PlySs2 did not reduce the OD600 ratio below 0.5 for any Streptococcus pneumoniae strains. Streptococcus gordonii was the only commensal against which PlySs2 exhibited activity (Fig. 1).
PlySs2 showed some activity against genera outside Staphylococcus and Streptococcus. While two strains of Listeria sp. were sensitive to PlySs2, other strains were not. In the Enterococcus genus, which is associated with high levels of antibiotic resistance, E. faecalis was sensitive to PlySs2 (although less so than staphylococci or streptococci), but E. faecium was not. No activity was seen against any of the different species of Bacillus, strains of lactobacilli, or Gram-negative organisms.
Efficacy of PlySs2 against Gram-positive pathogens.
PlySs2 was tested for the log fold killing of several species and strains of susceptible organisms that were tested based on the OD600 decrease (see above). At 128 μg/ml PlySs2 and with a 60-min exposure, the VISA strain was only reduced by 2 logs. However, PlySs2 reduced the viability of L. monocytogenes, S. agalactiae, S. aureus, and S. pyogenes from ∼3 to >6 logs (Fig. 2).
Fig 2.

PlySs2 was bactericidal across multiple species of bacteria. Log-phase bacteria were treated in 96-well plates with 128 μg/ml PlySs2 in buffer A for 60 min, then serially diluted and plated onto BHI agar for CFU enumeration. The log kill was calculated by comparing the difference between vehicle-treated and PlySs2-treated CFU results.
When the MIC of PlySs2 was tested against these strains, most of the values qualitatively correlated directly to the lytic and killing activities. MICs ranged from 8 to 256 μg/ml for all strains, except the VISA strain, which was not inhibited at >1,024 μg/ml (Table 1).
Table 1.
MIC of PlySs2 for various Gram-positive speciesa
| Species | Strain | MIC (μg/ml) |
|
|---|---|---|---|
| Visual | Colorimetric | ||
| L. monocytogenes | HER 1184 | 8 | 16 |
| HER 1083 | 8 | 16 | |
| S. aureus | MSSA 8325 | 16 | 16 |
| MRSA MW2 | 16 | 32 | |
| LyrA | 32 | 32 | |
| VISA III | 32 | 64 | |
| GrAS | SF370 | 128 | 128 |
| MGAS 5005 | 128 | 256 | |
| GBS | 090R | 256 | 256 |
| Type II | 512 | 512 | |
| E. coli | TOP10 | >1,024 | >1,024 |
All MICs were evaluated visually for bacterial growth and with alamarBlue vital dye (colorimetrically) at concentrations from 0.5 to 1,024 μg/ml of PlySs2 for each strain of each species listed. There was a low MIC for MRSA MW2, as expected, and a higher MIC for GrAS strain MGAS 5005. The MIC of PlySs2 for the negative control, E. coli, was above the limits of the assay. For a reference, ClyS, LysK, and CHAPK lysins have registered MICs against S. aureus strains from 30 to 80 μg/ml.
Resistance to PlySs2.
By using a published, standardized method for calculating resistance, both staphylococcal and streptococcal strains were analyzed for the development of resistance against PlySs2 by serial exposure to incrementally doubling concentrations of the lysin. Under these testing conditions, none of the S. aureus or S. pyogenes strains exposed to PlySs2 over 8 days developed resistance (defined as a 4-fold increase from the original MIC) (Fig. 3). Following the same procedure, both S. aureus strains MW2 and 8325 developed resistance to the antibiotic mupirocin (Fig. 3).
Fig 3.
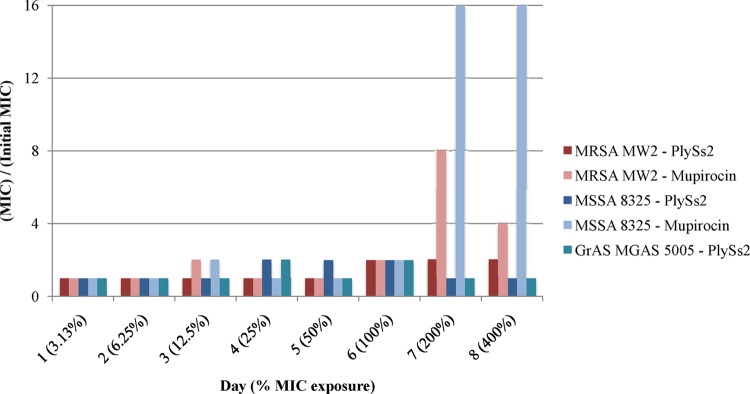
MRSA, MSSA, and GrAS did not acquire resistance to PlySs2 in vitro. MRSA strain MW2, MSSA strain 8325, and GrAS strain MGAS 5005 were exposed to 1/32× to 4× the MIC of PlySs2 and mupirocin (S. aureus strains) over 8 days. The daily MICs of PlySs2 were compared to the starting MIC of PlySs2 for each strain of bacteria to ascertain resistance. None developed resistance to PlySs2. Both MW2 and 8325 developed resistance to the positive control, mupirocin.
Biochemical characterization and stability.
PlySs2 activity was tested through a range of pH values to determine its optimum physiological buffering conditions. The lysin was most active in citrate/phosphate buffer at pH 8.0 (see Fig. S5A in the supplemental material) and in bis-Tris propane buffer at pH 9.7 (see Fig. S5B). PlySs2 could be optimally active at higher pH levels, but those levels would not be physiologically relevant. In the acidic range, there was strong activity at pH 6.0. Unlike certain other lysins, salt did not augment PlySs2 activity (see Fig. S6 in the supplemental material). Conversely, DTT did not inhibit PlySs2 function (see Fig. S7 in the supplemental material). Treatment with >4 μM EDTA had an inhibitory effect on PlySs2-induced lysis of S. suis (see Fig. S8 in the supplemental material).
When PlySs2 was incubated at different temperatures from 22°C to 85°C for 30 min, its activity was principally unaffected until 60°C (see Fig. S9A in the supplemental material). Activity was retained during incubation at 37°C for 24 h, with activity starting to diminish after 48 h (Fig. 9B). There was no observable decrease in activity after 15 days at 4°C (see Fig. S9C) or storage at −80°C for >7 months (see Fig. S9D). The lysin also endured 10 consecutive freeze-thaws cycles (−80°C to room temperature) without any observable effects (see Fig. S10 in the supplemental material).
Protection from a mixed bacterial infection.
To determine if the broad lytic activity of PlySs2 could provide in vivo protection from Gram-positive pathogens, mice were infected i.p. with either MRSA (MW2) or S. pyogenes (MGAS 5005) independently or simultaneously and subsequently treated with lysin(s). The results from 4 separate experiments were combined, and mouse survival data were plotted on a Kaplan-Meier survival curve (Fig. 4).
Fig 4.

PlySs2 protected mice from death caused by mixed MRSA and GrAS infection. FVB/NJ mice were injected i.p. with 5% mucin containing the pathogen of interest. Three hours postinfection, mice received one i.p. injection of either 20 mM phosphate buffer (control) or lysin treatment. (A) Survival data for the MRSA infection. Mice were infected with ∼5 × 105 CFU of MRSA strain MW2 and treated with either 1 mg of ClyS, 1 mg of PlyC, or 1 mg of PlySs2. (B) Survival data for the GrAS infection. Mice were infected with ∼1 × 107 GrAS strain MGAS 5005 and treated with either 1 mg of ClyS, 1 mg of PlyC, or 2 mg of PlySs2. (C) Survival data for the mixed MRSA and GrAS infection. Mice were infected with a combination of both bacteria from the above inoculums at the same concentrations. Mice were treated with either 1 mg of ClyS, 1 mg of PlyC, a combination of 1 mg of ClyS plus 1 mg of PlyC, or 2 mg of PlySs2. In all tests (A to C), mice were monitored for survival over 10 days. The results from 4 independent experiments were combined, and the data are plotted as a Kaplan-Meier survival curve; bars indicate standard errors. All the PlySs2 treatment groups (A to C) showed statistically significant differences (P < 0.0001) compared to the nonspecific single lysin or buffer controls, based on the log rank (Mantel-Cox) test.
Mice infected with MRSA alone were protected from death with either PlySs2 (89%; 16/18) or ClyS (86%; 24/28) lysins, but not the Streptococcus-specific PlyC (17%; 2/12) (Fig. 4A), while mice infected with S. pyogenes alone could only be protected with either PlySs2 (94%; 15/16) or PlyC (100%; 12/12) but not the Staphylococcus-specific ClyS (0%; 0/12) (Fig. 4B). All deaths from the MRSA infection occurred within the first 2 days; deaths from the S. pyogenes infection continued into the fourth day. In the first 24 h of infection, 4/18 (22%) control mice survived MRSA, and 11/15 (73%) control mice survived S. pyogenes, with only 1/18 (6%) and 1/15 (7%) surviving to the end of the experiment, respectively. Likewise, only 1/23 (4%) of the buffer-treated control mice survived when simultaneously infected with both MRSA and S. pyogenes. In these mixed infection experiments, single-agent therapy with either ClyS (11%; 2/18) or PlyC (17%; 3/18) was also not significantly protective. However, mice were protected from death with either a combination of ClyS and PlyC (80%; 16/20) or PlySs2 alone (92%; 22/24) over the 10-day course of the experiment (Fig. 4C).
DISCUSSION
A novel S. suis lysin, PlySs2, has demonstrated broad lytic activity against multiple Gram-positive pathogens, including S. pyogenes and S. aureus, in vitro. All previously characterized lysins, by contrast, have demonstrated activity against a narrow spectrum of species. Likewise, no lysin has been used to clear a bacteremic infection in vivo by more than one pathogenic organism; mixed infections, to our knowledge, have not been previously tested.
A noted strength of many lysins is their target specificity. Antibiotics may kill commensal organisms along with target pathogens, potentially leading to adverse sequelae (e.g., diarrhea or more serious Clostridium difficile complications). In contrast, lysins might be used to treat a single pathogen without disrupting the normal bacterial flora (17), although this specificity could admittedly be a limitation in treating multiple pathogens. Our results show it is possible that a single lysin like PlySs2 could be used to treat multiple Gram-positive pathogens, leaving many Gram-positive and all Gram-negative commensals unaffected. Furthermore, this study also demonstrated that a combination of lysins (PlyC and ClyS) with different specificities could be used effectively to treat mixed infections (Fig. 4C).
PlySs2 exhibits activity against members of two distinct phylogenetic orders: Bacillales (Staphylococcus, Listeria, etc.) and Lactobacillales (Streptococcus, Enterococcus, etc.). The peptidoglycan structures of these two orders are quite similar except for their cross-bridges, which vary widely in composition and length (30). Phage lysins have not previously displayed activity on different families or genera (and rarely on different species) (21). Furthermore, other lysins usually retain greater activity against the species infected by the phage from which the lysin was cloned, whereas PlySs2 demonstrated more activity against S. aureus than against S. suis.
All tested strains of S. aureus were highly susceptible to lysis by PlySs2 (Fig. 1), including strains resistant to methicillin, vancomycin, daptomycin, mupirocin, and lysostaphin. Its lytic activity against VISA and Newman strains was somewhat less than its lytic activity against other staphylococcal strains. The reduced activity against the VISA strains could be the result of the thicker cell wall in these organisms (31), which would increase the time necessary to result in lysis. The strong activity of PlySs2 against S. simulans and S. epidermidis supports its use in treatment of a wide array of other staphylococcal infections.
Many streptococci were susceptible to in vitro PlySs2 lysis. PlySs2 exhibited potent lytic activity against its native species, S. suis, as well as S. equi, S. equi zooepidemicus, and S. pyogenes. Of note, there was no difference in PlySs2 activity against unencapsulated or highly encapsulated variants of S. pyogenes. PlySs2 also demonstrated comparable activity against strains of GBS with or without a virulence-enhancing capsule (S. agalactiae type II and S. agalactiae 090R, respectively). Moderate activity was observed against group C, E, and G streptococci, suggesting in vivo experiments will be necessary to determine if PlySs2 can be used to treat infections by these organisms. It is unlikely that PlySs2 could be used therapeutically for S. mutans, S. oralis, S. rattus, and S. sobrinus infections, or for some strains of S. pneumoniae, because of its low activity against these pathogens. PlySs2 did not lyse any commensal lactobacilli. PlySs2 showed significant activity against other genera, killing some strains of Listeria and E. fecaelis, but not E. faecium or strains of bacilli. The ability of PlySs2 to lyse a wide range of pathogens, without lysing many commensals, suggests that certain pathogens per se share a binding receptor for this lysin.
We found that the bactericidal assays quantitatively confirmed PlySs2's ability to effectively kill strains that vary in drug resistance and encapsulation, with correlations between the lytic, bactericidal, and MIC assays. Thus, even though they were not tested, it is likely that pathogens that were found to be more sensitive to PlySs2 lysis than S. pyogenes MGAS 5005 in vitro may also be sensitive to PlySs2 in vivo. The MIC of PlySs2 against several S. aureus strains was less than or equal to the MICs of other staphylococcal lysins, including ClyS, LysK, and CHAPK (21, 29, 32, 33).
The binding domain of PlySs2 may contribute to its unique activity profile. Phage lysin binding domains have been shown to determine lysin specificity (34). As such, an SH3b (bacterial homolog of SH3) domain of a Bacillus cereus endopeptidase has been shown to bind the free amino group of the N-terminal alanine in the stem peptide of the peptidoglycan (35). Because of its ubiquity in peptidoglycans, this amine could also be the substrate for the PlySs2 SH3b domain. Accordingly, experiments are in progress to determine the binding and cleavage substrates for PlySs2.
An important finding in this study was our ability to clear mixed MRSA and S. pyogenes bacteremic infections from 92% of mice with a single dose of PlySs2. This survival rate was better than the 80% survival of mice treated with the combination of ClyS and PlyC (Fig. 4C). In a mixed infection, use of either PlyC or ClyS alone eliminated only one of the infecting pathogens, resulting in death by the other; this death occurred within the same time frame and rate as the corresponding singly infected, nontreated controls, i.e., MRSA-infected animals died within 24 h and S. pyogenes-infected animals died within 3 days. These results strongly support the idea that PlySs2 cures the animals of their infection by simultaneously killing both pathogens.
Recent studies have indicated that secondary infections caused by colonizing MRSA, S. pyogenes, or S. pneumococcus account for up to 90% of deaths from influenza pandemics (36–39). Mupirocin and polysporin are the only anti-infectives approved to reduce colonizing pathogenic bacteria on mucous membranes, but S. aureus can develop resistance to each (40). Lysins with specific activities against either staphylococci, streptococci, or pneumococci have each been shown to decolonize these pathogens in animal models of oral and nasal mucosal colonization (18, 19, 21, 22, 24, 41); however, a mixture of the three enzymes would need to be used to remove these pathogens. PlySs2 alone could be used to decolonize susceptible populations of staphylococci, streptococci, and perhaps certain pneumococcal isolates during flu season to reduce the possibility of a secondary infection.
The inability of pathogenic targets (MRSA and S. pyogenes) to establish resistance to PlySs2 (under conditions leading to mupirocin resistance) is consistent with findings for other lysins, such as PlyG (41). Antibiotic resistance may occur when bacteria either inactivate the drug or alter the target site. An extracellular lysin protease has yet to be identified, and it is less likely that a PlySs2-susceptible pathogen could easily alter the PlySs2 peptidoglycan target. To date, the only known resistance to a lysin-like molecule involves the insertion of a serine residue into an S. aureus pentaglycine cross-bridge to establish resistance to lysostaphin (a nonphage endopeptidase from S. simulans). However, it is unlikely that PlySs2 is a canonical cross-bridge endopeptidase, because of its activity against disparate bacterial species with diverse cross-bridge structures (42), including lysostaphin-resistant staphylococci.
PlySs2 is more tractable and stable than previously reported lysins (21, 25, 26). Its preparation is straightforward, yielding very pure, high yields of product in just a few steps. It remains soluble in concentrations exceeding 20 mg/ml (data not shown) and can be subjected to high or low temperatures for prolonged periods with little effect on its activity, even when repeatedly freeze-thawed. These features support PlySs2 as a suitable lysin for further development.
PlySs2 represents a novel breakthrough in the field of bacteriophage lysin technology. It is now possible to envision other lysins with broad therapeutic activities that retain specificity to a subset of mostly pathogenic Gram-positive organisms. For PlySs2, this novel capability possibly lies in the divergent PlySs2 CHAP domain and unique SH3 binding domain. PlySs2 occupies a vital space along the spectrum between strict lysin specificity and unselective antibiotic activity. Ideally, a therapeutic agent should have activity against major pathogens without affecting commensals; this report of PlySs2 is the first to indicate that a lysin could serve that function.
In summary, while pursuing a novel treatment for S. suis infection, we discovered a lysin with broad lytic activity against strains of MRSA, VISA, S. suis, Listeria, S. simulans, S. equi zooepidemicus, S. equi, S. agalactiae, S. pyogenes, S. sanguinis, S. gordonii, group G streptococci, group E streptococci, E. faecalis, and S. pneumoniae. PlySs2 was relatively simple to produce, tractable, and very stable. We demonstrated here the ability of PlySs2 to protect mice with mixed infection by MRSA and S. pyogenes. Neither of these pathogens was observed to develop resistance to PlySs2 in vitro. PlySs2 could therefore become a vital addition to the armamentarium against multidrug-resistant S. aureus, S. pyogenes, and various other Gram-positive pathogens.
Supplementary Material
ACKNOWLEDGMENTS
For their adroit technical assistance, we thank Nathan Franck, Anna Serrano, and Magid Mohamed. We are grateful to Olaf Schneewind for S. simulans, S. aureus Newman, and S. aureus lyrA; Barry Kreiswirth for S. epidermidis; Alexander Tomasz for MRSA, VISA, and other staphylococcal strains; ATCC for MRSA and streptococcal strains; Raymond Schuch of ContraFect Corporation for lactobacilli strains; Marcelo Gottschalk for S. suis 89/1591; Jaap Wagenaar for S. suis 7997; and Joseph Ferretti for S. pyogenes SF370 and NZ131.
Daniel Gilmer is a Gilliam fellow of the Howard Hughes Medical Institute. This work was supported by U.S. PHS grant AI11822 to V. A. Fischetti.
V. A. Fischetti has equity in ContraFect Corp., a company that has licensed the rights to PlySs2.
Footnotes
Published ahead of print 9 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02526-12.
REFERENCES
- 1. Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5:685–694 [DOI] [PubMed] [Google Scholar]
- 2. Swedo SE, Leonard HL, Mittleman BB, Allen AJ, Rapoport JL, Dow SP, Kanter ME, Chapman F, Zabriskie J. 1997. Identification of children with pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections by a marker associated with rheumatic fever. Am. J. Psychiatry 154:110–112 [DOI] [PubMed] [Google Scholar]
- 3. Parker MT, Tomlinson AJ, Williams RE. 1955. Impetigo contagiosa; the association of certain types of Staphylococcus aureus and of Streptococcus pyogenes with superficial skin infections. J. Hyg. (Lond.) 53:458–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bisno AL, Brito MO, Collins CM. 2003. Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3:191–200 [DOI] [PubMed] [Google Scholar]
- 5. White A, Smith J. 1963. Nasal reservoir as the source of extranasal staphylococci. Antimicrob. Agents Chemother. 161:679–683 [PubMed] [Google Scholar]
- 6. Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5:751–762 [DOI] [PubMed] [Google Scholar]
- 7. Klein E, Smith DL, Laxminarayan R. 2007. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg. Infect. Dis. 13:1840–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tiemersma EW, Bronzwaer S, Lyytikainen O, Degener JE, Schrijnemakers P, Bruinsma N, Monen J, Witte W, Grundman H. 2004. Methicillin-resistant Staphylococcus aureus in Europe, 1999–2002. Emerg. Infect. Dis. 10:1627–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laupland KB, Ross T, Gregson DB. 2008. Staphylococcus aureus bloodstream infections: risk factors, outcomes, and the influence of methicillin resistance in Calgary, Canada, 2000–2006. J. Infect. Dis. 198:336. [DOI] [PubMed] [Google Scholar]
- 10. Centers for Disease Control and Prevention 1999. From the Centers for Disease Control and Prevention. Four pediatric deaths from community-acquired methicillin-resistant Staphylococcus aureus: Minnesota and North Dakota, 1997–1999. JAMA 282:1123–1125 [PubMed] [Google Scholar]
- 11. Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle-Vavra S, Leitch CD, Daum RS. 1998. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279:593–598 [DOI] [PubMed] [Google Scholar]
- 12. Zetola N, Francis JS, Nuermberger EL, Bishai WR. 2005. Community-acquired methicillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect. Dis. 5:275–286 [DOI] [PubMed] [Google Scholar]
- 13. David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23:616–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller LG, Perdreau-Remington F, Rieg G, Mehdi S, Perlroth J, Bayer AS, Tang AW, Phung TO, Spellberg B. 2005. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N. Engl. J. Med. 352:1445–1453 [DOI] [PubMed] [Google Scholar]
- 15. Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, Chambers HF, Lu Y, Otto M. 2009. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 106:5883–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Flaherty S, Ross RP, Coffey A. 2009. Bacteriophage and their lysins for elimination of infectious bacteria. FEMS Microbiol. Rev. 33:801–819 [DOI] [PubMed] [Google Scholar]
- 17. Fischetti VA. 2008. Bacteriophage lysins as effective antibacterials. Curr. Opin. Microbiol. 11:393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nelson D, Loomis L, Fischetti VA. 2001. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. U. S. A. 98:4107–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Loeffler JM, Nelson D, Fischetti VA. 2001. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 294:2170–2172 [DOI] [PubMed] [Google Scholar]
- 20. Fischetti VA. 2005. Bacteriophage lytic enzymes: novel anti-infectives. Trends Microbiol. 13:491–496 [DOI] [PubMed] [Google Scholar]
- 21. Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA. 2010. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 54:1603–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng Q, Nelson D, Zhu S, Fischetti VA. 2005. Removal of group B streptococci colonizing the vagina and oropharynx of mice with a bacteriophage lytic enzyme. Antimicrob. Agents Chemother. 49:111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Flaherty S, Coffey A, Meaney W, Fitzgerald GF, Ross RP. 2005. The recombinant phage lysin LysK has a broad spectrum of lytic activity against clinically relevant staphylococci, including methicillin-resistant Staphylococcus aureus. J. Bacteriol. 187:7161–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rashel M, Uchiyama J, Ujihara T, Uehara Y, Kuramoto S, Sugihara S, Yagyu K, Muraoka A, Sugai M, Hiramatsu K, Honke K, Matsuzaki S. 2007. Efficient elimination of multidrug-resistant Staphylococcus aureus by cloned lysin derived from bacteriophage ϕMR11. J. Infect. Dis. 196:1237–1247 [DOI] [PubMed] [Google Scholar]
- 25. Nelson D, Schuch R, Chahales P, Zhu S, Fischetti VA. 2006. PlyC: a multimeric bacteriophage lysin. Proc. Natl. Acad. Sci. U. S. A. 103:10765–10770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y, Sun JH, Lu CP. 2009. Purified recombinant phage lysin LySMP: an extensive spectrum of lytic activity for swine streptococci. Curr. Microbiol. 58:609–615 [DOI] [PubMed] [Google Scholar]
- 27. Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163–175 [DOI] [PubMed] [Google Scholar]
- 28. Rouse MS, Rotger M, Piper KE, Steckelberg JM, Scholz M, Andrews J, Patel R. 2005. In vitro and in vivo evaluations of the activities of lauric acid monoester formulations against Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3187–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA. 2011. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob. Agents Chemother. 55:738–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schleifer KH, Kandler O. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36:407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sieradzki K, Tomasz A. 2003. Alterations of cell wall structure and metabolism accompany reduced susceptibility to vancomycin in an isogenic series of clinical isolates of Staphylococcus aureus. J. Bacteriol. 185:7103–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fenton M, Ross RP, McAuliffe O, O'Mahony J, Coffey A. 2011. Characterization of the staphylococcal bacteriophage lysin CHAP(K). J. Appl. Microbiol. 111:1025–1035 [DOI] [PubMed] [Google Scholar]
- 33. Becker SC, Dong S, Baker JR, Foster-Frey J, Pritchard DG, Donovan DM. 2009. LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol. Lett. 294:52–60 [DOI] [PubMed] [Google Scholar]
- 34. Hermoso JA, Monterroso B, Albert A, Galan B, Ahrazem O, Garcia P, Martinez-Ripoll M, Garcia JL, Menendez M. 2003. Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp-1. Structure 11:1239–1249 [DOI] [PubMed] [Google Scholar]
- 35. Xu Q, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Cai X, Carlton D, Chen C, Chiu HJ, Chiu M, Clayton T, Das D, Deller MC, Duan L, Ellrott K, Farr CL, Feuerhelm J, Grant JC, Grzechnik A, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Lam WW, Marciano D, Miller MD, Morse AT, Nigoghossian E, Nopakun A, Okach L, Puckett C, Reyes R, Tien HJ, Trame CB, van den Bedem H, Weekes D, Wooten T, Yeh A, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. 2010. Structure of the gamma-D-glutamyl-L-diamino acid endopeptidase YkfC from Bacillus cereus in complex with L-Ala-γ-D-Glu: insights into substrate recognition by NlpC/P60 cysteine peptidases. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 66:1354–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morens DM, Taubenberger JK, Fauci AS. 2009. The persistent legacy of the 1918 influenza virus. N. Engl. J. Med. 361:225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brundage JF, Shanks GD. 2007. What really happened during the 1918 influenza pandemic? The importance of bacterial secondary infections. J. Infect. Dis. 196:1717–1718 [DOI] [PubMed] [Google Scholar]
- 38. Brundage JF, Shanks GD. 2008. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg. Infect. Dis. 14:1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hussell T, Wissinger E, Goulding J. 2009. Bacterial complications during pandemic influenza infection. Future Microbiol. 4:269–272 [DOI] [PubMed] [Google Scholar]
- 40. Hudson IR. 1994. The efficacy of intranasal mupirocin in the prevention of staphylococcal infections: a review of recent experience. J. Hosp. Infect. 27:81–98 [DOI] [PubMed] [Google Scholar]
- 41. Schuch R, Nelson D, Fischetti VA. 2002. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889 [DOI] [PubMed] [Google Scholar]
- 42. Robinson JM, Hardman JK, Sloan GL. 1979. Relationship between lysostaphin endopeptidase production and cell wall composition in Staphylococcus staphylolyticus. J. Bacteriol. 137:1158–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.