

Abstract
Elvitegravir (EVG) is an effective HIV-1 integrase (IN) strand transfer inhibitor (INSTI) in advanced clinical development. Primary INSTI resistance-associated mutations (RAMs) at six IN positions have been identified in HIV-1-infected patients failing EVG-containing regimens in clinical studies: T66I/A/K, E92Q/G, T97A, S147G, Q148R/H/K, and N155H. In this study, the effect of these primary IN mutations, alone and in combination, on susceptibility to the INSTIs EVG, raltegravir (RAL), and dolutegravir (DTG); IN enzyme activities; and viral replication fitness was characterized. Recombinant viruses containing the six most common mutations exhibited a range of reduced EVG susceptibility: 92-fold for Q148R, 30-fold for N155H, 26-fold for E92Q, 10-fold for T66I, 4-fold for S147G, and 2-fold for T97A. Less commonly observed primary IN mutations also showed a range of reduced EVG susceptibilities: 40- to 94-fold for T66K and Q148K and 5- to 10-fold for T66A, E92G, and Q148H. Some primary IN mutations exhibited broad cross-resistance between EVG and RAL (T66K, E92Q, Q148R/H/K, and N155H), while others retained susceptibility to RAL (T66I/A, E92G, T97A, and S147G). Dual combinations of primary IN mutations further reduced INSTI susceptibility, replication capacity, and viral fitness relative to either mutation alone. Susceptibility to DTG was retained by single primary IN mutations but reduced by dual mutation combinations with Q148R. Primary EVG RAMs also diminished IN enzymatic activities, concordant with their structural proximity to the active site. Greater reductions in viral fitness of dual mutation combinations may explain why some primary INSTI RAMs do not readily coexist on the same HIV-1 genome but rather establish independent pathways of resistance to EVG.
INTRODUCTION
Advances in antiretroviral (ARV) therapy have resulted in the maintenance and improvement of quality of life for HIV-1-infected patients. Current recommended regimens of highly active antiretroviral therapy (HAART) include a combination of two nucleoside/nucleotide reverse transcriptase (RT) inhibitors (NRTIs) plus a protease inhibitor (PI), nonnucleoside reverse transcriptase inhibitor (NNRTI), or integrase strand transfer inhibitor (INSTI) (1). However, successful control of HIV-1 infection requires long-term therapy and continues to be hampered by inadequate drug adherence, poor tolerability, and the emergence of drug-resistant variants. Thus, new effective ARV drugs and greater availability of convenient, well-tolerated, single-tablet regimens are needed to provide sustained efficacy with favorable tolerability and safety profiles for patients with HIV-1 infection.
Integration of the double-stranded DNA product of reverse transcription into the host cell chromosome is an essential step in the life cycle of HIV-1 and is a validated target for therapeutic intervention (2). The tetrameric HIV-1 integrase enzyme (IN) catalyzes integration in a two-step reaction. First, 3′ processing results in the removal of a guanosine-thymidine (GT) dinucleotide from the 3′ end of the viral DNA long terminal repeat (LTR) sequences. Second, strand transfer results in the concerted, yet staggered, insertion of both viral DNA ends into the host cell genome (3). Both reactions require divalent cations (Mg2+ or Mn2+) in the IN active site coordinated by a catalytic triad of carboxylate amino acid residues (D64, D116, and E152).
INSTIs have demonstrated efficacy in vitro and in clinical trials in both ARV-naive patients and treatment-experienced patients, the latter carrying HIV-1 strains resistant to other ARV drug classes. Raltegravir (RAL) was the first INSTI approved for combination therapy with other ARVs for treatment-naive and -experienced HIV-1-infected patients, requiring twice-daily dosing (4, 5). Elvitegravir (EVG) and dolutegravir (DTG) are new and investigational INSTIs, respectively, with similar virologic activity in advanced clinical studies for the treatment of HIV-1 infection (6–8). As part of a once-daily ritonavir-boosted regimen, or as a component of a single-tablet regimen along with cobicistat (a pharmacoenhancer) plus the NRTI backbone of emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF), EVG has shown durable efficacy and favorable safety and tolerability (6, 7, 9).
Consistent with other ARV drug classes, the emergence of resistance to INSTIs is associated with virologic failure in a subset of patients. Viral variants that develop phenotypic loss of susceptibility to INSTIs contain primary resistance-associated mutations (RAMs) within the IN catalytic core domain (10–15). Based on cell-based virus passage experiments, the evolution pathway toward greater INSTI resistance with ongoing drug-selective pressure often involves different patterns of RAMs and/or genotypic switching among primary IN RAMs. For RAL, three major primary resistance pathways have been recognized, involving mutations at IN residues Y143, Q148, and N155 (14, 16–18), while for DTG, the pathway toward primary resistance has not been readily defined but likely involves the accumulation of multiple mutations (15). The resistance profile for EVG has shown that T66I/A develops early, E92Q and/or S147G develops concurrently or subsequently, and Q148R/K develops late, sometimes as a mixture with the preceding primary IN mutations (10–12). Collective data from clinical trials corroborate these findings for EVG and now indicate that resistance to EVG is attributable primarily to the emergence of mutations at six amino acid positions in HIV-1 IN (Fig. 1) (6, 7, 9). These primary EVG RAMs include T66I, E92Q, T97A, S147G, Q148R, and N155H and less commonly T66A/K, E92G, and Q148H/K, detected by population-based sequencing alone or occasionally as a complex dynamic mixture of up to 4 mutations. In patients with continued virologic failure, a stepwise accumulation and switch from one primary INSTI RAM to another have been observed and are frequently associated with additional secondary amino acid substitutions (19, 20). These secondary changes typically have little impact on their own but in combination with primary EVG RAMs further reduce EVG susceptibility and/or increase viral replication fitness (21). In this study, we constructed a series of recombinant viruses and IN enzymes carrying clinically observed primary EVG RAMs so as to determine and compare their individual and combined effects on drug susceptibility, viral fitness, replication capacity, and enzymatic activities.
Fig 1.
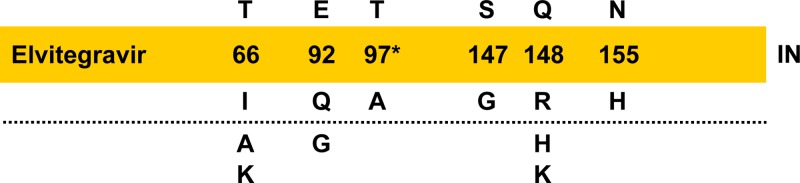
Clinical development of EVG primary INSTI RAMs. Most commonly observed IN mutations are indicated above the dotted line, while less commonly observed mutations are indicated below. *, T97A may require additional mutations for reduced phenotypic susceptibility.
MATERIALS AND METHODS
Reagents and cell lines.
Elvitegravir (EVG), raltegravir (RAL), dolutegravir (DTG), emtricitabine (FTC), and tenofovir (TFV) were synthesized at Gilead Sciences, Inc. (Foster City, CA). MT-2 cells were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Germantown, MD). HEK 293T cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). All previously described deoxyoligonucleotides (ST1, ST2, ST5, ST6, ST10, and ST11) (22) were purchased from Trilink Biotechnologies (San Diego, CA) and annealed in the pairs ST1/ST2, ST5/ST6, and ST10/ST11 at 50 μM in 20 mM Tris (pH 7.4)–100 mM NaCl.
Construction of site-directed IN mutants.
Single or double primary EVG RAMs were generated by using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) and introduced into (i) full-length infectious HIV-1HXB2 and HIV-1LAI/IIIB vectors and (ii) an HIV-1LAI/IIIB integrase expression vector (pET3B-N6H) by using standard cloning techniques (12). Viruses containing IN mutations were generated by transient transfection of HEK 293T cells using TransIT-293 (Mirus Bio LLC, Madison, WI). Recombinant mutant IN enzymes containing an N-terminal six-histidine tag (6His-IN) were expressed in Escherichia coli BL21(DE3)/pLysS cells (Life Technologies, Grand Island, NY) and purified by Ni-nitrilotriacetic acid (NTA) affinity chromatography after extraction under high-salt conditions, as previously described (23).
Replication capacity and antiviral drug susceptibility assays.
Replication capacities and phenotypic susceptibilities of wild-type (WT) and IN mutant viruses to INSTIs (EVG, RAL, and DTG) and NRTIs (FTC and TFV) were determined by Monogram Biosciences (South San Francisco, CA), using the single-cycle PhenoSense IN HIV assay (24). Briefly, the IN coding regions of HIV-1HXB2 vectors were cloned into an HIV-1NL4-3-derived test vector containing a luciferase gene and an env deletion. Pseudotyped virus stocks were generated by cotransfection of test vectors with an amphotropic murine leukemia virus (A-MLV) env-containing vector and used to infect fresh HEK 293 cells in the presence of drug dilution series. Luciferase gene expression in HEK 293 cells was measured after 2 days, and fold changes in susceptibility values were normalized to the values generated by using the wild-type HIV-1HXB2 IN coding region. Phenotypic susceptibilities of some IN mutant viruses against INSTIs (EVG and RAL) and NRTIs (FTC and TFV) were also determined in a multiple-round replication assay that measured cell viability, as previously described (12). Briefly, MT-2 cells infected by nutation for 3 h (multiplicity of infection [MOI] = 0.01) were incubated with drug in triplicate. After 5 days of growth, cytopathic effect was measured by using CellTiter-Glo luminescent cell viability detection reagent (Promega, Madison, WI). Effective drug concentrations needed to inhibit 50% of viral replication (EC50) were determined by nonlinear regression of data converted to percent cell death (GraphPad Prism Software, La Jolla, CA).
Determination of viral replication fitness by growth competition assay.
Viral growth competition and MultiCode reverse transcriptase (RTx) PCR assays were performed essentially as previously described (25). Competing recombinant viruses carrying opposing silent markers at amino acid residues 6 and 7 within the RT coding region (xx-LAI and F-xx-LAI) were mixed together at a 50:50, 60:40, or 80:20 ratio and used to inoculate MT-2 cells (MOI = 0.01), followed by washing with phosphate-buffered saline (PBS) to remove unbound virus. On days 3, 6, and 9, cultures were passaged by inoculating fresh MT-2 cells with the virus-containing supernatant (1:1,000 dilution). At each time point, viral RNA was extracted from 200 μl of the supernatant by using EZ1 virus minikit v2.0 and the BioRobot EZ1 workstation (Qiagen, Valencia, CA) and digested with DNase I. By using the extracted viral RNA and MultiCode RTx allele-specific PCR (EraGen, Madison, WI), the percentages of the two competing viruses at each time point were determined (25, 26). PCR mixtures (20 μl) contained the forward allele-specific primers (200 nM), reverse primer (400 nM), 1× ISOlution buffer (EraGen), 5 mM dithiothreitol (DTT), Titanium Taq DNA polymerase (Clontech, Palo Alto, CA), and SuperScript III RT (Invitrogen, Carlsbad, CA) at the recommended concentrations. MultiCode RTx PCRs were performed on a Roche LightCycler 480 instrument (Roche, Indianapolis, IN) under the following conditions: 5 min at 54°C for reverse transcription; 2 min at 95°C for initial denaturation; and then 1 cycle of 5 s at 95°C, 5 s at 47°C, and 20 s at 72°C followed by 45 cycles of 5 s at 95°C, 5 s at 57°C, and 20 s at 72°C with optical recording. A thermal melt from 60°C to 95°C with 2.5 optical recordings per degree Celsius was performed immediately after the last cycle. Percentages of competing viruses were determined from standard curves generated by using SigmaPlot (Systat Software, San Jose, CA).
Quantitative estimates of relative viral fitness were calculated with the following equation:
| (1) |
where s is the selection coefficient, t is time in days, and Mt0 and Wt0 are initial fractions and Mt and Wt are time of measurement fractions of mutant and wild-type viruses, respectively (27). Relative fitness (1 + s) values of 1.00 indicate that both viruses grew with equivalent fitness. Relative fitness (1 + s) values of <1.00 indicate less efficient growth of the second virus relative to the first virus competitor.
HTRF-based IN strand transfer activity assay.
IN strand transfer activity in the absence and presence of INSTIs (EVG, RAL, and DTG) was measured by using a homogenous time-resolved fluorescence resonance energy transfer (HTRF)-based assay, as previously described (22, 28). Reactions were performed with 25 μl strand transfer buffer (20 mM Tris [pH 7.4], 25 mM NaCl, 7.5 mM MgCl2, 0.05% Brij 35, 10% glycerol, 1 mM DTT, 1% dimethyl sulfoxide [DMSO], 0.1 mg/ml bovine serum albumin [BSA]) in 96-well half-area white flat-bottom plates (Corning, Lowell, MA). Briefly, 9 μl of 6His-IN (250 nM final concentration) were preincubated in triplicate with 1 μl of 25× drug dilution series (final concentration, 1% [vol/vol] DMSO) for 5 min at room temperature. Reactions were initiated by the addition of a 15-μl mixture containing Cy5-labeled 3′-processed ST1/ST2 donor DNA substrate (12.5 nM final concentration) that mimics the HIV-1 LTR terminus and biotin-labeled ST5/ST6 target DNA substrate (5 nM final concentration). After 120 min at 37°C, reactions were stopped by the addition of 25 μl of 2× Stop/Development buffer (25 mM EDTA, 1 M NaCl, 1 mg/ml BSA, 0.05% Brij 35, 2 nM europium chelate-streptavidin) (Perkin-Elmer, Boston, MA). Plates were read after 16 h of incubation at room temperature on an Envision 2102 multilabel reader (Perkin-Elmer) (excitation at 320 nm and emission at 620 nm and at 665 nm, with a D400 dichroic mirror). An increase in the fluorescence resonance energy transfer (FRET) signal (calculated as a ratio of fluorescence intensity at 665 nm to fluorescence intensity at 620 nm) was associated with an increased proportion of salt-stable noncovalently linked donor-target DNA integration product due to an energy transfer to the Cy5 fluorophore acceptor. In the absence of INSTIs, enzymatic activity was assessed by using a time course (kinetic) format, whereby reactions were stopped every 15 to 30 min up to 240 min.
Dose-response data were analyzed with the following curve-fitting equation:
| (2) |
where y is the 665/620-nm ratio, IC50 is the 50% inhibitory concentration, [I] is the inhibitor concentration, n is the Hill coefficient, M is the signal in the absence of inhibitor, and H is the signal at full inhibition.
Strand transfer kinetic data were analyzed with the following curve-fitting equation:
| (3) |
where y and ymax are the 665/620-nm ratio, k is the enzymatic rate constant, and x is time.
HTRF-based IN 3′-processing activity assay.
IN 3′-processing activity in the absence of INSTIs was measured by using an HTRF-based assay with a format similar to the one described above for IN strand transfer activity, except for the use of a nonprocessed donor DNA and no target DNA (22, 28). This specially designed nonprocessed blunt-ended ST10/ST11 donor DNA substrate (12.5 nM final concentration) was Cy5 labeled at the dinucleotide-removable 3′ end and biotin labeled at the other 3′ end. A decrease in the FRET signal (665/620 nm) was associated with an increased proportion of 3′-processed DNA due to removal of the dinucleotide containing the Cy5 fluorophore acceptor.
Kinetic data for 3′ processing were analyzed with the following curve-fitting equation:
| (4) |
where y, yo, and ymin are the ratios of fluorescence intensities at 665 nm and 620 nm at each time point, at start of the experiment, and at reaction completion (background), respectively, k is the enzymatic rate constant, and x is time.
Molecular homology modeling.
A three-dimensional HIV-1 IN homology model was built by using PyMol software v1.4.1 (Schrödinger LLC, Portland, OR) and a previously reported crystal structure of full-length tetrameric IN from human prototype foamy virus (PFV) in complex with Mg2+, 3′-processed donor DNA, and EVG (Protein Data Bank [PDB] accession number 3L2U) (29). A secondary-structure-based alignment of HIV-1 and PFV IN was used to guide the construction of the HIV-1 homology model via amino acid substitution. Potential interactions of primary IN residues with the constituents of the IN active site were determined based on proximity (<4 Å). These constituents included Mg2+, donor DNA, the flexible loop (G140 to G149), and/or EVG.
RESULTS
Primary IN mutations exhibit broad cross-resistance to EVG and RAL while maintaining susceptibility to DTG.
To evaluate the impact of each primary EVG RAM on antiviral drug susceptibilities, site-directed HIV-1 mutants were generated and characterized by using the commercially available single-cycle PhenoSense IN HIV assay (Table 1). HIV-1 with Q148R/K mutations exhibited the greatest reduction in EVG susceptibility (>92-fold). Among the most common IN mutations, reduced susceptibility to EVG followed the order of Q148R (92-fold) > N155H ≈ E92Q (26- to 30-fold) > T66I (9.7-fold) > S147G (4.1-fold) > T97A (2.4-fold). Less common IN mutations showed various degrees of reduced susceptibility, ranging from <10-fold reductions (T66A and Q148H) to >10-fold reductions (T66K, E92G, Q148K, and N155S). Some IN mutations conferred reduced susceptibility to EVG but retained susceptibility to RAL (T66I/A, E92G, and S147G), while others conferred reduced susceptibility to both EVG and RAL (T66K, E92Q, Q148R/H/K, and N155H). Among the most frequent primary RAL RAMs (Y143R/H/C, Q148R/H/K, and N155H), all but Y143R/H/C conferred significant reductions in susceptibility to EVG (>5-fold). In contrast, antiviral susceptibility to DTG was retained for all primary INSTI RAMs. In addition, all viruses remained fully susceptible to NRTIs (FTC and TFV) and other classes of antiretrovirals (Table 1 and data not shown). Finally, where tested, these results were confirmed in acutely infected MT-2 cells by using a 5-day multiple-round replication assay, where cell viability was assessed (Table 2).
Table 1.
Antiviral susceptibilities of INSTIs and NRTIs against HIV-1 determined by PhenoSense IN single-cycle infectivity assays
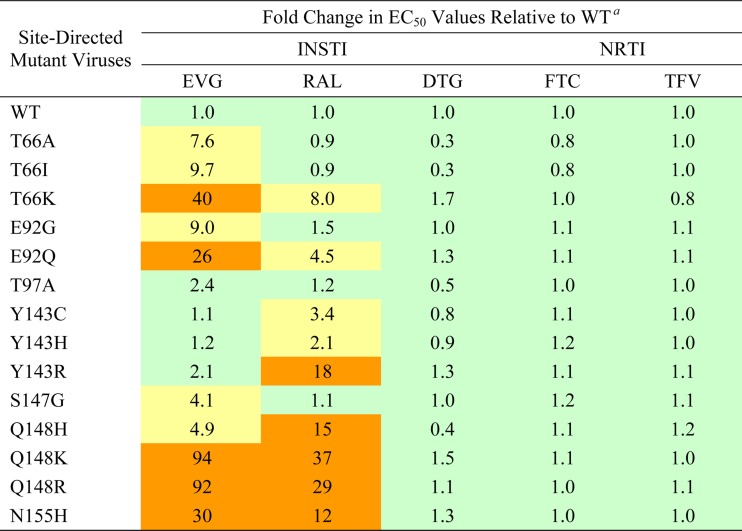
a Data shown represent the results of an independent experiment performed with replicate drug dilutions by Monogram Biosciences (South San Francisco, CA). Color coding indicates the degree of reduced susceptibility relative to established biological cutoffs (BCOs) for EVG (2.5-fold change) and RAL (1.5-fold change): less than or equal to the BCO (green), greater than the BCO and less than 10 (yellow), and greater than 10 (orange). An arbitrary biological cutoff for DTG was set at 2.0.
Table 2.
Antiviral susceptibilities of INSTIs and NRTIs against HIV-1 in multiple-round replication assays
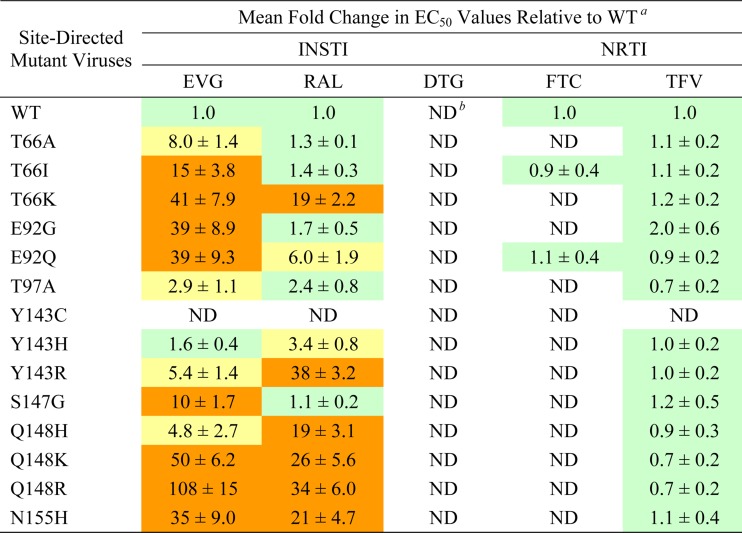
a Data shown represent the means and standard deviations from at least three independent experiments. Color coding indicates the degree of reduced susceptibility: less than or equal to 2.5 (green), greater than 2.5 and less than 10 (yellow), and greater than 10 (orange). All fold change values of >2.5 demonstrated a statistically significant difference compared to the WT by using a two-tailed Student t test, assuming equal variance (P < 0.01).
b ND, not determined.
Primary IN mutations reduce viral replication fitness.
Data generated by using the PhenoSense IN HIV assay also provided a general indication of the impact of each primary EVG RAM on viral replication capacity (Table 3). With one exception (S147G), all primary EVG RAMs (13 of 14) exhibited reduced IN-mediated viral replication capacity values compared to wild-type (WT) values (range, 6 to 84%; median, 69%). Interestingly, primary EVG RAMs with the most attenuated replication capacities were some of the least commonly observed (T66K and E92G exhibited 9% and 6% replication capacities, respectively). The relative viral fitness of the most common primary EVG RAMs was also evaluated by direct competition with WT virus in multiple-round replication assays. Viruses were mixed in ratios of infectious units (50:50, 60:40, or 80:20) and used to inoculate MT-2 cells. The relative proportion of each IN mutant in growth competition with WT was quantified by real-time allele-specific PCR at regular time intervals (see Fig. S1 in the supplemental material), and mean relative viral fitness (1 + s) values were determined from replicate experiments (Table 4). Most primary EVG RAMs showed substantial reductions in viral replication fitness. Only T66I and S147G mutations exhibited no significant effect on viral replication fitness, consistent with their minimally reduced replication capacity values determined by the PhenoSense IN HIV assay. Overall, the relative viral fitness of primary IN mutants in competition with the WT increased in the following order: Q148R < N155H < E92Q < T66I < S147G. Plotting these viral replication fitness values against fold resistance values to EVG (Fig. 2) revealed a strong correlation (r2 = 0.88), suggesting that higher levels of resistance are associated with greater reductions in viral fitness in the absence of additional compensatory mutations.
Table 3.
Replication capacities of recombinant HIV-1 determined by PhenoSense IN single-cycle infectivity assays
| Site-directed IN mutant virus | RC (% of WT)a |
|---|---|
| WT | 100 |
| T66A | 69 |
| T66I | 84 |
| T66K | 9 |
| E92G | 6 |
| E92Q | 64 |
| T97A | 68 |
| Y143C | 49 |
| Y143H | 78 |
| Y143R | 75 |
| S147G | 108 |
| Q148H | 54 |
| Q148K | 36 |
| Q148R | 70 |
| N155H | 72 |
| T66I + E92Q | 25 |
| T66I + Q148R | 20 |
| T66I + N155H | 16 |
| E92Q + Q148R | 4 |
| E92Q + N155H | 40 |
| Q148R + N155H | 15 |
Data shown represent the results of an independent experiment performed in the absence of drug by Monogram Biosciences (South San Francisco, CA). RC, replication capacity.
Table 4.
Relative viral fitness of recombinant HIV-1 in growth competition assays
| Competing site-directed IN mutant virusesa | Mean relative fitness value (1 + s) ± SDb | P valued |
|---|---|---|
| WT vs WTc | 1.00 ± 0.01 | |
| WT vs T66I | 0.92 ± 0.06 | 0.131 |
| WT vs E92Q | 0.82 ± 0.06 | 0.007 |
| WT vs S147G | 1.01 ± 0.01 | 0.395 |
| WT vs Q148R | 0.62 ± 0.08 | 0.001 |
| WT vs N155H | 0.76 ± 0.01 | 0.001 |
| WT vs E92Q + N155H | 0.55 ± 0.06 | 0.001 |
| E92Q vs E92Q + N155H | 0.68 ± 0.05 | 0.001 |
| N155H vs E92Q + N155H | 0.82 ± 0.04 | 0.004 |
| Q148R vs E92Q + Q148R | <0.50 ± 0.01 | <0.001 |
| Q148R vs Q148R + N155H | <0.50 ± 0.01 | <0.001 |
Growth competition assays to compare relative viral fitness of the indicated pairs of viruses were performed. Viruses at a 50:50, 60:40, or 80:20 ratio were used to inoculate MT-2 cells, as described in Materials and Methods.
Relative fitness value versus WT HIV-1 was calculated as (1 + s) = exp[1/t × ln(Mt/Wt × Wt0/Mt0)], where t is time in days and Mt0 and Wt0 are initial fractions and Mt and Wt are time of measurement fractions of mutant and wild-type viruses, respectively (27). Relative fitness (1 + s) values of 1.00 indicate that both viruses grew with equivalent fitness. Relative fitness (1 + s) values of <1.00 indicate less efficient growth of the second virus relative to the first virus 1 competitor. Data shown represent the means and standard deviations from at least three independent experiments.
A control experiment was performed to verify that isogenic HIV-1 recombinants differing only in their sequence tags would grow with equivalent fitness.
P values compared to WT versus WT were determined by using a two-tailed Student t test.
Fig 2.

Correlation between phenotypic EVG resistance determined by the PhenoSense IN HIV assay and relative viral replication fitness.
Primary IN mutations impact the enzymatic activities of IN.
The impact of primary EVG RAMs on antiviral susceptibility and viral replication fitness could be attributable to a direct effect on IN enzymatic functions required for the integration process. To address this question, 3′-processing and strand transfer catalytic activities of recombinant mutant IN enzymes were investigated by using fluorescence-resonance energy transfer (FRET)-based technology. In the presence of EVG, most primary mutant IN enzymes showed variably reduced sensitivity to inhibition of strand transfer activity relative to the WT (Table 5). HIV-1 IN with the Q148R mutation showed a substantial 11-fold reduction in sensitivity to inhibition by EVG, while other common primary IN mutations (T66I, E92Q, and N155H) showed <5-fold reductions. Both the T97A and S147G mutations retained sensitivity to EVG in strand transfer IN activity assays. Cross-resistance between EVG and RAL was also observed among primary EVG RAMs; some IN mutations (Q148R and N155H) conferred a greater reduction in sensitivity to inhibition by EVG (>5-fold), while others (T66I and E92Q) exhibited similar slightly reduced sensitivities to inhibition by both EVG and RAL. Sensitivity to strand transfer inhibition by DTG was retained against all primary IN mutations. In the absence of EVG, most primary IN mutant enzymes showed substantial impairment of 3′-processing and/or strand transfer activities (Fig. 3). HIV-1 IN enzymes with the Q148R or N155H mutation were the most attenuated in both 3′-processing and strand transfer activities, consistent with previous biochemical assessments of recombinant IN enzymes possessing primary RAL RAMs (11, 13, 14, 30–32). Other primary EVG RAMs (T66I, E92Q, and S147G) exhibited marginal impairment of 3′-processing activity (80 to 87% of WT activity), with substantial impairment of strand transfer activity (∼40 to 75% of WT activity). Only T97A exhibited a marginal impairment of 3′-processing activity (84% of WT activity), with no significant effect on strand transfer IN activities. These results show that, similar to their impact on the virus, primary EVG RAMs are associated with reduced EVG sensitivity and IN activities in an enzyme-based system.
Table 5.
Activities of INSTIs against HIV-1 IN enzymes in strand transfer activity assays

a Data shown represent the means and standard deviations from at least three independent experiments. Color coding indicates the degree of reduced sensitivity: less than or equal to 2.0 (green), greater than 2.0 and less than 10 (yellow), and greater than 10 (orange).
Fig 3.

Relative catalytic activities of HIV-1 IN enzymes. IN-mediated 3′-processing (A) and strand transfer (B) activities were determined by using unprocessed and preprocessed donor DNA substrates, respectively. Data shown represent the means and standard deviations of data from an independent experiment performed in triplicate.
Multiple primary IN mutations further reduce INSTI susceptibility and viral replication fitness.
Recent clonal analyses of plasma from patients failing EVG-containing regimens have indicated that while most virus populations consist of a complex mixture of primary EVG RAMs, as assessed by population sequencing, these IN mutations are rarely linked on the same genome (20). Therefore, we sought to understand the underlying characteristics that may force their independent development. To determine the impact of multiple primary EVG RAMs, dual combinations of the most common primary IN mutations were introduced into viral vectors and characterized by using the PhenoSense IN HIV assay (Table 6). In general, the addition of a second primary IN mutation resulted in a much greater reduction in both EVG and RAL susceptibility than either mutation alone. Although T66I is a primary INSTI RAM specific to EVG, the combination of T66I and E92Q resulted in a strong reduction in RAL susceptibility (13-fold). Interestingly, the addition of the Q148R mutation to any primary EVG RAM resulted in a 3- to 6-fold reduction in DTG susceptibility that was not observed for any IN mutation alone. Other dual combinations of primary EVG RAMs had no effect on DTG susceptibility.
Table 6.
Antiviral susceptibilities of INSTIs against HIV-1 dual primary IN mutants determined by PhenoSense IN single-cycle infectivity assays
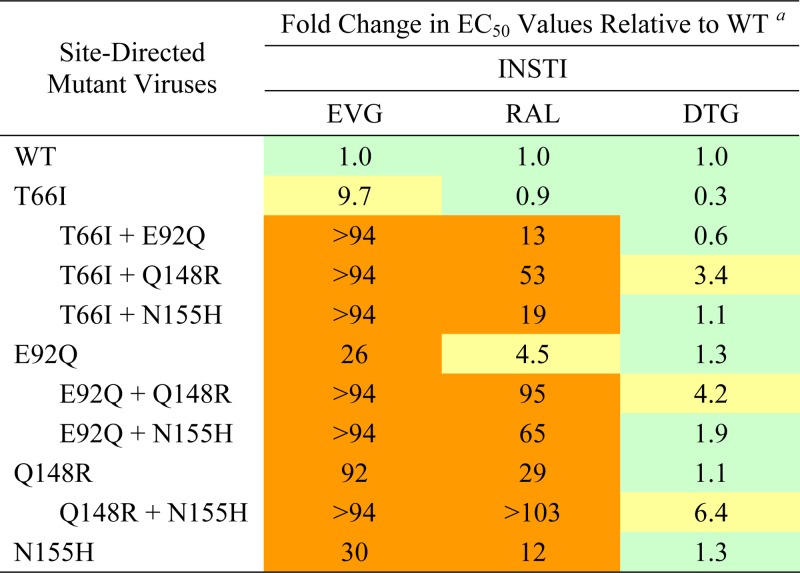
a Data shown represent the results of an independent experiment performed with replicate drug dilutions by Monogram Biosciences (South San Francisco, CA). Color coding indicates the degree of reduced susceptibility relative to established BCOs for EVG (2.5-fold change) and RAL (1.5-fold change): less than or equal to the BCO (green), greater than the BCO and less than 10 (yellow), and greater than 10 (orange). An arbitrary biological cutoff for DTG was set at 2.0.
The addition of a second primary IN mutation was also found to further reduce viral replication capacity values (1.6- to 16-fold) relative to values for the WT or either IN mutation alone (Table 3). In pairwise growth competition experiments, the addition of Q148R onto the backbone of either E92Q or N155H was found to substantially reduce the viral fitness of Q148R more than 2-fold (Table 4; see also Fig. S1 in the supplemental material). In comparison, the dual combination of E92Q and N155H (E92Q+N155H) reduced both replication capacity and viral fitness in competition with WT only slightly (≈1.5-fold). Further growth competition analyses showed that the dual combination E92Q+N155H mutant had a lesser impact on the relative fitness of the N155H virus (1 + s = 0.82) than on the relative fitness of the E92Q virus (1 + s = 0.68). Taken together, these results show that multiple primary EVG RAMs confer greater reductions in INSTI susceptibility at the expense of further reducing viral fitness.
Structural proximity of EVG RAMs within the IN active site may impact both drug susceptibility and enzymatic activity.
While the structure of the full-length HIV-1 IN tetramer has remained unsolved, the full-length structure of the related human PFV IN has served as a reliable surrogate for structural analyses due to active-site similarity. Crystal structures of the PFV IN in complex with its cognate viral DNA, referred to as the intasome, have been solved in the absence and presence of INSTIs (EVG, RAL, or DTG) (29, 33). These studies have led to significant insight into the molecular mechanism of retroviral IN inhibition. The current model highlights at least four key functional components of IN: (i) the catalytic DDE triad (D64, D116, and E152) of active-site residues, (ii) the metal ion cofactors (Mg2+ or Mn2+), (iii) the terminus of the viral DNA where key contacts are made, and (iv) the flexibility or contacts with surrounding secondary structures (e.g., α4 helix and β4-α2 loop). The fundamental basis for the mechanism of IN activation by the binding of an INSTI is believed to be (i) displacement of the reactive end of the 3′-processed viral DNA from the IN active site and (ii) chelation of a metal ion away from the active-site residues (29, 34).
To explore the structural impact of primary IN mutations on reduced EVG susceptibility and IN activities, homology modeling of HIV-1 IN was performed. First, a model of WT HIV-1 IN was created by substitution of the corresponding amino acid residues into the published structure of PFV IN (29). In this structure, EVG is bound within a hydrophobic pocket, and the positions of amino acid residues associated with primary EVG RAMs are located proximal to the catalytic active site (Fig. 4). Second, we systematically introduced substitutions corresponding to primary EVG RAMs to assess a plausible structural impact. No primary EVG RAMs were found to reside within 4 Å of bound EVG, suggesting that reduced susceptibility to EVG is associated with a lack of direct interaction. Conversely, most primary EVG RAMs were proximally located to a variety of active-site components, including Mg2+ ions, viral DNA, and/or secondary structural components of the IN enzyme. Based on these interpretations and the findings of others, the structural impact of primary EVG RAMs is discussed in detail below.
Fig 4.

Proximal location of EVG primary INSTI RAMs in a homology model of the HIV-1 IN active site. Amino acid residues of HIV-1 IN were substituted in a crystal structure model of PFV IN with bound EVG and its cognate DNA substrate (PDB accession number 3L2U) (29). The protein backbone in a ribbon representation is shown in green. The viral DNA substrate sugar-phosphate backbone and nitrogenous bases are shown in brown and blue, respectively. Residues associated with primary EVG RAMs are shown as light blue spheres, and Mg2+ ions are shown as pink spheres. Elvitegravir (EVG) is labeled and shown as yellow spheres. White labels indicate HIV-1 IN residue numbers and mutations.
DISCUSSION
Elvitegravir (EVG) is a new ARV with demonstrated clinical efficacy in both treatment-naive and -experienced HIV-1-infected patients (6, 7, 9). However, as shown in vitro and in some patients failing EVG-containing regimens, primary IN mutations can develop, leading to reduced EVG susceptibility. HIV-1 IN mutations T66I, E92Q, S147G, and Q148R/K were first identified as primary EVG RAMs in cell-based resistance selection experiments (10, 11, 19, 35, 36). In patients experiencing virologic failure on EVG-containing regimes, additional RAMs, including T66A/K, E92G, T97A, Q148H, and N155H, have been observed (7, 9, 19, 36). Thus, overall, primary EVG RAMs may develop at six amino acid positions in HIV-1 IN (Fig. 1). Recombinant forms of HIV-1 and IN were used to determine the effect of each primary EVG RAM on phenotypic susceptibilities to INSTIs. Fold change values relative to the WT were comparable in both types of HIV-1 drug susceptibility assays used (single cycle and multiple round) and were further confirmed in enzymatic strand transfer IN activity assays. These results demonstrated a ranked order among primary EVG RAMs whereby the most common clinically observed primary IN mutations (E92Q, Q148R, and N155H) exhibited the greatest reduction in EVG susceptibility, consistent with previous reports (10, 11, 30). The finding that T97A showed only very-low-level reduced EVG susceptibility in the multicycle assay is similar to its effect on RAL susceptibility in patients experiencing virologic failure on RAL-containing regimens (32, 37). This special case of a primary EVG RAM suggests that additional amino acid substitutions may serve an accessory role for T97A to further reduce phenotypic susceptibility.
Structural analyses have shown that both EVG and RAL share similar scaffolds and binding modes within the IN active site leading to inhibition by disengaging the DDE catalytic triad from the viral DNA (29). Thus, it is not surprising that both EVG and RAL exhibit largely overlapping primary INSTI resistance profiles in HIV-infected patients experiencing virologic failure on either INSTI. For RAL, three independent common primary RAMs, Y143R/H/C, Q148R/H/K, and N155H, have been identified (14, 16, 17). Both N155H and Q148R/H/K are associated with the development of EVG cross-resistance, whereas Y143R/H/C mutations do not lead to decreased EVG susceptibility, likely due to the absence of a RAL-specific oxadiazole group to interact with (17, 29, 31, 38). Importantly, E92Q and T97A have also been observed in HIV-1-infected patients experiencing virologic failure on RAL, either independently or in association with N155H and Y143R/H/C, respectively (18, 39–42). While E92Q is often referred to as a secondary RAL RAM (16, 32, 37), our data indicate a significant impact on EVG and RAL resistance, independent of other mutations. Small geometric differences between the interactions of EVG and those of RAL within IN may account for the observed variability in cross-resistance (T66K, E92Q, and Q148R/H/K) and why certain primary EVG RAMs (T66I/A, E92G, and S147G) retain susceptibility to RAL while certain primary RAL RAMs (Y143R/H/C) retain susceptibility to EVG.
Structural analyses have suggested that DTG shares a similar interfacial mechanism of inhibition with EVG and RAL but is able to make more intimate contacts with the viral DNA. In addition, DTG may effectively be able to readjust its position and conformation to structural changes in the active sites of EVG- or RAL-resistant IN enzymes and avoid some cross-resistance due to slower dissociation (15, 33, 43). This may explain why primary high-level resistance to DTG does not readily occur in vitro and appears difficult to establish de novo clinically. However, recent findings indicated that the Q148R/H/K resistance pathway observed for EVG and RAL can impact cross-resistance to DTG as the number of secondary mutations increases (i.e., E138K, G140S, and N155H) (15, 44). The data presented in this study extend these observations by demonstrating that DTG retains activity against all individual primary EVG and RAL RAMs. Taken together, these results support clinical evidence that subjects who experience virologic failure on EVG-containing regimens respond poorly to treatment with RAL (45) and that DTG may have utility as a second-line regimen in cases of virologic failure with EVG or RAL, although multiple mutations that include Q148R/H/K may reduce responses (46).
The selective advantage of primary EVG RAMs in patients with virologic failure while on EVG-containing regimens drives both the initial appearance of these mutations and a pathway toward higher levels of resistance. While the development and prevalence of primary IN mutations may be ascribed to their effect on EVG susceptibility, their effect on viral replication fitness is equally relevant. HIV-1 with reduced EVG susceptibility from patients experiencing virologic failure on EVG typically exhibits a reduced replication capacity (36, 47). The results of the PhenoSense IN HIV assay showed that most primary EVG RAMs are associated with a reduction in replication capacity. Previous studies of primary RAL RAMs in recombinant forms of HIV-1 have reported similar findings (10, 11, 16, 48–50). Growth competition analyses further demonstrated reduced viral fitness of mutants expressing EVG RAMs and suggested a correlation between increased EVG resistance and decreased viral fitness. These results suggest that certain viral variants with primary EVG RAMs may emerge first because they are more fit and not necessarily more resistant. Indeed, previous studies of primary RAL RAMs suggested that an N155H mutant is likely to emerge before Q148R/H/K mutants because it has a substantial fitness advantage (50, 51). The results reported here extend this interpretation to include other common primary IN mutations (T66I, E92Q, and S147G). Moreover, of the most commonly observed viral mutants in patients experiencing virologic failure on EVG-containing regimens (6, 7, 9), both the E92Q and N155H mutations appear to exhibit a selective advantage, with reasonable viral fitness and significantly reduced susceptibility to EVG.
Patients exhibiting virologic failure on EVG or RAL therapy typically harbor a complex dynamic population of primary IN mutations that are generally not found on the same HIV-1 genome. The following combinations of primary INSTI RAMs have, however, been found to colocalize in rare exceptions: T97A+Y143R/C in RAL failures, S147G+Q148R/H/K in EVG failures, and E92Q+N155H in both RAL and EVG failures (14, 16, 20, 21, 39, 40). To understand the underlying typical mutual exclusivity among primary EVG RAMs, dual combinations were introduced into recombinant viral vectors. As expected, the addition of a second primary IN mutation further reduced EVG and RAL susceptibility relative to that of either mutation alone. In contrast, DTG retained susceptibility against most double primary IN mutants but showed significantly reduced susceptibility in dual combinations with Q148R. While the Q148R+N155H mutant has been shown to reduce DTG susceptibility (15), the finding that other primary EVG RAMs (T66I or E92Q) in combination with Q148R also have the same effect has not been reported. The addition of a second primary IN mutation also further reduced replication capacity and, where studied, viral fitness. The extent of this reduction for the E92Q+N155H mutant was least among all dual combinations, in agreement with previous assessments of replication capacity (16, 50) and the observed coexistence of these primary INSTI RAMs in resistant viral variants. While an earlier report found the E92Q+N155H mutant to be more fit than the N155H mutant in growth competition experiments (51), the discordance in these results could be attributable to differences in the viral backbone utilized (HIV-1LAI/IIIB and HIV-1NL4-3, respectively). With prolonged selective pressure to EVG or RAL, Q148R/H/K with compensatory secondary mutations (E138K or G140S/A) tends to replace earlier primary INSTIs RAMs as the dominant species (19, 48, 52, 53). The results presented here suggest that viruses bearing an additional primary IN mutation fail to gain a similar selective advantage despite a further reduction in phenotypic susceptibility. Thus, genotypic switching among primary EVG RAMs over a period of virologic failure likely represents active emergence of distinct and progressively dominant viral variants.
Not surprisingly, most primary EVG RAMs surround the catalytic pocket of IN and are proximal to the catalytic DDE triad and other active-site components (29). Whereas T66 and T97 lie slightly outside the active site, D64 and E152 surround N155, D64 and D116 surround E92, and all three acidic residues of the catalytic triad are close to S147 and Q148. Since the Q148 residue lies within the catalytic β4-α2 loop, Q148R/H/K mutations would change the flexibility of this loop, which may (i) hamper metal ion binding by D64 and E152 and chelation by EVG (54), (ii) sterically affect binding of the viral DNA 5′ end (55–57), and (iii) weaken the backbone-backbone H-bond interaction between Q148 and E152 (34). In contrast, an S147G mutation within the same catalytic loop may have a less dramatic impact on loop flexibility, potentially affecting viral DNA binding, strand separation, and interactions of EVG with adjacent residues. Residue 155 lies within the α4 helix, and an N155H mutation here has been predicted to (i) perturb a salt bridge with the phosphate of the terminal 3′ adenosine, (ii) affect coordination of the metal ion by E152, and (iii) widen the base of the catalytic pocket (34, 43). Although T66 lies within the β2 sheet distal from the DDE triad, its proximity to the viral DNA 3′ end and N155 suggests that T66I/A/K mutations may (i) sterically affect viral DNA binding and/or (ii) metal ion coordination through N155. The E92 residue lies within a β3-α1 loop less than 4 Å from a metal ion and another 1 to 2 Å from the isobutyl substituent of EVG. An E92Q/G mutation, which results in the loss of a negative charge, may (i) stabilize the metal ion by making hydrogen bonding with a coordinating water molecule less favorable and (ii) remove any repulsion charge with the hydroxyl in the isobutyl substituent of EVG (10). Finally, although T97 lies within the α1 helix farthest away from the IN active site, a T97A mutation would eliminate a hydroxyl moiety and the H-bond interaction with N120, thus unblocking the α2 helix and creating an empty space in the active site. This structural change may (i) permit small movement of the viral DNA, (ii) affect proper placement of the β4-α2 catalytic loop (32), and/or (iii) perturb coordination of a metal ion with D116 in a manner similar to that of E92Q. Taken together, all primary EVG RAMs are hypothesized to subtly change the IN active-site environment by affecting local secondary structure and network interactions involved in coordination of the metal ions and/or the viral DNA. These interpretations are in agreement with the higher koff rates for INSTI dissociation from IN-DNA complexes and reductions in phenotypic susceptibility to INSTIs (43).
In summary, the key primary EVG RAMs observed in HIV-1-infected patients experiencing virologic failure on EVG-containing regimens (T66I/A/K, E92Q/G, T97A, S147G, Q148R/H/K, and N155H) have been defined and characterized. All primary IN mutations showed both reduced EVG susceptibility and reduced viral fitness. Moreover, these attributes correlated with both reduced activities and inhibition sensitivity of the HIV-1 IN enzyme. Multiple primary EVG RAMs detected by population sequencing commonly represent mixtures of multiple primary IN mutations that are not capable of coexisting on the same HIV-1 genome, likely due to more severely attenuated viral fitness. Patients who experience initial virologic failure on EVG-containing regimens typically transition toward EVG RAMs that confer greater phenotypic resistance and greater viral fitness with the accumulation of compensatory secondary IN mutations. Thus, as with other classes of ARVs, prolonged virologic failure under continuous EVG-selective pressure should be avoided in order to minimize resistance development within the class of INSTIs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gregg Jones, Manuel Tsiang, Elaine Kan, Dharmaraj Samuel, Magdeleine Hung, Debi Jin, and Xiaohong Liu from Gilead Sciences and Tim Persyn and Wei Huang from Monogram Biosciences for their contributions.
Footnotes
Published ahead of print 25 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02568-12.
REFERENCES
- 1. Thompson MA, Aberg JA, Hoy JF, Telenti A, Benson C, Cahn P, Eron JJ, Gunthard HF, Hammer SM, Reiss P, Richman DD, Rizzardini G, Thomas DL, Jacobsen DM, Volberding PA. 2012. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA Panel. JAMA 308:387–402 [DOI] [PubMed] [Google Scholar]
- 2. Sakai H, Kawamura M, Sakuragi J, Sakuragi S, Shibata R, Ishimoto A, Ono N, Ueda S, Adachi A. 1993. Integration is essential for efficient gene expression of human immunodeficiency virus type 1. J. Virol. 67:1169–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Engelman A, Mizuuchi K, Craigie R. 1991. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67:1211–1221 [DOI] [PubMed] [Google Scholar]
- 4. Eron JJ, Jr, Rockstroh JK, Reynes J, Andrade-Villanueva J, Ramalho-Madruga JV, Bekker LG, Young B, Katlama C, Gatell-Artigas JM, Arribas JR, Nelson M, Campbell H, Zhao J, Rodgers AJ, Rizk ML, Wenning L, Miller MD, Hazuda D, DiNubile MJ, Leavitt R, Isaacs R, Robertson MN, Sklar P, Nguyen BY. 2011. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect. Dis. 11:907–915 [DOI] [PubMed] [Google Scholar]
- 5. Steigbigel RT, Cooper DA, Teppler H, Eron JJ, Gatell JM, Kumar PN, Rockstroh JK, Schechter M, Katlama C, Markowitz M, Yeni P, Loutfy MR, Lazzarin A, Lennox JL, Clotet B, Zhao J, Wan H, Rhodes RR, Strohmaier KM, Barnard RJ, Isaacs RD, Nguyen BY. 2010. Long-term efficacy and safety of raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 phase III trials. Clin. Infect. Dis. 50:605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, Gallant JE, Liu HC, Zhong L, Yale K, White K, Kearney BP, Szwarcberg J, Quirk E, Cheng AK. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 379:2439–2448 [DOI] [PubMed] [Google Scholar]
- 7. DeJesus E, Rockstroh J, Henry K, Molina J-M, Gathe J, Ramanathan S, Wei X, Yale K, Szwarcberg J, White K, Cheng AK, Kearney BP. 2012. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet 379:2429–2438 [DOI] [PubMed] [Google Scholar]
- 8. van Lunzen J, Maggiolo F, Arribas JR, Rakhmanova A, Yeni P, Young B, Rockstroh JK, Almond S, Song I, Brothers C, Min S. 2012. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naive adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect. Dis. 12:111–118 [DOI] [PubMed] [Google Scholar]
- 9. Molina JM, Lamarca A, Andrade-Villanueva J, Clotet B, Clumeck N, Liu YP, Zhong L, Margot N, Cheng AK, Chuck SL. 2012. Efficacy and safety of once daily elvitegravir versus twice daily raltegravir in treatment-experienced patients with HIV-1 receiving a ritonavir-boosted protease inhibitor: randomised, double-blind, phase 3, non-inferiority study. Lancet Infect. Dis. 12:27–35 [DOI] [PubMed] [Google Scholar]
- 10. Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, Dockx K, Strijbos R, Smits V, Vos A, Meersseman G, Jochmans D, Vermeire K, Schols D, Hallenberger S, Hertogs K. 2008. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 82:10366–10374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. 2008. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 82:764–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, White KL, Miller MD. 2012. In vitro resistance selections using elvitegravir, raltegravir, and two metabolites of elvitegravir M1 and M4. Antiviral Res. 93:288–296 [DOI] [PubMed] [Google Scholar]
- 13. Fikkert V, Van Maele B, Vercammen J, Hantson A, Van Remoortel B, Michiels M, Gurnari C, Pannecouque C, De Maeyer M, Engelborghs Y, De Clercq E, Debyser Z, Witvrouw M. 2003. Development of resistance against diketo derivatives of human immunodeficiency virus type 1 by progressive accumulation of integrase mutations. J. Virol. 77:11459–11470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, Tchertanov L, Peytavin G, Reynes J, Mouscadet JF, Katlama C, Calvez V, Marcelin AG. 2008. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 52:1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 55:813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fransen S, Gupta S, Danovich R, Hazuda D, Miller M, Witmer M, Petropoulos CJ, Huang W. 2009. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 83:11440–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cooper DA, Steigbigel RT, Gatell J, Rockstroh J, Katlama C, Yeni P, Lazzarin A, Clotet B, Kumar P, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Zhao J, Chen J, Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Teppler H, Nguyen B. 2008. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N. Engl. J. Med. 359:355–365 [DOI] [PubMed] [Google Scholar]
- 18. Fransen S, Gupta S, Danovich R, Hazuda D, Miller M, Witmer M, Petropoulos CJ, Parkin NT, Huang W. 2008. Abstr. XVII Int. HIV Drug Resist. Workshop, Sitges, Spain, abstr 7 [Google Scholar]
- 19. Waters JM, Margot N, Hluhanich R, Svarovskaia J, Harris J, Borroto-Esoda K, Miller MD, McColl DJ. 2009. Abstr. XVIII Int. HIV Drug Resist. Workshop, Fort Myers, FL, abstr 116 [Google Scholar]
- 20. Winters MA, Lloyd RM, Jr, Shafer RW, Kozal MJ, Miller MD, Holodniy M. 2012. Development of elvitegravir resistance and linkage of integrase inhibitor mutations with protease and reverse transcriptase resistance mutations. PLoS One 7:e40514 doi:10.1371/journal.pone.0040514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goodman DD, Hluhanich R, Waters JM, Margot NA, Fransen S, Gupta S, Huang W, Parkin N, Borroto-Esoda K, Svarovskaia ES, Miller MD, McColl DJ. 2008. Abstr. XVII Int. HIV Drug Resist. Workshop, Stiges, Spain, abstr 13 [Google Scholar]
- 22. Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, Mitchell M, Guo H, Babaoglu K, Liu X, Geleziunas R, Sakowicz R. 2012. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 287:21189–21203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsiang M, Jones GS, Hung M, Mukund S, Han B, Liu X, Babaoglu K, Lansdon E, Chen X, Todd J, Cai T, Pagratis N, Sakowicz R, Geleziunas R. 2009. Affinities between the binding partners of the HIV-1 integrase dimer-lens epithelium-derived growth factor (IN dimer-LEDGF) complex. J. Biol. Chem. 284:33580–33599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, Tian H, Smith D, Winslow GA, Capon DJ, Whitcomb JM. 2000. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 44:920–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Svarovskaia ES, Feng JY, Margot NA, Myrick F, Goodman D, Ly JK, White KL, Kutty N, Wang R, Borroto-Esoda K, Miller MD. 2008. The A62V and S68G mutations in HIV-1 reverse transcriptase partially restore the replication defect associated with the K65R mutation. J. Acquir. Immune Defic. Syndr. 48:428–436 [DOI] [PubMed] [Google Scholar]
- 26. Moser MJ, Ruckstuhl M, Larsen CA, Swearingen AJ, Kozlowski M, Bassit L, Sharma PL, Schinazi RF, Prudent JR. 2005. Quantifying mixed populations of drug-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 49:3334–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu H, Huang Y, Dykes C, Liu D, Ma J, Perelson AS, Demeter LM. 2006. Modeling and estimation of replication fitness of human immunodeficiency virus type 1 in vitro experiments by using a growth competition assay. J. Virol. 80:2380–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y, Klock H, Yin H, Wolff K, Bieza K, Niswonger K, Matzen J, Gunderson D, Hale J, Lesley S, Kuhen K, Caldwell J, Brinker A. 2005. Homogeneous high-throughput screening assays for HIV-1 integrase 3beta-processing and strand transfer activities. J. Biomol. Screen. 10:456–462 [DOI] [PubMed] [Google Scholar]
- 29. Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. 2008. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 47:9345–9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Metifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, Marchand C, Pommier Y. 2011. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS 25:1175–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reigadas S, Masquelier B, Calmels C, Laguerre M, Lazaro E, Vandenhende M, Neau D, Fleury H, Andreola ML. 2011. Structure-analysis of the HIV-1 integrase Y143C/R raltegravir resistance mutation in association with the secondary mutation T97A. Antimicrob. Agents Chemother. 55:3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hare S, Smith SJ, Metifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, Cherepanov P. 2011. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 80:565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. 2010. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U. S. A. 107:20057–20062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McColl DJ, Fransen S, Gupta S, Parkin N, Margot N, Hluhanich R, Chen X, Chuck S, Cheng AK, Miller MD. 2007. Abstr. 11th Eur. AIDS Conf., Madrid, Spain, abstr P7.1/03 [Google Scholar]
- 36. McColl DJ, Fransen S, Gupta S, Parkin N, Margot N, Ledford R, Chen J, Chuck S, Cheng AK, Miller MD. 2007. Abstr. 16th Int. HIV Drug Resist. Workshop, Bridgetown, Barbados, abstr 9 [Google Scholar]
- 37. Ceccherini-Silberstein F, Van Baelen K, Armenia D, Trignetti M, Rondelez E, Fabeni L, Scopelliti F, Pollicita M, Van Wesenbeeck L, Van Eygen V, Dori L, Sarmati L, Aquaro S, Palamara G, Andreoni M, Stuyver LJ, Perno CF. 2010. Secondary integrase resistance mutations found in HIV-1 minority quasispecies in integrase therapy-naive patients have little or no effect on susceptibility to integrase inhibitors. Antimicrob. Agents Chemother. 54:3938–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Charpentier C, Karmochkine M, Laureillard D, Tisserand P, Belec L, Weiss L, Si-Mohamed A, Piketty C. 2008. Drug resistance profiles for the HIV integrase gene in patients failing raltegravir salvage therapy. HIV Med. 9:765–770 [DOI] [PubMed] [Google Scholar]
- 39. Canducci F, Sampaolo M, Marinozzi MC, Boeri E, Spagnuolo V, Galli A, Castagna A, Lazzarin A, Clementi M, Gianotti N. 2009. Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS 23:455–460 [DOI] [PubMed] [Google Scholar]
- 40. Reigadas S, Anies G, Masquelier B, Calmels C, Stuyver LJ, Parissi V, Fleury H, Andreola ML. 2010. The HIV-1 integrase mutations Y143C/R are an alternative pathway for resistance to raltegravir and impact the enzyme functions. PLoS One 5:e10311 doi:10.1371/journal.pone.0010311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cooper DA, Gatell J, Rockstroh J, Katlama C, Yeni P, Lazzarin A, Chen J, Isaacs R, Teppler H, Nguyen B-Y. 2007. Abstr. 14th Conf. Retroviruses Opportun. Infect., Los Angeles, CA, abstr 105aLB [Google Scholar]
- 42. Steigbigel R, Kumar P, Eron JJ, Schechter M, Markowitz M, Loutfy M, Zhao J, Isaacs R, Nguyen B-Y, Teppler H. 2007. Abstr. 14th Conf. Retroviruses Opportun. Infect., Los Angeles, CA, abstr 105bLB [Google Scholar]
- 43. Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, Tomberlin GH, Carter HL, III, Broderick T, Sigethy S, Seki T, Kobayashi M, Underwood MR. 2011. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob. Agents Chemother. 55:4552–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seki T, Kobayashi M, Wakasa-Morimoto C, Yoshinaga T, Sato A, Fujiwara T, Underwood MR, Garvey EP, Johns BA. 2010. Abstr. 17th Conf. Retroviruses Opportun. Infect., San Francisco, CA, abstr 555 [Google Scholar]
- 45. DeJesus E, Cohen C, Elion R, Ortiz R, Maroldo L, Franson S, Pesano R. 2007. Abstr. 4th Int. AIDS Soc. Conf. HIV Pathog. Treat., Sydney, Australia, abstr TUPEB032 [Google Scholar]
- 46. Vavro CL, Dudas KC, Hasan S, Huang JO, Yeo JM, Underwood MR. 2012. Dolutegravir treatment of HIV subjects with raltegravir resistance: integrase resistance evolution in cohort II of the VIKING study. Antivir. Ther. 17:A13 [Google Scholar]
- 47. Weber J, Rose JD, Wylie D, Vazquez AC, Rhea AM, Winner D, Margot N, McColl D, Miller M, Quinones-Mateu ME. 2010. Resistance mutations in protease, reverse transcriptase and integrase genes: do they have an epistatic effect on drug susceptibility and/or HIV-1 replicative fitness? Antivir. Ther. 15:A94 [Google Scholar]
- 48. Nakahara K, Wakasa-Morimoto C, Kobayashi M, Miki S, Noshi T, Seki T, Kanamori-Koyama M, Kawauchi S, Suyama A, Fujishita T, Yoshinaga T, Garvey EP, Johns BA, Foster SA, Underwood MR, Sato A, Fujiwara T. 2009. Secondary mutations in viruses resistant to HIV-1 integrase inhibitors that restore viral infectivity and replication kinetics. Antiviral Res. 81:141–146 [DOI] [PubMed] [Google Scholar]
- 49. Buzon MJ, Marfil S, Puertas MC, Garcia E, Clotet B, Ruiz L, Blanco J, Martinez-Picado J, Cabrera C. 2008. Raltegravir susceptibility and fitness progression of HIV type-1 integrase in patients on long-term antiretroviral therapy. Antivir. Ther. 13:881–893 [PubMed] [Google Scholar]
- 50. Quercia R, Dam E, Perez-Bercoff D, Clavel F. 2009. Selective-advantage profile of human immunodeficiency virus type 1 integrase mutants explains in vivo evolution of raltegravir resistance genotypes. J. Virol. 83:10245–10249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu Z, Kuritzkes DR. 2010. Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J. Acquir. Immune Defic. Syndr. 55:148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mukherjee R, Jensen ST, Male F, Bittinger K, Hodinka RL, Miller MD, Bushman FD. 2011. Switching between raltegravir resistance pathways analyzed by deep sequencing. AIDS 25:1951–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Delelis O, Malet I, Na L, Tchertanov L, Calvez V, Marcelin AG, Subra F, Deprez E, Mouscadet JF. 2009. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 37:1193–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dicker IB, Samanta HK, Li Z, Hong Y, Tian Y, Banville J, Remillard RR, Walker MA, Langley DR, Krystal M. 2007. Changes to the HIV long terminal repeat and to HIV integrase differentially impact HIV integrase assembly, activity, and the binding of strand transfer inhibitors. J. Biol. Chem. 282:31186–31196 [DOI] [PubMed] [Google Scholar]
- 55. Johnson AA, Santos W, Pais GC, Marchand C, Amin R, Burke TR, Jr, Verdine G, Pommier Y. 2006. Integration requires a specific interaction of the donor DNA terminal 5′-cytosine with glutamine 148 of the HIV-1 integrase flexible loop. J. Biol. Chem. 281:461–467 [DOI] [PubMed] [Google Scholar]
- 56. Gerton JL, Ohgi S, Olsen M, DeRisi J, Brown PO. 1998. Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J. Virol. 72:5046–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Khan E, Mack JP, Katz RA, Kulkosky J, Skalka AM. 1991. Retroviral integrase domains: DNA binding and the recognition of LTR sequences. Nucleic Acids Res. 19:851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.