

Abstract
Multidrug-resistant tuberculosis (MDR-TB) threatens global TB control. The lengthy treatment includes one of the injectable drugs kanamycin, amikacin, and capreomycin, usually for the first 6 months. These drugs have potentially serious toxicities, and when given as intramuscular injections, dosing can be painful. Advances in particulate drug delivery have led to the formulation of capreomycin as the first antituberculosis drug available as a microparticle dry powder for inhalation and clinical study. Delivery by aerosol may result in successful treatment with lower doses. Here we report a phase I, single-dose, dose-escalating study aimed at demonstrating safety and tolerability in healthy subjects and measuring pharmacokinetic (PK) parameters. Twenty healthy adults (n = 5 per group) were recruited to self-administer a single dose of inhaled dry powder capreomycin (25-mg, 75-mg, 150-mg, or 300-mg nominal dose) using a simple, handheld delivery device. Inhalations were well tolerated by all subjects. The most common adverse event was mild to moderate transient cough, in five subjects. There were no changes in lung function, audiometry, or laboratory parameters. Capreomycin was rapidly absorbed after inhalation. Systemic concentrations were detected in each dose group within 20 min. Peak and mean plasma concentrations of capreomycin were dose proportional. Serum concentrations exceeded 2 μg/ml (MIC for Mycobacterium tuberculosis) following the highest dose; the half-life (t1/2) was 4.8 ± 1.0 h. A novel inhaled microparticle dry powder formulation of capreomycin was well tolerated. A single 300-mg dose rapidly achieved serum drug concentrations above the MIC for Mycobacterium tuberculosis, suggesting the potential of inhaled therapy as part of an MDR-TB treatment regimen.
INTRODUCTION
Multidrug-resistant tuberculosis (MDR-TB) is difficult to treat and readily spread and threatens global tuberculosis control, especially where HIV coinfection is common. According to the WHO, an estimated 440,000 new cases of MDR-TB occur each year (1, 2), but only a small fraction are receiving quality-assured treatment (1). In most public health treatment programs, 18 to 24 months of therapy with four or more second-line drugs are required. Second-line drugs are less efficacious, more toxic, and associated with treatment success rates of only 40 to 80% (1, 3–6). According to current WHO MDR-TB treatment guidelines, at least one injectable antibiotic (kanamycin, amikacin, or capreomycin) is an essential part of the 6-month intensive phase of treatment. These 3- to 7-times-per-week injections are painful for patients, especially for children and those with little muscle mass. Injections require skilled health care staff and place them at risk of needle stick injuries and infections (3). Treatment with injectable drugs is associated with systemic toxicity, including nephrotoxicity, which is typically mild and reversible, and ototoxicity, which can be significant and irreversible. Dosing must also be adjusted in individuals with preexisting renal insufficiency. Lastly, injectable drugs require a cold chain for storage and sterile conditions for reconstitution.
Despite these drawbacks, injectable drugs remain an important part of the treatment of MDR-TB because of their in vitro activity against actively replicating M. tuberculosis, their ability to inhibit the development of drug resistance when used in combination with other drugs (7), and the lack of alternative drugs to replace them. There are two important classes of injectable drugs: the aminoglycosides (e.g., amikacin and kanamycin) and the polypeptide agent capreomycin. Capreomycin has also been shown to have activity against nonreplicating forms of M. tuberculosis (8). The parenteral administration of aminoglycosides in humans, however, is associated with relatively low lung concentrations of these antibiotics, and high peak serum concentrations (Cmax) are needed to obtain microbiologically active concentrations in the alveoli, in the pulmonary interstitium, and intracellularly within macrophages, the most common sites of M. tuberculosis infection. In the few published clinical studies specifically evaluating alveolar lining fluid concentrations of aminoglycosides after systemic administration, alveolar concentrations ranged from 32% to 50% of the peak systemic concentration (9–11). Furthermore, preclinical studies of capreomycin pharmacokinetics in mice have shown that parenteral doses penetrate poorly into the lung (12). Since clinical effectiveness of the aminoglycosides and capreomycin is closely correlated with the ratio of Cmax to MIC, based on in vitro and in vivo studies with Gram-negative bacteria and with M. tuberculosis (13–15), one of the major challenges in TB chemotherapy is the ability to achieve both adequate lung compartment-specific Cmax and systemic Cmax (for extrapulmonary sites of infection) while limiting or avoiding systemic toxicity.
Inhaled drug delivery offers the potential to achieve high concentrations of pharmacologic agents in the lungs while producing adequate systemic concentrations through alveolar-capillary absorption to treat extrapulmonary sites of infection. It also offers the potential of reduced manufacturing costs and administration of lower doses. Inhaled nebulized antimicrobials are used in the treatment of pulmonary infections (both viral and bacterial) in various patient populations, such as in individuals with cystic fibrosis and bronchiectasis and for antimicrobial prophylaxis (fungal and bacterial) of the lung following pulmonary transplantation (16–18). An important requirement in the management of tuberculosis, especially for HIV-infected individuals, is to achieve therapeutic systemic drug concentrations in order to treat extrapulmonary foci of infection. Fortunately, the science of drug delivery continues to evolve, converting parenteral, oral, or nebulized drugs into chemically or physically modified inhaled formulations that enhance delivery to specific distal lung tissue compartments, with or without systemic absorption, as required (19, 20). Inhaled dry powder formulations of drugs are particularly attractive because they may help avoid the requirements for electricity, cold chains for storage, and materials for reconstitution needed to administer nebulized formulations of drugs, especially in resource-limited settings. One such unique, dry powder, microparticle formulation of capreomycin exhibited good aerosolization properties, physical-chemical stability, efficient lung deposition, and absorption in preclinical testing (21).
Capreomycin sulfate is a polypeptide antibiotic indicated for infections caused by drug-resistant strains of M. tuberculosis. Capreomycin is administered intramuscularly (i.m.) or intravenously (i.v.) at a usual dose of 15 to 20 mg/kg of body weight/day for at least 6 months, but it may be needed for as long as 2 years if tolerated. Capreomycin is currently used in combination with at least three or four other antituberculosis agents to which the patient's strain is susceptible (22). In two clinical studies of 10 patients each, peak serum concentrations following administration of 1 g of capreomycin given intramuscularly reached 28 and 32 μg/ml, respectively (range, 20 to 47 μg/ml), 1 to 2 h after administration, and by 24 h postdose, concentrations were very low (Eli Lilly, unpublished data). Among 722 patients treated with parenterally administered capreomycin, 3% developed clinical hearing loss (FDA label) (23).
Preceding the current clinical study, preclinical toxicological studies of the inhaled dry powder formulation in rats and dogs given the drug for 14 days and monitored for up to 14 days postexposure showed mild, reversible, dose-dependent irritation of the respiratory tract histopathologically and helped establish a no-adverse-effect level (NOAEL) of 7 mg/kg in the dog model (unpublished data). In this first-in-human study, we used capreomycin in inhaled doses ranging from doses 22-fold lower than the NOAEL in dogs (for the 25-mg group) to 1.8-fold lower than the NOAEL in dogs (for the 300-mg group). Preclinical pharmacokinetic (PK) studies of inhaled capreomycin in the guinea pig have shown efficient drug absorption from the lung into blood and a linear terminal phase of the plasma concentration-time curve (24). Together, these preclinical data suggested that the inhalable microparticle formulation of capreomycin used in this clinical study could safely produce both pulmonary and systemic concentrations above the MIC for M. tuberculosis. The objectives of this phase I study were therefore to determine the safety, tolerability, and systemic pharmacokinetics of inhaled dry powder capreomycin in healthy adult volunteers.
MATERIALS AND METHODS
Study design.
We conducted a phase I clinical study among 20 healthy adult volunteers. Inclusion criteria included an age of 18 to 65 years; no history of current or recent (within the past 6 months) tobacco use; no current medication usage; normal renal, hematologic, and liver tests; and FEV1 (forced expiratory volume in 1 second), FVC (forced vital capacity), and DLCO (diffusing capacity of the lung for carbon monoxide) greater than or equal to the lower limit of the 95% confidence interval for the predicted value for the subject based on age and height. Exclusion criteria included pregnancy and current breastfeeding; receipt of capreomycin, aminoglycosides, or any other investigational drug within the 30-day period before study drug administration; or a history of intolerance of capreomycin or aminoglycoside antibiotics. Additional exclusion criteria included use of prescription medications (except hormonal contraception) in the 30-day period before study drug administration, use of nonprescription medication within 7 days before study drug administration, abnormalities on any screening examination or test, or history of asthma.
Baseline and 24-h-postdose (final) pulmonary function testing was performed by using pulmonary function testing equipment (Morgan Scientific, Haverhill, MA). At 1, 2, 3, 4, 6, 8, 12, 30, and 36 h postdose, a handheld spirometer (KoKo Legend; nSpire Health) was used to measure FEV1, FVC, and FEV1/FVC. Upon enrollment, subjects were sequentially assigned to one of four escalating dosage groups (25 mg, 75 mg, 150 mg, and 300 mg capreomycin; n = 5 per group).
Each subject received a single dose of inhaled capreomycin according to the dose group, which was self-administered by using a handheld inhaler (Cyclohaler; Plastiape, Italy) (Fig. 1). Capsules (each containing 25 mg of capreomycin and 5 mg of the excipient l-leucine, at an 80:20 ratio) were inserted sequentially into the handheld inhaler for self-administration. Capsules were punctured within the handheld inhaler to release the drug prior to inhalation. Group 1 (25-mg dose) self-administered one capsule, while group 4 (300-mg dose) self-administered 12 capsules.
Fig 1.

Handheld inhaler for self-administration of capreomycin. (Left) Inhaler in full view; (middle) inhaler opened, showing the chamber that holds the capsule; (right) inhaler with capsule inserted.
Each subject underwent blood sampling for capreomycin pharmacokinetic analysis at 13 time points: predose; 10, 20, 30, and 45 min postdose; as well as 1, 2, 3, 4, 6, 8, 12, and 24 h postdose. Drug concentrations were assayed by using a highly sensitive high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) methodology. The lower limit of quantitation of capreomycin in human plasma was 100 ng/ml, with an accuracy of 88.2%, and the linear range of quantitation was 100 to 5,000 ng/ml. The HPLC-MS/MS assay was validated by using viomycin as an internal standard. In addition, subjects underwent blood sampling, urinalysis, audiometry, and chest X ray (CXR) at 24 h postdose for safety assessments. The study scheme is illustrated in Fig. 2.
Fig 2.

Study flow chart/scheme. CXR, chest X ray; U/A, urinalysis; PK, pharmacokinetic.
Study drug formulation.
Capreomycin inhalation powder was developed by using spray-drying technology (21) and manufactured and quality control released by using fully good manufacturing practices (GMP)-compliant processes and facilities. For the 80:20 (capreomycin-leucine excipient) dry powder formulation, prior work using scanning electron microscopy demonstrated the mean geometric particle diameter (at a pressure of 1 Pa) to be 3.69 ± 1.68 μm (21). Particles with lower capreomycin content (e.g., 50 to 70 wt%) had higher mean geometric diameters than the 80:20 particles (21). Mass mean aerodynamic diameters (MMADs) of the 80:20 formulation were 4.74 ± 0.07 μm, as determined by cumulative mass distribution curves of aerosol collections on cascade impactors (21), suggesting some aggregation of particles, as has been observed for dry powders (25). In vitro estimation of particle distribution based on its size suggested that the 80:20 formulation would yield both bronchial and alveolar deposition of drug. The study drug was stored at 2°C to 8°C (note that, with additional stability data, the product may now be stored at controlled room temperature).
Statistical analyses.
Clinical and demographic data on study subjects are reported using summary measures. Pharmacokinetic data were also summarized descriptively and stratified by dosing group by using SAS version 8.2. The PK population consisted of all subjects who enrolled in the study, received the full dose of study drug, and completed the study. Subjects for whom select PK parameters could not be adequately characterized (e.g., half-life [t1/2] was not reported, per internal standard operating procedures, if the adjusted r2 value was ≤0.75 and the time points used in estimating the half-life spanned less than 1 half-life) were excluded from the calculation of overall means and statistics for those select parameters. PK analyses were performed by using standard, noncompartmental methods.
Human subject research approval.
All research was approved by the institutional review board of the Brigham and Women's Hospital and was conducted in accordance with good clinical practice and good laboratory practice guidelines.
RESULTS
Subject characteristics.
Twenty adult subjects were enrolled in this study. Their characteristics are summarized in Table 1.
Table 1.
Clinical characteristics of the study subjects
| Clinical parametera | Value |
|---|---|
| Male/female ratio | 17:3 |
| Mean age (yr) (range) | 39.4 (18–59) |
| Mean wt (lb) ± SD | 174.5 ± 38.4 |
| Mean height (in.) ± SD | 69.3 ± 3.8 |
| Mean BMI (lb/in2) ± SD | 25.6 ± 5.7 |
| Mean FEV1 (baseline) (liters) ± SD | 4.04 ± 0.78 |
| Mean FVC (baseline) (liters) ± SD | 4.06 ± 1.20 |
| Mean FEV1/FVC ratio (baseline) ± SD | 0.78 ± 0.09 |
| Mean creatinine concn (baseline) (mg/dl) ± SD | 0.97 ± 0.17 |
BMI, body mass index.
Pharmacokinetic data.
Capreomycin was detectable in serum samples within 20 min of inhalation in 17/20 (85%) subjects. In three subjects, the initial detection of capreomycin occurred at between 1 and 3 h postdose. These three subjects were all in the 25-mg (lowest) dose group. In the 25-mg dose group, the mean area under the concentration-time curve from 0 h to the last measurable concentration (AUC0–t) was 969 h · ng/ml, while in the 300-mg dose group, the mean AUC0–t was 19,959 h · ng/ml. The mean Cmax values for each successive dose group were 169 ng/ml, 569 ng/ml, 972 ng/ml, and 2,315 ng/ml. The average times to maximal concentration (Tmax) in each group were 2.4 h (25-mg group), 2.1 h (75-mg group), 3.2 h (150-mg group), and 2.8 h (300-mg group). The apparent t1/2 values were 4.2 ± 1.1 h (75-mg group), 4.4 ± 1.3 h (150-mg group), and 4.8 ± 1.0 h (300-mg group). Table 2 summarizes these values.
Table 2.
PK data for each dose groupa
| Measure | Value for group |
|||
|---|---|---|---|---|
| 1 (25-mg dose) | 2 (75-mg dose) (n = 5 subjects) | 3 (150-mg dose) (n = 5 subjects) | 4 (300-mg dose) (n = 5 subjects) | |
| AUC0–t (h · ng/ml) | ||||
| Mean (no.) | 969 (5) | 3,555 (5) | 7,019 (5) | 19,959 (5) |
| Range (min, max) | 51, 2,176 | 1,450, 5,274 | 1,761, 14,187 | 7,554, 35,855 |
| Median | 1,083 | 4,035 | 4,984 | 16,843 |
| AUC0–∞ (h · ng/ml) | ||||
| Mean (no.) | NR | 5,683 (3) | 9,333 (4) | 22,425 (5) |
| Range (min, max) | 4,785, 6,516 | 5,488,15,253 | 9,505, 37,194 | |
| Median | 5,747 | 8,296 | 22,503 | |
| Cmax (ng/ml) | ||||
| Mean (no.) | 169 (5) | 569 (5) | 972 (5) | 2,315 (5) |
| Range (min, max) | 102, 250 | 218, 970 | 250, 1,947 | 939, 4,579 |
| Median | 178 | 513 | 786 | 2,105 |
| Tmax (h) | ||||
| Mean (no.) | 2.4 (5) | 2.1 (5) | 3.2 (5) | 2.8 (5) |
| Range (min, max) | 2.0, 3.0 | 0.5, 4.0 | 1.0, 6.0 | 2.0, 3.0 |
| Median | 2.0 | 2.0 | 3.0 | 3.0 |
| kel (1/h) | ||||
| Mean (no.) | NR | 0.171 (3) | 0.167 (4) | 0.151 (5) |
| Range (min, max) | 0.127, 0.206 | 0.111, 0.197 | 0.116, 0.214 | |
| Median | 0.180 | 0.180 | 0.138 | |
| t1/2 (h) | ||||
| Mean (no.) | NR | 4.2 (3) | 4.4 (4) | 4.8 (5) |
| Range (min, max) | 3.4, 5.4 | 3.5, 6.3 | 3.2, 6.0 | |
| Median | 3.9 | 3.8 | 5.0 | |
| CL/F (ml/min/kg) | ||||
| Mean (no.) | NR | 3.13 (3) | 4.57 (4) | 3.46 (5) |
| Range (min, max) | 2.99, 3.38 | 1.99, 7.17 | 1.61, 6.63 | |
| Median | 3.02 | 4.55 | 2.37 | |
| V/F (liters/kg) | ||||
| Mean (no.) | NR | 1.14 (3) | 1.60 (4) | 1.35 (5) |
| Range (min, max) | 0.87, 1.42 | 1.06, 2.4 | 0.70, 2.65 | |
| Median | 1.13 | 1.47 | 1.11 | |
AUC0–t, area under the plasma concentration-time curve computed up to the last measurable concentration; AUC0–∞, area under the plasma concentration-time curve from time of dosing to infinity with extrapolation of the terminal phase; Cmax, peak drug concentration; Tmax, time to reach maximum plasma concentration; kel, terminal elimination rate constant; t1/2, terminal elimination half-life; CL/F, apparent total body clearance uncorrected for fraction absorbed after extravascular administration; V/F, apparent volume of distribution uncorrected for fraction absorbed based on the terminal elimination phase; NR, not calculated for reasons described in Materials and Methods.
The mean plasma drug concentrations versus time for each dosage group are shown in Fig. 3. Superimposed upon Fig. 3 is an MIC of 2 μg/ml (or 2,000 ng/ml) for M. tuberculosis based on values reported in other studies (26). Both the maximal plasma concentration (Cmax) and the AUC from 0 h to infinity (AUC0–∞) were dose proportional, as shown in Fig. 4.
Fig 3.

Graph of plasma concentration versus time for each group. The horizontal line represents an MIC of 2 μg/ml for M. tuberculosis inhibition by capreomycin.
Fig 4.
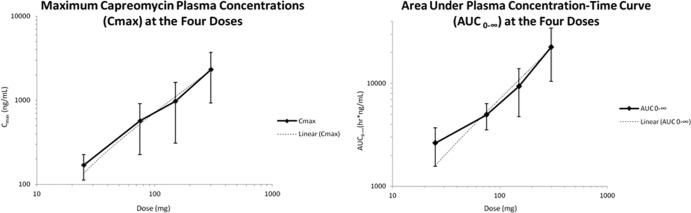
Graphs of dose proportionality of Cmax (left) and AUC (right).
Safety data.
Inhalation of capreomycin was not associated with alterations in clinical or laboratory parameters. Pulmonary function was unchanged postinhalation (Table 3). Renal function was similarly unchanged after study drug administration. There were no abnormalities in liver function tests; leukocyte, red blood cell, or platelet counts; or urine sediment in any patients. Audiometry was stable after dose administration.
Table 3.
Pulmonary function results pre- and postdosea
| Group | Mean FEV1 (liters) ± SD |
Mean FVC (liters) ± SD |
Mean FEV1/FVC (%)± SD |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Predose | Immediately postdose | 24 h postdose | Predose | Immediately postdose | 24 h postdose | Predose | Immediately postdose | 24 h postdose | |
| 1 | 4.00 ± 0.66 | 3.98 ± 0.65 | 4.09 ± 0.67 | 5.58 ± 1.56 | 5.42 ± 1.28 | 5.64 ± 1.31 | 73.8 ± 11.3 | 75.0 ± 11.0 | 73.8 ± 9.47 |
| 2 | 4.00 ± 0.80 | 4.35 ± 2.16 | 4.02 ± 1.00 | 4.94 ± 0.45 | 5.75 ± 1.87 | 5.06 ± 0.51 | 80.2 ± 9.96 | 73.4 ± 18.4 | 77.8 ± 13.4 |
| 3 | 3.70 ± 1.05 | 3.63 ± 1.01 | 3.66 ± 1.00 | 4.74 ± 1.29 | 4.75 ± 1.21 | 4.74 ± 1.23 | 78.6 ± 7.02 | 76.6 ± 6.69 | 77.6 ± 7.06 |
| 4 | 4.45 ± 0.59 | 4.30 ± 0.56 | 4.40 ± 0.59 | 5.64 ± 0.82 | 5.43 ± 0.84 | 5.58 ± 0.81 | 79.4 ± 9.21 | 79.8 ± 6.83 | 79.2 ± 8.17 |
Values for FEV1 and FVC are means ± standard deviations for each group, in liters. Values for FEV1/FVC are percentages. Group 1, 25-mg dose; group 2, 75-mg dose; group 3, 150-mg dose; group 4, 300-mg dose.
Adverse events.
There were no serious or severe adverse events in this study. Several subjects experienced mild/moderate symptoms, the most common one being cough, occurring in 5 subjects. Table 4 lists all the adverse events occurring in the study and their relationship to the study drug. All adverse events were self-limited. When coughing occurred, it started upon administration of study drug and ended within 5 min of the last capsule that was actuated. Coughing was reported by subjects as being mild/moderate.
Table 4.
List of adverse events
| Subject | Dose (mg) | Adverse event (severity) | Relationship to study drug |
|---|---|---|---|
| 001 | 25 | Cough (mild) | Related |
| 006 | 75 | Cough (mild) | Related |
| 008 | 75 | Headache (mild) | Probably unrelated |
| Chest tightness (mild) | Possibly related | ||
| 011 | 150 | Cough (moderate) | Related |
| i.v. infiltration (mild) | Unrelated | ||
| 012 | 150 | Cough (moderate) | Related |
| 017 | 300 | Cough (moderate) | Related |
| Migraine headache (moderate) | Probably unrelated | ||
| Vomiting (moderate) | Probably unrelated |
DISCUSSION
In this study, we evaluated the safety and systemic pharmacokinetics of a novel dry powder formulation of the antituberculous drug capreomycin, a polypeptide antibiotic that inhibits mycobacterial protein biosynthesis. After administration of a single 300-mg inhaled dose to healthy volunteers, systemic concentrations were equal to or higher than reported MICs (2 μg/ml) for M. tuberculosis (26). From a safety and tolerability perspective, inhalation was easy to self-administer, well tolerated, and not associated with any significant alterations in pulmonary function, renal function, or other clinical parameters.
Capreomycin elimination after single-dose inhalation showed biphasic kinetics in all dose groups (Fig. 3). Absorption of the drug through the lungs was rapid, as illustrated by detection of drug within 20 min among all subjects receiving 75 mg or more of capreomycin. The apparent t1/2 after inhalation ranged from 4.2 to 4.8 h. Maximal drug concentrations (Cmax) and the AUC were dose proportional (Fig. 4), and since the highest nominal dose used in this phase I study (300 mg) was below the conservative estimate of the NOAEL determined through preclinical testing in a canine model, higher doses may be reasonably expected to safely produce even higher Cmax and AUC. However, 300 mg required sequential inhalation of 12 25-mg capsules. While higher-dose capsules are feasible, achieving the usual 15-mg/kg dose in a 70-kg adult (1,000 mg) may require a logistically unacceptably large powder load and divided doses, a practice usually avoided in the ambulatory treatment of drug-resistant tuberculosis, where full supervision of ingestion is standard practice.
When administered parenterally as part of current MDR-TB treatment regimens, capreomycin must be given in doses that result in very high peak serum concentrations. Although these high Cmax concentrations inhibit mycobacterial growth, they are often accompanied by the risk of toxicities (FDA label). Moreover, some of these effects are potentiated when capreomycin is used concurrently with other nephrotoxic and ototoxic drugs, further limiting its use. It appears that inhaled capreomycin could be a promising option in MDR-TB management. Future studies of the pharmacokinetic and pharmacodynamic profiles of higher nominal doses and multiple-daily-dosing schemes in healthy volunteers and MDR-TB patients can help support the potential incorporation of inhaled capreomycin into treatment regimens.
The findings of this clinical study are more significant in light of preclinical studies of inhaled dry powder capreomycin in guinea pigs, which highlight an important critical advantage of the inhaled route of delivery over parenteral administration. The systemic concentrations achieved after a 14.5-mg/kg inhaled dose in the guinea pig were comparable to the systemic concentrations seen 2 h after a 14.5-mg/kg single dose given i.v. or i.m. (24). Furthermore, the inhaled route of delivery did not produce the high initial spike in systemic concentrations seen within the first hour of the i.v. or i.m. dose (24). After a single 20-mg/kg inhaled dose, lung tissue homogenate concentrations reached 40 to 100 times the MIC for M. tuberculosis, while bronchoalveolar lavage fluid concentrations reached 40 to 50 times the MIC (27). Eight hours after administration of a 20-mg/kg inhaled dose, lung tissue drug concentrations averaged 100 μg/g tissue and far exceeded the concentrations achieved by the i.m. route of administration (27). In the multiple-dose portion of these studies, the AUC, half-life, and lung concentrations were higher and longer-lasting after the second and third doses (27). In addition, daily administration of the inhaled drug over 4 weeks (at 14.5 mg/kg) resulted in significant reductions in bacterial counts and lung pathology in M. tuberculosis-infected guinea pigs and the effect was greater than the pharmacodynamic effect on bacterial counts in the lung seen with the i.m. route of delivery of the 20-mg/kg dose (24).
Taken together, the pharmacokinetic and pharmacodynamic data on capreomycin in guinea pigs show that the pulmonary route of delivery of a single dose achieved systemic concentrations above an MIC of 2 μg/ml for M. tuberculosis and produced a longer systemic half-life of the drug than that following parenteral administration (21, 24). In humans, the systemic half-life of capreomycin after parenteral administration has been shown to be on the order of 2 to 3 h (28, 29). This represents only 60% of the apparent systemic half-life observed in the current study after single-dose pulmonary inhalation. In the guinea pig model, inhaled drug delivery also resulted in higher concentrations in the lung tissue and greater systemic mean residence times than when administered parenterally (21, 27). This finding in an animal model provides evidence that inhaled capreomycin is superior to parenterally administered capreomycin in terms of achieving therapeutic pulmonary drug concentrations. Further clinical evaluation, including bronchoscopy and lung lavage sampling, could be helpful in expanding this finding to patients. A lingering concern is the amount of powder and number of doses needed to achieve therapeutic concentrations. It remains to be seen whether inhaled capreomycin will be a practical therapeutic option in adults. Regular injections are a particular hardship for children, and the substitution of inhaled capreomycin in that population seems especially desirable and feasible. Studies done with asthmatic children in Europe, the Americas, and Asia have shown the feasibility and acceptability of the use of a metered dose and a dry powder inhaler device of asthma drugs by children (30–33).
The results should also be considered in light of some of the limitations of this study. First, subjects were predominantly male and healthy. Second, subjects had normal pulmonary function, but capreomycin distribution within the airways and lungs after inhalation may be different in patients with underlying obstructive lung disease. The pharmacokinetics of inhaled capreomycin in TB patients with or without lung disease and other comorbidities may be quite different. In some dose groups, subjects whose half-life data did not meet inclusion criteria for analysis (as described above) were excluded. This in turn affected the calculation of all of the half-life-dependent PK parameters for that group.
In conclusion, the results of this first-in-human study of inhaled capreomycin provide both a sound basis and a need for further testing of inhaled capreomycin for therapeutic efficacy. Given the safety profile of the doses used in this study, future studies should evaluate higher doses of dry powder capreomycin with the goal of further increasing the Cmax/MIC and AUC/MIC ratios. The doses evaluated in the current study were below the NOAEL seen in the preclinical canine model, suggesting that testing of higher doses could be safely undertaken. If proven efficacious, the use of dry powder capreomycin has the potential to significantly improve the treatment of MDR-TB by extending its ability to be administered in a wider range of clinical settings, including in community-based treatment models, in settings with limited resources, and through self-administration under supervision. Its use in pediatric drug-resistant tuberculosis is also attractive. Further studies to develop and test dry powder formulations of this and other TB drugs should be encouraged.
ACKNOWLEDGMENTS
We thank Eli Lilly, the Gates Foundation, and the NIAID. We also thank Hisun for donation of capreomycin plus other resources and expertise. Finally, we greatly appreciate the efforts of the Brigham and Women's clinical research staff, including Margaret Oliner.
Funding for this study came from the Gates Foundation.
The funding source had no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; or in the decision to submit the paper for publication.
A.S.D., M.K., B.G., P.B.F., and E.N. were responsible for the design and conduct of the study and data interpretation. A.S.D., M.K., B.G., A.J.H., P.B.F., and E.N. wrote, reviewed, and revised the manuscript.
Footnotes
Published ahead of print 25 March 2013
REFERENCES
- 1. World Health Organization 2011. WHO report 2011: global tuberculosis control. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/2011/gtbr11_full.pdf [Google Scholar]
- 2. Institute of Medicine 2012. Facing the reality of drug-resistant tuberculosis in India: challenges and potential solutions. Summary of a joint workshop. National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- 3. World Health Organization 2009. WHO report 2009: management of MDR-TB. A field guide. World Health Organization, Geneva, Switzerland: http://whqlibdoc.who.int/publications/2009/9789241547765_eng.pdf [Google Scholar]
- 4. Jeon DS, Shin DO, Park SK, Seo JE, Seo HS, Cho YS, Lee JY, Kim DY, Kong SJ, Kim YS, Shim TS. 2011. Treatment outcome and mortality among patients with multidrug-resistant tuberculosis in tuberculosis hospitals of the public sector. J. Korean Med. Sci. 26:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orenstein EW, Basu S, Shah NS, Andrews JR, Friedland GH, Moll AP, Gandhi NR, Galvani AP. 2009. Treatment outcomes among patients with multidrug-resistant tuberculosis: systematic review and meta-analysis. Lancet Infect. Dis. 9:153–161 [DOI] [PubMed] [Google Scholar]
- 6. Tabarsi P, Chitsaz E, Baghaei P, Shamaei M, Farnia P, Marjani M, Kazempour M, Amiri M, Mansouri D, Masjedi MR, Velayati AA, Caminero JA. 2010. Impact of extensively drug-resistant tuberculosis on treatment outcome of multidrug-resistant tuberculosis patients with standardized regimen: report from Iran. Microb. Drug Resist. 16:81–86 [DOI] [PubMed] [Google Scholar]
- 7. Donald PR, Sirgel FA, Venter A, Smit E, Parkin DP, Van de Wal BW, Mitchison DA. 2001. The early bactericidal activity of amikacin in pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 5:533–538 [PubMed] [Google Scholar]
- 8. Heifets L, Simon J, Pham V. 2005. Capreomycin is active against non-replicating M. tuberculosis. Ann. Clin. Microbiol. Antimicrob. 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braude AC, Hornstein A, Klein M, Vas S, Rebuck AS. 1983. Pulmonary disposition of tobramycin. Am. Rev. Respir. Dis. 127:563–565 [DOI] [PubMed] [Google Scholar]
- 10. Carcas AJ, Garcia-Satue JL, Zapater P, Frias-Iniesta J. 1999. Tobramycin penetration into epithelial lining fluid of patients with pneumonia. Clin. Pharmacol. Ther. 65:245–250 [DOI] [PubMed] [Google Scholar]
- 11. Valcke YJ, Vogelaers DP, Colardyn FA, Pauwels RA. 1992. Penetration of netilmicin in the lower respiratory tract after once-daily dosing. Chest 101:1028–1032 [DOI] [PubMed] [Google Scholar]
- 12. Reisfeld B, Metzler CP, Lyons MA, Mayeno AN, Brooks EJ, Degroote MA. 2012. A physiologically based pharmacokinetic model for capreomycin. Antimicrob. Agents Chemother. 56:926–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heifets L, Lindholm-Levy P. 1989. Comparison of bactericidal activities of streptomycin, amikacin, kanamycin, and capreomycin against Mycobacterium avium and M. tuberculosis. Antimicrob. Agents Chemother. 33:1298–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deziel-Evans LM, Murphy JE, Job ML. 1986. Correlation of pharmacokinetic indices with therapeutic outcome in patients receiving aminoglycosides. Clin. Pharm. 5:319–324 [PubMed] [Google Scholar]
- 15. Moore RD, Lietman PS, Smith CR. 1987. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J. Infect. Dis. 155:93–99 [DOI] [PubMed] [Google Scholar]
- 16. Ryan G, Singh M, Dwan K. 2011. Inhaled antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2011:CD001021 doi:10.1002/14651858.CD001021.pub2 [DOI] [PubMed] [Google Scholar]
- 17. Rubin BK. 2008. Aerosolized antibiotics for non-cystic fibrosis bronchiectasis. J. Aerosol Med. Pulm. Drug Deliv. 21:71–76 [DOI] [PubMed] [Google Scholar]
- 18. Corcoran TE. 2009. Aerosol drug delivery in lung transplant recipients. Expert Opin. Drug Deliv. 6:139–148 [DOI] [PubMed] [Google Scholar]
- 19. Crowder TM, Rosati JA, Schroeter JD, Hickey AJ, Martonen TB. 2002. Fundamental effects of particle morphology on lung delivery: predictions of Stokes' law and the particular relevance to dry powder inhaler formulation and development. Pharm. Res. 19:239–245 [DOI] [PubMed] [Google Scholar]
- 20. Muttil P, Wang C, Hickey AJ. 2009. Inhaled drug delivery for tuberculosis therapy. Pharm. Res. 26:2401–2416 [DOI] [PubMed] [Google Scholar]
- 21. Fiegel J, Garcia-Contreras L, Thomas M, VerBerkmoes J, Elbert K, Hickey A, Edwards D. 2008. Preparation and in vivo evaluation of a dry powder for inhalation of capreomycin. Pharm. Res. 25:805–811 [DOI] [PubMed] [Google Scholar]
- 22. Elsevier 2008. Capreomycin. Tuberculosis (Edinb.) 88:89–91 [DOI] [PubMed] [Google Scholar]
- 23. Shin S, Furin J, Alcantara F, Hyson A, Joseph K, Sanchez E, Rich M. 2004. Hypokalemia among patients receiving treatment for multidrug-resistant tuberculosis. Chest 125:974–980 [DOI] [PubMed] [Google Scholar]
- 24. Garcia-Contreras L, Fiegel J, Telko MJ, Elbert K, Hawi A, Thomas M, VerBerkmoes J, Germishuizen WA, Fourie PB, Hickey AJ, Edwards D. 2007. Inhaled large porous particles of capreomycin for treatment of tuberculosis in a guinea pig model. Antimicrob. Agents Chemother. 51:2830–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louey MD, Van Oort M, Hickey AJ. 2004. Aerosol dispersion of respirable particles in narrow size distributions produced by jet-milling and spray-drying techniques. Pharm. Res. 21:1200–1206 [DOI] [PubMed] [Google Scholar]
- 26. Rastogi N, Labrousse V, Goh KS. 1996. In vitro activities of fourteen antimicrobial agents against drug susceptible and resistant clinical isolates of Mycobacterium tuberculosis and comparative intracellular activities against the virulent H37Rv strain in human macrophages. Curr. Microbiol. 33:167–175 [DOI] [PubMed] [Google Scholar]
- 27. Garcia-Contreras L, Muttil P, Fallon JK, Kabadi M, Gerety R, Hickey AJ. 2012. Pharmacokinetics of sequential doses of capreomycin powder for inhalation in guinea pigs. Antimicrob. Agents Chemother. 56:2612–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Black HR, Griffith RS, Brickler JF. 1963. Preliminary laboratory studies with capreomycin. Antimicrob. Agents Chemother. (Bethesda) 161:522–529 [PubMed] [Google Scholar]
- 29. Black HR, Griffith RS, Peabody AM. 1966. Absorption, excretion and metabolism of capreomycin in normal and diseased states. Ann. N. Y. Acad. Sci. 135:974–982 [DOI] [PubMed] [Google Scholar]
- 30. Konig P, Gayer D, Kantak A, Kreutz C, Douglass B, Hordvik NL. 1988. A trial of metaproterenol by metered-dose inhaler and two spacers in preschool asthmatics. Pediatr. Pulmonol. 5:247–251 [DOI] [PubMed] [Google Scholar]
- 31. O'Callaghan C, Everard ML, Bush A, Hiller EJ, Ross-Russell R, O'Keefe P, Weller P. 2002. Salbutamol dry powder inhaler: efficacy, tolerability, and acceptability study. Pediatr. Pulmonol. 33:189–193 [DOI] [PubMed] [Google Scholar]
- 32. Vichyanond P, Phanichyakarn P, Omar AH, Tam A, Wong E. 1994. Ease of handling and efficacy of Bricanyl Turbuhaler in Asian asthmatic children. Asian Pac. J. Allergy Immunol. 12:1–6 [PubMed] [Google Scholar]
- 33. Williams J, Richards KA. 1997. Ease of handling and clinical efficacy of fluticasone propionate Accuhaler/Diskus inhaler compared with the Turbohaler inhaler in paediatric patients. UK Study Group. Br. J. Clin. Pract. 51:147–153 [PubMed] [Google Scholar]