

Abstract
We investigated the efficacy and safety of liposomal clarithromycin formulations with different surface charges against clinical isolates of Pseudomonas aeruginosa from the lungs of cystic fibrosis (CF) patients. The liposomal clarithromycin formulations were prepared by the dehydration-rehydration method, and their sizes were measured using the dynamic-light-scattering technique. Encapsulation efficiency was determined by microbiological assay, and the stabilities of the formulations in biological fluid were evaluated for a period of 48 h. The MICs and minimum bactericidal concentrations (MBCs) of free and liposomal formulations were determined with P. aeruginosa strains isolated from CF patients. Liposomal clarithromycin activity against biofilm-forming P. aeruginosa was compared to that of free antibiotic using the Calgary Biofilm Device (CBD). The effects of subinhibitory concentrations of free and liposomal clarithromycin on bacterial virulence factors and motility on agar were investigated on clinical isolates of P. aeruginosa. The cytotoxicities of the liposome preparations and free drug were evaluated on a pulmonary epithelial cell line (A549). The average diameter of the formulations was >222 nm, with encapsulation efficiencies ranging from 5.7% to 30.4%. The liposomes retained more than 70% of their drug content during the 48-h time period. The highly resistant strains of P. aeruginosa became susceptible to liposome-encapsulated clarithromycin (MIC, 256 mg/liter versus 8 mg/liter; P < 0.001). Liposomal clarithromycin reduced the bacterial growth within the biofilm by 3 to 4 log units (P < 0.001), significantly attenuated virulence factor production, and reduced bacterial twitching, swarming, and swimming motilities. The clarithromycin-entrapped liposomes were less cytotoxic than the free drug (P < 0.001). These data indicate that our novel formulations could be a useful strategy to enhance the efficacy of clarithromycin against resistant P. aeruginosa strains that commonly affect individuals with cystic fibrosis.
INTRODUCTION
Cystic fibrosis (CF) is a fatal inherited disease that is common among the Caucasian population and affects 30,000 and 3,000 newborns/year in the United States and Canada, respectively (1, 2). Cystic fibrosis is a multiorgan disease affecting the liver, pancreas, gastrointestinal tract, and lungs; however, pulmonary injury is the main cause of death among CF patients (3–5).
The underlying molecular mechanism of CF is mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on chromosome 7 (4, 6). The CFTR molecule is a 1,480-amino-acid membrane-bound chloride channel (7). The structure and function of the channel in CF are compromised by over 1,800 types of mutations (2). The most prevalent mutation, delta F 508, is a deletion of phenylalanine at position 508 and is responsible for 70% of CF cases worldwide (6, 7). The CFTR glycoprotein regulates salt and water transport across epithelial cells (8, 9). Accumulation of the chloride ion inside the cells carrying defective CFTR protein results in dehydration of the epithelial lining fluid and overproduction of thick and sticky mucus (10). The condition, in part, provides a suitable environment for microbial growth, including bacteria, such as Staphylococcus aureus, Haemophilus influenzae, Burkholderia cepacia, and Pseudomonas aeruginosa (11). P. aeruginosa, however, persists in the lungs of over 80% of adults suffering from CF and causes recurrent infection and inflammation (1, 4, 12).
P. aeruginosa is a ubiquitous aerobic Gram-negative bacterium that affects individuals with compromised immune systems and has a high intrinsic resistance to most antibiotics (13, 14). P. aeruginosa possesses a large array of virulence factors, such as flagellum, pili, elastase, chitinase, lipase, and proteases (15–17). The flagellum and pili can bind to the overexpressed asialoganglioside (GM1) in CF epithelial cells and help bacteria to twitch, swarm, and swim toward nutritional signals, as well as in biofilm formation (18–21). Elastase, chitinase, lipase, and proteases can cause degradation and damage of elastin, collagen, and immunoglobulins, affecting alveolar epithelial permeability (22).
There are several molecular mechanisms by which bacteria, including P. aeruginosa, resist the action of macrolide, including target site modification by methylation and/or mutation that prevents the binding of antibiotics to their target molecules and inactivation of the drugs or efflux (23–25).
Forming biofilm is one of the strategies for bacteria to evade chemotherapy, as well as the host immune response (26). Biofilm formation, however, starts with attachment of the microorganism to a surface, followed by production of extracellular matrix composed of polysaccharides and proteins, which mediate bacterial attachment during the initial biofilm community formation process (27–29). Biofilm protects bacteria from phagocytosis, opsonization by antibodies, and their removal by the ciliary action of tracheal epithelium (30, 31). Furthermore, the extracellular polymeric matrix delays the diffusion of some antibiotics into the community (27, 32), and thus, bacteria might be exposed to a drug concentration below the MIC, leading to increased mutation and resistance of the bacteria (33). P. aeruginosa in biofilms was found to be resistant to macrolide due to mutation in nfxB, which encodes the negative transcriptional regulator protein NfxB for the efflux pump, leading to increased expression of the efflux pump MexCD-OprJ and resistance of P. aeruginosa to macrolide (34).
Pseudomonal lung infections are treated with antibiotics, such as aminoglycosides and macrolides, to reduce infection (35, 36). Macrolide antibiotics are usually characterized by a large lactone ring within their structure (23). They are classified according to the number of lactone ring components: 14-membered (erythromycin and clarithromycin [CAM]), 15-membered (azithromycin) (37), and 16-membered (roxithromycin) (17) groups. Macrolides are effective against most aerobic and anaerobic Gram-positive organisms and many Gram-negative bacteria (35, 37). They are used for treating respiratory tract and soft tissue infections (23, 38). Macrolides, such as clarithromycin, inhibit protein synthesis in bacteria by reversibly binding to the 50S ribosomal subunits (38). Clarithromycin is also known as the most effective chemotherapy against Mycobacterium avium complex (MAC) (39). The effective doses of oral clarithromycin are 200 to 500 mg/ml in adult humans; long exposure and high doses are required for treating chronic respiratory P. aeruginosa infection (40, 41). A group of investigators reported the beneficial effect of clarithromycin on treatment of biofilm-associated chronic respiratory P. aeruginosa infection in a murine model (26). Clarithromycin, however, is a known inhibitor of the hepatic microsomal cytochrome CYP3A4 (42), which has a significant role in metabolizing macrolides. The loss of CYP3A4 catalytic ability resulted in elevated serum drug levels and hepatotoxicity (43). Due to the high resistance of P. aeruginosa to most antimicrobial agents, including macrolides (35), and the appearance of toxicity of some drugs (44), there is a strong demand for novel drugs, as well as new and safe delivery systems, such as liposomes, to combat P. aeruginosa-induced chronic infection (45).
Liposomes are round vesicles consisting of one or more phospholipid bilayers surrounding an aqueous solution space (46, 47). Hydrophobic drugs, such as macrolides, can be entrapped in the lipid bilayers of the biocompatible and biodegradable liposomes, while hydrophilic drugs can be incorporated into their aqueous compartments (36, 48, 49). Liposomes, as a drug carrier system, have the ability to improve antibiotic therapy by decreasing antibiotic toxicity and enhancing bactericidal efficacy through fusion with the bacterial membrane (48, 50–52). Liposomes have the ability to protect their loads from the host cellular elements and the action of bacterial enzymes (50, 53–55). Another study indicated that the liposomal formulation was effective in enhancing polymyxin B antimicrobial activity against Gram-negative bacteria compared to the free drugs (56). A previous in vivo study performed in a rat model demonstrated that liposomal tobramycin administered intratracheally improved the pharmacokinetic parameters and significantly reduced P. aeruginosa bacteria after multiple treatments (57). Another study showed that liposome-encapsulated clarithromycin significantly increased the uptake of human macrophages into the encapsulated agent and reduced M. avium complex infection compared to the free drug (58). Furthermore, combination therapy using liposomal amikacin in the initial phase of chemotherapy in M. avium infection enhanced the efficacy of a clarithromycin/ethambutol regimen (59).
The aim of this work was to investigate whether the lack or the type of surface charges in liposomal formulations containing clarithromycin—negatively charged liposomal clarithromycin (NEG-Lipo-CAM), positively charged liposomal clarithromycin (POS-Lipo-CAM), and uncharged liposomal clarithromycin (NEU-Lipo-CAM)—would enhance clarithromycin antimicrobial activity. We also evaluated clarithromycin cell toxicity and measured the liposomal formulation size, stability, and antibacterial activity (MIC and minimum bactericidal concentration [MBC]) in vitro. Furthermore, we investigated the formulations' ability to prevent biofilm formation, virulence factor production, and motility of clarithromycin-resistant strains of P. aeruginosa.
MATERIALS AND METHODS
Chemicals and media.
Clarithromycin was obtained from Sigma-Aldrich (Oakville, ON, Canada). Dipalmitoylphosphatidylcholine (DPPC) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Other chemicals, such as didecyldimethylammonium bromide (DDAB), dicetyl phosphate (DCP), Triton X-100, trypan blue, elastin-Congo red, chitin azure, and agarose, were also obtained from Sigma-Aldrich (Oakville, ON, Canada). For antibiotic susceptibility tests, Mueller-Hinton agar, trypsin-EDTA, and the Cell Titer Blue Cell Viability Assay kit were purchased from Fisher Scientific (Ottawa, ON, Canada). Tryptic soy agar, tryptic soy broth, Luria-Bertani (LB) broth, and Luria-Bertani agar were purchased from Becton Dickinson Microbiology Systems (Oakville, ON, Canada). Cationic-adjusted Mueller-Hinton broth for culturing microorganisms was purchased from BD (Franklin Lakes, NJ). Normal pooled plasma was purchased from Precision Biologic (Dartmouth, NS, Canada). ABt medium consisted of 27 mM (NH4)2SO4, 30 mM Na2HPO4 · 2H2O, 20 mM KH2PO4, 47 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 0.01 mM FeCl2, 0.5% (wt/vol) glucose, 0.5% (wt/vol) Casamino Acids, and 0.00025% (wt/vol) thiamine.
Microorganisms.
A laboratory strain of Bacillus subtilis (ATCC 6633) was used as an indicator organism for clarithromycin activity. Laboratory strains of P. aeruginosa (ATCC 10145 and ATCC 25619) and clinical isolates of P. aeruginosa (PA-M13639-1, PA-M13641-2, PA-1, PA-11, PA-12, PA-13572, and PA-M13640) were purchased from PML Microbiologicals (Mississauga, ON, Canada) or obtained from the Clinical Microbiology Laboratory of Memorial Hospital (Sudbury, ON, Canada). All strains were stored at −80°C in cationic-adjusted Mueller-Hinton broth supplemented with 10% glycerol.
Liposome preparation.
Clarithromycin was encapsulated into liposomes composed of different lipids (DPPC, DDAB, and DCP) and cholesterol (CHOL). The positively charged liposomal formulation was composed of DDAB, DPPC, and cholesterol in a ratio of 4:2:1; the negatively charged liposomal formulation was composed of DCP, DPPC, and cholesterol in a ratio of 4:2:1; and the uncharged liposomal formulation was composed of DPPC and cholesterol in a ratio of 6:1. The liposomal formulations were prepared by the dehydration-rehydration method (60, 61). Briefly, the lipids were dissolved in the chloroform-methanol solution (2:1 [vol/vol]). A rotary evaporator (Rotavapor; Büchi Labortechnik AG) was used to evaporate the organic solvent (62). Once a thin, dry lipid layer was formed, clarithromycin solution (1 mg/ml) was added, followed by a series of sonications using the Sonic Dismembrator (FS20H; Fisher Scientific, Ottawa, Canada) (45).
Microbiological assay.
After preparation of the liposomes, the mixture was centrifuged at 16,000 × g for 20 min at 4°C. Triton X-100 was added to the pellet to release the drug, as previously reported (60, 63). The concentrations of clarithromycin incorporated into liposomes were measured by agar diffusion assay (56). A B. subtilis laboratory strain (ATCC 6633) was used as the indicator organism for clarithromycin activity, as recommended by the Clinical and Laboratory Standards Institute (CLSI). B. subtilis was cultured overnight in cationic Mueller-Hinton broth, and a bacterial solution was prepared equivalent to a 0.5 McFarland standard (1.5 × l08 CFU/ml) (60). The cells were added to an agar solution at 41°C and quickly poured into a sterile glass plate (440 mm by 340 mm) to form a thin layer of agar and bacteria. Wells 5 mm in diameter made with a well puncher were filled with 25 μl of samples or standard solutions (62), and the plate was incubated for 18 h at 37°C (47). After the incubation period, inhibition zones in the plate were measured in triplicate. The averaged values for each triplicate sample were used to analyze the encapsulation efficiency of the liposomal formulations for clarithromycin.
The sensitivity of the assay was 0.002 mg/liter. The quantifiable limit for clarithromycin was 0.002 mg/liter. At concentrations from 0.002 to 0.0125 mg/liter, the coefficients of variation ranged between 1 and 2%. Over the same concentrations, the intraday coefficients of variation ranged between 2 and 3%. For 10 samples of spiked clarithromycin, the standard curve linearity extended over the range of 0.002 to 0.0125 mg/liter and gave a correlation coefficient greater than 0.99. The concentration measurements are the means of at least three independent experiments, with each experiment measured in triplicate.
Encapsulation efficiency determination.
The encapsulation efficiency of liposomal clarithromycin was determined as the percentage of clarithromycin entrapped in the liposomes relative to the initial total amount of the drug in solution (54). The concentration of the entrapped clarithromycin was determined by the microbiological assay outlined above (62).
Size determination and polydispersity index.
The polydispersity index (PI) and the mean diameter of liposomes were determined by using a Submicron Particle Sizer Model 270 (Nicomp, Santa Barbara, CA) (47, 54, 64).
Stability of liposomal clarithromycin.
The stability of liposomal clarithromycin was assessed in phosphate-buffered saline (PBS) at 4°C and 37°C. The stability of liposomal clarithromycin was determined as the percentage of retention of the initial encapsulated drug after a period of time under different conditions (54, 65). Briefly, liposomal clarithromycin was suspended in PBS and incubated in a water bath shaker with mild agitation at 100 rpm (Julabo SW22 Incubator Shaker; Labortechnik, Seelbach, Germany). After incubation times of 0.25, 0.5, 1, 3, 6, 12, 24, and 48 h (64), samples were centrifuged at 18,300 × g for 15 min at 4°C to remove the released drugs (52, 66). The supernatants of the liposomal samples were collected, and 25 μl was transferred into holes on a plate containing agar prepared with an appropriate bacterial culture (B. subtilis ATCC 6633). The plates were then incubated at 37°C for 18 h, and the inhibition zones were measured. Free-clarithromycin concentrations were also determined by agar diffusion assay (60).
MIC and MBC.
A broth dilution method was used to determine the MICs of liposomal clarithromycin. Overnight cultures of the clinical strains of P. aeruginosa were diluted in cationic Mueller-Hinton broth to achieve 0.5 McFarland standard (56). The bacterial cell populations were then exposed to several dilutions of liposomal or free clarithromycin ranging from 0.031 to 256 mg/liter and thoroughly mixed with Mueller-Hinton agar. The plates were incubated for 18 h at 37°C (45). For MBC assays, bacterial suspensions were mixed with subinhibitory concentrations, MICs, and two times the MICs of free CAM or Lipo-CAM, and the plates were incubated for 24 h at 37°C (63). Broth medium alone and free clarithromycin bacterial cultures were used as negative and positive controls, respectively.
Bactericidal activity of liposomal clarithromycin against P. aeruginosa in biofilm (MBEC).
In order to assess the minimum biofilm eradication concentration (MBEC), P. aeruginosa strain PA-13572 was allowed to form a biofilm in a Calgary biofilm plate (Innovotech, Edmonton, AB, Canada) as previously reported (67). Briefly, strain PA-13572 (1.5 × 106 CFU/ml; 24 ml) was added to the Calgary biofilm device plates. The plates were placed in an incubator shaker rotating at 50 rpm (Innova 4000 Incubator Shaker; New Brunswick Scientific, NJ) at 37°C for 4 days, ensuring equal distribution of medium in the troughs and adhesion of PA-13572 to the pegs (fresh broth was added every 24 h to remove the nonadherent bacteria). After 5 days, the biofilms on the pegs were washed twice with medium, and sterile forceps were used to transfer the biofilm pegs into microcentrifuge tubes containing 1 ml of PBS. The pegs were then sonicated for 1 min to detach and disperse bacteria, followed by vortexing for 2 min. The bacterial suspension was subjected to 10-fold serial dilutions for a bacterial count to serve as the control. Aliquots of 100 μl of each dilution were plated on Mueller-Hinton agar and incubated for 24 h at 37°C. The rest of the pegs were then submerged in a 96-well pate containing 200 μl of different dilutions of free CAM, NEG-Lipo-CAM, POS-Lipo-CAM, and NEU-Lipo-CAM. The plate was incubated at 37°C for 24 h. The peg lid was washed with medium twice, and the pegs were removed and transferred to microcentrifuge tubes, sonicated, and serially diluted (10-fold) for bacterial counts (CFU) after 24 h of incubation at 37°C.
Virulence factor assays.
To test which concentrations below the MIC are subinhibitory, free or liposomal formulations (NEG-Lipo-CAM, NEU-Lipo-CAM, and POS-Lipo-CAM) at concentrations of 1/2 to 1/8 the MIC were introduced into P. aeruginosa PA-13572. The bacterial growth was repetitively monitored (optical density at 600 nm [OD600]) up to 8 h. For experiments involving lipase, chitinase, elastase, and protease assays, P. aeruginosa PA-13572 was cultured in ABt medium for 18 h at 37°C (68), and then the cell density of the bacteria in a 100-ml flask was adjusted to match 0.5 McFarland standard (OD600 = 0.132) following incubation for 1 h at 37°C. When the bacterial cell density doubled to an OD600 of 0.26 (67), the cells were exposed to equal volumes of free and liposomal formulations at 1/8 the MIC. After 24 h of incubation, bacterial concentrations were measured at OD600 and the suspension was centrifuged for 15 min (16,000 × g at 4°C) and filtered sterilized (0.22 μm) for biochemical assays.
(i) Lipase assay.
The reaction mixture for the lipase assay consisted of 0.6 ml of 10% Tween 20 in Tris buffer, 0.1 ml of 1 M CaCl2, 0.6 ml of filtered supernatant, and 1.6 ml of double-distilled water; we used medium alone (blank) as a control (68, 69). The reaction mixtures were incubated at 37°C for 24 h (69) with agitation at 200 rpm (Innova 4000 Incubator Shaker; New Brunswick Scientific, NJ). Lipase uses Tween 20 as a substrate and converts it into fatty acid and alcohol. The resulting fatty acid bound to the calcium and formed an insoluble complex the absorbance of which was measured in a spectrophotometer at 400 nm (67, 70). Lipase experiments were done three times in triplicate.
(ii) Chitinase assay.
Insoluble chitin azure (5 mg) was properly mixed with 1 ml of filtered suspended supernatant or medium alone (blank) in 1 ml of PBS. The reaction mixture was incubated for 24 h at 37°C. Chitinase breaks chitin azure and produces a blue compound whose absorbance was determined at 290 nm. Experiments were performed three times with two replicates. The resulting data were normalized by dividing the optical density by the cell density (OD600) (68, 71). The experiment was repeated three times in triplicate.
(iii) Elastase assay.
Insoluble elastin-Congo red (20 mg) (72) was mixed with 1 ml of PBS and 1 ml of filtered suspended supernatant or medium alone (blank) as a control (68). The mixture was incubated for 24 h at 37°C with agitation at 200 rpm (Innova 4000 Incubator Shaker; New Brunswick Scientific, NJ). Elastase breakdown of insoluble elastin-Congo red produced a red compound whose absorbance was measured at OD459 (73) after centrifugation at 16,000 × g. All these experiments were repeated at least three times in triplicate (68).
(iv) Protease assay.
Filtered supernatants or medium alone (100 μl) was transferred into the wells of a petri dish containing 2% agarose and 2% skim milk, following incubation for 48 h at 37°C. Zones of clearance due to the proteolytic activity of protease could be easily observed (74) and were measured (in mm) using digital calipers (67, 68). All these experiments were repeated at least three times in triplicate.
(v) Effect of liposomal clarithromycin on P. aeruginosa motility.
The motility of P. aeruginosa was investigated by methods described previously (67). Briefly, P. aeruginosa PA-13572 grown overnight was diluted to 1.5 × 108 CFU/ml, and 1 μl was inoculated onto a 3-mm depth of ABt-agarose plates containing a subinhibitory concentration of free or liposomal clarithromycin (1/8 the MIC). Inoculation into the bottom of ABt medium with agarose (1% [wt/vol]) was used for twitching, and point inoculation onto the medium with agarose (0.3% [wt/vol]) was used for swimming and swarming (0.5% [wt/vol]). After 12 h of incubation at 37°C, swimming and swarming diameters were measured. For twitching at the agarose-petri dish interface, after 24 h of incubation at 37°C, the medium was gently removed and the petri dish was air dried. A 1% crystal violet solution was used to stain the petri dish for 10 min. The petri dish was rinsed, and the crystal violet-stained twitching pattern was measured. All experiments were performed in three independent experiments in triplicate.
Determination of liposomal clarithromycin cytotoxicity.
We used a human lung carcinoma epithelial cell line (A549; ATCC, Manassas, VA) for the cell viability assay. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (75). The cells were allowed to grow until they attained 85% confluence in 5% CO2 at 37°C (50).
The viability of the cells was determined by Cell Titer Blue assay. The cells were seeded into 24-well plates (75) at a density of 5 × 105 cells/ml and left to adhere to the surfaces of the wells overnight (50). The cell culture medium was then replenished with 500 μl of fresh medium containing free clarithromycin or liposomal clarithromycin at four different concentrations (2×, 1.5×, 1×, and 0.5× the MIC) and transferred into the 24-well plates. In this assay, different liposomal formulations of clarithromycin—positively charged liposomal clarithromycin (DPPC, DDAB, and CHOL), negatively charged liposomal clarithromycin (DPPC, DCP, and CHOL), and uncharged liposomal clarithromycin (DPPC and CHOL)—were used. The concentrations of free or liposomal clarithromycin that had been introduced to the A549 cells were calculated based on the MIC value for each formulation, as stated above. All of these liposomal preparations were exposed to these concentrations for three different periods—24, 48, and 72 h—following incubation in 5% CO2 at 37°C. The wells containing only cell culture medium without the drug were used as controls (50, 51). Once the required exposure period was over, they were removed, and the cells were washed once with PBS and subsequently with cell culture medium to remove any residual Lipo-CAM or free CAM. Then, the existing medium was replaced with 500 μl of fresh medium, and 100 μl of resazurin dye was added to each of the 24-well plates. The cells were then incubated overnight in the dark at 37°C in 5% CO2 (50). The absorbance was measured at 570 nm, using 600 nm as a reference wavelength in a spectrophotometer. A blank well containing Cell Titer Blue reagent without cells was used as a reference (75). All experiments were performed independently three times in triplicate.
Data analysis.
The data are represented as means ± standard errors of the mean (SEM) of three independent experiments. For comparisons of multiple groups, one-way analysis of variance (ANOVA) was performed using GraphPad Prism, followed by a post-t test. P values of <0.05, <0.01, and <0.001 were considered statistically significant.
RESULTS
Encapsulation efficiency and size.
The percent encapsulation efficiency (EE%), size, and size distribution for liposomal formulations are summarized in Table 1. A PI of <0.1 indicates a homogeneous population. The results are given as means ± SEM of three separate experiments.
Table 1.
Encapsulation efficiencies, particle sizes, and polydispersity indexes of macrolide antibiotics
| Liposomal clarithromycin | Size (nm) (mean ± SEM) | PI (mean ± SEM) | EE% (mean ± SEM) |
|---|---|---|---|
| Neutral DPPC-CHOL | 221.60 ± 14.98 | 0.776 ± 0.004 | 15.96 ± 0.05 |
| Negative DPPC-DAP-CHOL | 199.63 ± 6.87 | 0.786 ± 0.024 | 30.37 ± 0.10 |
| Positive DPPC-DDAB-CHOL | 169.20 ± 16.75 | 0.518 ± 0.035 | 5.70 ± 0.01 |
Stability of liposomal clarithromycin.
The stability of liposomal clarithromycin was evaluated in PBS at 4°C (storage temperature) and at 37°C (body temperature) for a study period of 48 h. It was evident from the data that the liposomal clarithromycin stored at 4°C was more stable than that incubated at 37°C. The negatively charged liposomal clarithromycin retained 94.15% ± 0.34% of the drug at 4°C, and its retention rate was 92.03% ± 0.78% at 37°C. Antibiotic retention of the positively charged liposomes followed the same pattern, though the retention rates were much lower (60.13% ± 0.92% at 4°C and 53.00% ± 0.95% at 37°C). The negatively charged liposome formulation was significantly more stable than the positively charged formulation (P < 0.001). The uncharged liposomal clarithromycin showed more stability at a lower temperature and retained more antibiotics than the liposomes at 37°C (95.07% ± 0.005% versus 91.00% ± 0.76%, respectively). There was no significant difference between the stabilities of negatively charged liposomes and uncharged liposomes; however, the uncharged formulation was significantly more stable than the positively charged formulation (P < 0.001).
MICs and MBCs.
The MICs of liposomal clarithromycin against P. aeruginosa strains were significantly lower than those of free clarithromycin, as illustrated in Table 2. The experiments were done with two highly clarithromycin-resistant mucoid and nonmucoid clinical strains of P. aeruginosa. As demonstrated in Table 2, the MICs of free clarithromycin for all P. aeruginosa strains were ≥256 mg/liter compared to 64 mg/liter for negatively charged liposomal clarithromycin. The MICs for uncharged liposomal clarithromycin against P. aeruginosa strains were reduced from over 256 mg/liter for free clarithromycin to 32 mg/liter, whereas positively charged liposomal clarithromycin was effective at 8 mg/liter. The MIC against one of the resistant strains (PA-1) was 4 mg/liter for positively charged liposomal clarithromycin compared to 16 mg/liter for uncharged liposomal clarithromycin and 32 mg/liter for negatively charged liposomal clarithromycin. The difference between the MICs and MBCs of liposomal clarithromycin and free clarithromycin against P. aeruginosa strains was remarkable (2 to 4 versus 256 to 512 mg/liter, respectively). The MBCs of free clarithromycin against P. aeruginosa strains were ≥512 mg/liter compared to 64 mg/liter for NEG-Lipo-CAM and NEU-Lipo-CAM. The positively charged liposomal clarithromycin was bactericidal at 16 mg/liter (Table 2).
Table 2.
Antimicrobial activities of free and liposomal clarithromycin on P. aeruginosa strains
| Bacterial strain | Descriptiona | Antimicrobial activity (mg/liter) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| NEU-Lipo-CAM |
NEG-Lipo-CAM |
POS-Lipo-CAM |
Free CAM |
||||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | ||
| ATCC 10145 | Non Muc | 32 | >64 | 64 | >64 | 8 | 16 | >256 | 512 |
| ATCC 25619 | Non Muc | 32 | 64 | 64 | >64 | 8 | 16 | 256 | 512 |
| PA-M13639-1 | Muc | 32 | 64 | 64 | 64 | 8 | >64 | 256 | 512 |
| PA-M13641-2 | Muc | 32 | >64 | 64 | >64 | 8 | 16 | 256 | 512 |
| PA-1 | Non Muc | 16 | >64 | 32 | 32 | 4 | 8 | >256 | 512 |
| PA-11 | Non Muc | 32 | >64 | 64 | >64 | 8 | 16 | >256 | 512 |
| PA-12 | Non Muc | 32 | >64 | 64 | >64 | 8 | 16 | >256 | 512 |
| PA-13572 | Non Muc | 32 | 64 | 64 | 64 | 8 | 16 | >256 | 512 |
| PA-M13640 | Non Muc | 32 | 64 | 64 | 64 | 8 | 16 | 256 | 512 |
Non Muc, nonmucoid; Muc, mucoid.
Liposomal clarithromycin bactericidal activity on P. aeruginosa biofilm.
POS-Lipo-CAM and NEG-Lipo-CAM completely eradicated P. aeruginosa 13572 in the biofilm, whereas NEU-Lipo-CAM and free CAM reduced bacterial numbers in biofilm communities by 5 and 3 log units (CFU/ml), respectively, at 128 mg/liter (Fig. 1). POS-Lipo-CAM still proved to have a potent effect on bacteria in biofilms: it was able to completely eliminate the bacteria within the biofilm at a lower concentration of 64 mg/liter. The other formulation could only decrease P. aeruginosa at 64 mg/liter by 3 log units for free and negatively charged liposomes and 4 log units for uncharged liposomes compared to the control. It was also observed that the liposomal formulations and free clarithromycin at 32 mg/liter reduced bacterial counts by 2 log units for free CAM and NEG-Lipo-CAM and 3 log units for POS-Lipo-CAM compared to the control.
Fig 1.

Liposomal clarithromycin activity on P. aureginosa PA-13572 biofilm (MBEC assay). Free (F-CAM) or liposomal formulations were introduced to mature biofilm at concentrations of 32 mg/liter (a), 64 mg/liter (b), and 128 mg/liter (c). Untreated biofilm acted as a control. The data represent three independent experiments in triplicate and are shown as means ± SEM. P values were considered significant compared with the control: ***, P < 0.001.
Effects of subinhibitory concentrations of free and liposomal clarithromycin on growth of P. aeruginosa.
Both free and liposomal clarithromycin affected the growth of P. aeruginosa PA-13572 (Fig. 2a and b). Subinhibitory concentrations (1/8 the MIC), however, did not inhibit bacterial growth (Fig. 2c). For this reason, all experiments involving virulence factors were performed using subinhibitory concentrations of 1/8 the MIC.
Fig 2.

Effects of subinhibitory concentrations of liposomal clarithromycin formulations on growth of PA-13572 at 1/2 the MIC (a), 1/4 the MIC (b), and 1/8 the MIC (c). Experiments were tested three times in triplicate, with means shown (the error bars were deleted for clarity of the graphs). Shown are control (filled circles), negatively charged liposomal clarithromycin (filled squares), positively charged liposomal clarithromycin (filled triangle), uncharged liposomal clarithromycin (open circle), and free clarithromycin (open squares).
Effects of liposomal clarithromycin on bacterial virulence factors.
The levels of lipase, chitinase, elastase, and protease in the free- or liposomal-clarithromycin-treated PA-13572 cultures were measured at 1/8 the MIC. Positively charged liposomal clarithromycin attenuated lipase production significantly compared to the control (P < 0.001), while uncharged liposomal CAM and negatively charged liposomal CAM were ineffective (Fig. 3a). Chitinase production in the supernatant was evaluated by quantifying the release or breakdown of chitin azure (see Materials and Methods). Liposomal CAM (uncharged and positively charged) reduced chitinase production. However, the negatively charged liposomal CAM reduced chitinase production significantly (P < 0.001) (Fig. 3b). All liposomal formulations reduced elastase and protease production significantly at 1/8 the MIC (P < 0.05) compared to the control (Fig. 3c and d, respectively). There were no significant differences in the attenuation of elastase and protease production between the free-clarithromycin and liposomal-clarithromycin formulations.
Fig 3.

Effects of subinhibitory concentrations (1/8 the MIC) of free and liposomal clarithromycin on PA-13572 virulence factor production. (a) Lipase. (b) Chitinase. (c) Elastase. (d) Protease. The results represent the means and SEM of three independent experiments in triplicate. For lipase, chitinase, and elastase experiments, the results were normalized by dividing the OD by the OD600 (cell density). P values were considered significant compared with the control: ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Effect of liposomal clarithromycin on bacterial motility.
We examined bacterial motility, including twitching, swarming, and swimming, in the presence of subinhibitory concentrations of either free or liposomal clarithromycin with different surface charges (neutral, positive, and negative). Liposome-loaded clarithromycin significantly reduced twitching of P. aeruginosa PA-13572 compared to the free-clarithromycin and control groups (P < 0.001) (Fig. 4a). However, positively charged liposomes exhibited more reduction in the twitching motility of P. aeruginosa (P < 0.01 and P < 0.001 compared to negatively charged and uncharged liposomal formulations, respectively). For swarming, liposomes (neutral, positive, and negative) were able to reduce swarming of P. aeruginosa PA-13572 at the subinhibitory concentration compared to the free-clarithromycin and control groups (Fig. 4b). However, positively charged liposomes reduced swarming more significantly than uncharged and negatively charged liposomes (P < 0.001). Liposomal formulations attenuated the swimming activity of P. aeruginosa compared to the free-clarithromycin and control groups (Fig. 4c). However, positively charged liposomes reduced swimming activity significantly (P < 0.001) compared to neutral and negatively charged liposomes.
Fig 4.
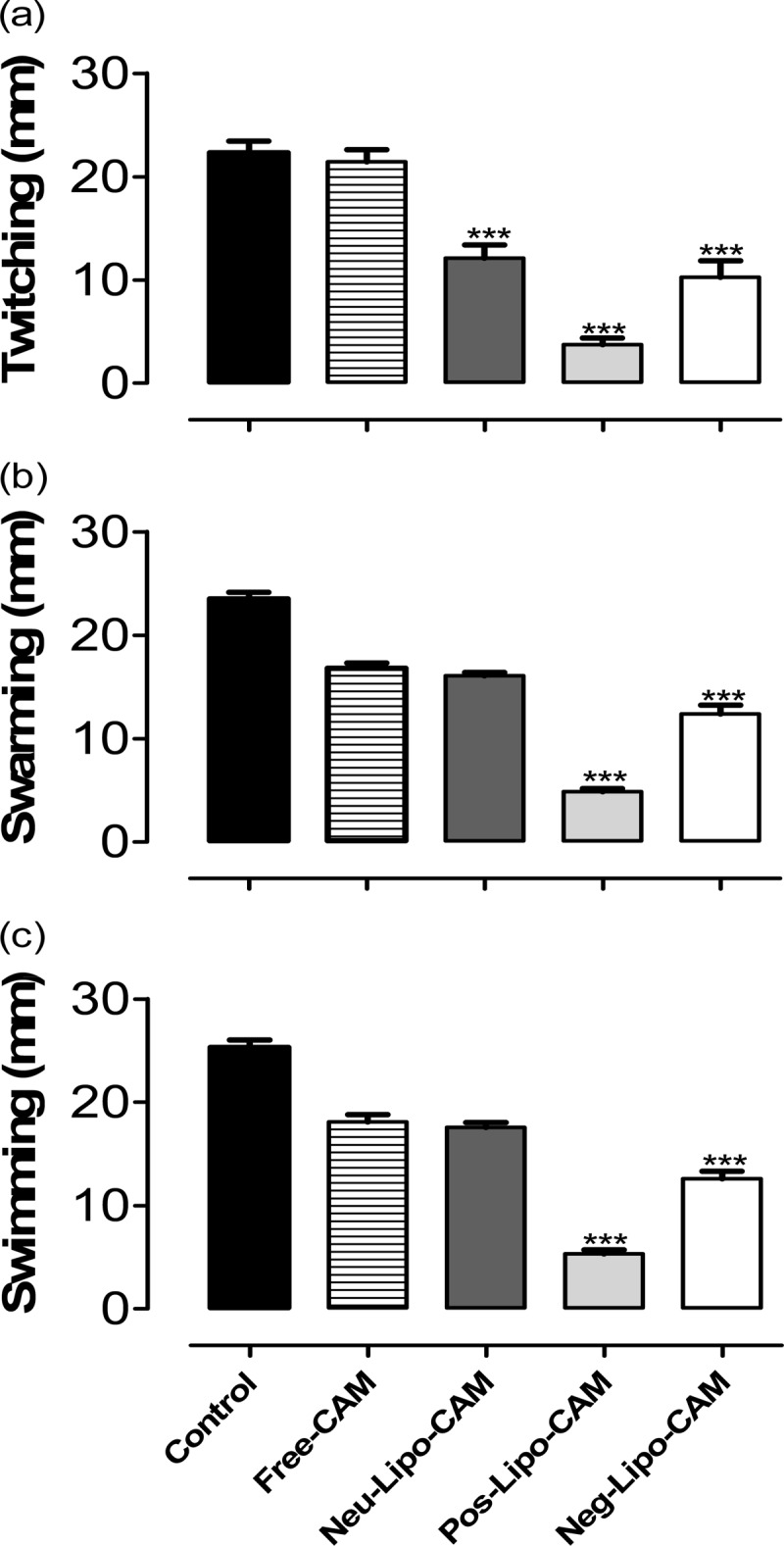
Effect of a subinhibitory concentration of liposomal clarithromycin on P. aeruginosa motility. Free or liposomal clarithromycin at 1/8 the MIC was added to agarose plates, and motility was examined. Twitching (1% agarose [wt/vol]) (a), swarming (0.5% agarose [wt/vol]) (b), and swimming (0.3% agarose [wt/vol]) (c) were measured with digital calipers. P values were considered significant compared with the control and between groups: ***, P < 0.001.
Toxicity of liposomal clarithromycin.
When lung cells were incubated for 24 h and exposed to 2× the MIC of treatments, cell viabilities were 100% for NEG-Lipo-CAM, 99% for NEU-Lipo-CAM, 1% for POS-Lipo-CAM, and 20% for free CAM. Following a 48-h period, the viabilities of lung cells exposed to treatment formulations at 2× the MIC were 97% for NEG-Lipo-CAM, 95% for NEU-Lipo-CAM, 1% for POS-Lipo-CAM, and 5% for free CAM. After 72 h of incubation, at 2× MIC, the percentages of cell viability were 93% for NEG-Lipo-CAM, 98% for NEG-Lipo-CAM, 0% for POS-Lipo-CAM, and 1% for free CAM (Table 3).
Table 3.
Cell viability of epithelial lung cells exposed to free or liposomal clarithromycin formulations
| Time (h) | Cell viability (%) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Free CAM |
NEG-Lipo-CAM |
NEU-Lipo-CAM |
POS-Lipo-CAM |
|||||
| 1× MIC | 2× MIC | 1× MIC | 2× MIC | 1× MIC | 2× MIC | 1× MIC | 2× MIC | |
| 0 | 100 ± 0.00 | 100 ± 0.00 | 100 ± 0.00 | 100 ± 0.00 | 100 ± 00 | 100 ± 00 | 100 ± 0.00 | 100 ± 0.00 |
| 24 | 98 ± 2.08 | 21 ± 7.60b | 99 ± 1.04 | 100 ± 0.63a,b | 100 ± 0.40 | 99 ± 0.40a,b | 0 ± 0.06a,b | 1 ± 0.04a,b |
| 48 | 95 ± 6.14 | 5 ± 0.41b | 97 ± 1.38 | 97 ± 1.79a,b | 96 ± 2.15 | 95 ± 3.38a,b | 0 ± 1.29a,b | 1 ± 1.97a,b |
| 72 | 97 ± 1.57 | 1 ± 1.48b | 96 ± 3.67 | 93 ± 9.04a,b | 93 ± 3.14 | 98 ± 3.26a,b | 0 ± 0.20a,b | 0 ± 0.02a,b |
P values were considered significant compared with free clarithromycin (P<0.001).
P values were considered significant for free or liposomal clarithromycin compared to the control (P<0.001).
DISCUSSION
In this study, we established that macrolide antibiotics can be efficiently encapsulated in liposomes composed of DPPC-cholesterol, DPPC-DDAB-cholesterol, or DPPC-DCP-cholesterol. Our data show that the efficiency of encapsulation of clarithromycin by negatively charged liposomes was significantly higher than that of other formulations due to attractive interaction with positively charged clarithromycin (76). These results were in agreement with a study that showed that an electrostatic attraction between the negatively charged drug ciprofloxacin and positively charged lipids increases the percent encapsulation efficacy compared to a negatively charged drug (77).
The size of a liposomal formulation is a determining factor in terms of its intended anatomical target. The particle diameter of negatively charged liposomal clarithromycin was larger than that of the positively charged formulation. This is due to the inclusion of charge, which results in an increased space between the adjacent bilayers. The phenomenon can be explained by the attraction of the drug to negatively charged particulates and pushing the phospholipid head group apart (54, 77).
An additional factor that must be considered in the development of an effective drug delivery system is the carrier stability. The liposome's stability was found to be largely independent of temperature, although liposomal formulations were more stable at 4°C. In order to enhance the stability further, we chose to incorporate cholesterol in our formulations. Cholesterol reduces the bilayer permeability of the liposomal membrane, allowing greater drug retention at higher temperatures (64, 66). We found that liposomes composed of DPPC-CHOL and DCP-DPPC-CHOL retained more drug in PBS than liposomes composed of DDAB-DPPC-CHOL at the end of a 48-h experimental period. This may be due to the electrostatic repulsion that occurs between the drug and the positively charged liposomes, which results in a higher rate of drug release and a change in the phase transition temperature (77). This may also be explained by highlighting important factors that contribute to liposome stability, such as the formulation of the saturated neutral phospholipids with different acyl chain lengths and the transition temperature of the phospholipids (78). An earlier report from our laboratory confirmed the notion that the higher transition temperatures of liposomes are more stable due to an increase in the acyl chain length of constituent lipids; this may explain the better stability of liposomes in formulations containing DPPC (64).
We have shown that the liposomal clarithromycin formulations enhance clarithromycin antimicrobial activity against a resistant clinical strain of P. aeruginosa. This is in agreement with previous reports indicating that liposomal formulations are highly effective against most strains of bacteria compared to free drug (52). The MICs and MBCs of liposomal clarithromycin were less than those of free clarithromycin. In most cases, the MIC of free clarithromycin was 256 mg/liter, which is consistent with a previous study (78). To our knowledge, our formulations are the first liposomal carriers that enhance clarithromycin antibacterial activity against antibiotic-resistant clinical strains of P. aeruginosa. Positively charged liposomal clarithromycin was highly effective against P. aeruginosa strains, reducing the MIC from a resistant level of 256 mg/liter to a sensitive level of 8 mg/liter. It is possible that electrostatic attraction and fusion occurring between positively charged liposomal clarithromycin and the cell membrane of P. aeruginosa enhances its activity in vitro. Similar results were obtained in previous studies, which showed other formulations of positively charged liposomes (PC-DOPE-DOTAP [phosphatidylcholine–dioleoyl-glycero-phosphoethanolamine–dioleyloxy trimethyl ammonium-propane] and PC-CHOL-DOTAP) exhibited better antimicrobial efficiency against P. aeruginosa than other formulations of liposomes (79).
Clarithromycin in its free form is known to be a bactericidal enhancer in the treatment of P. aeruginosa biofilms (80, 81). A previous study offered two explanations for the eradication of membranous structures of biofilms after treatment with clarithromycin: destruction of the polysaccharide glycocalyx by clarithromycin and inhibition of de novo polysaccharide synthesis (80). In terms of the improved efficacy of our liposomal formulations, continuous contact with the target and slow antibiotic release may accelerate biofilm penetration by the drug (50). Positively charged liposomal clarithromycin affects biofilm more than other formulations, as it completely eradicates the biofilm community at lower concentrations than the others. This might be due to the attraction between the opposite charges of the bacterial membrane and the liposomal formulation. This allows better penetration of the liposomes into the biofilm and release of antibiotics within the community (79). Different electrical charges affect biofilms differently: negatively charged liposomal clarithromycin eradicated biofilms completely at high concentrations, while uncharged liposomal clarithromycin produced an acceptable reduction of the biofilm community. This is in agreement with our earlier findings on the efficacy of uncharged liposomal antibiotic formulations (65, 82). Thus, formulations containing a variety of liposomes are superior in biofilm eradication and can be utilized to overcome bacterial resistance to antibiotics.
Previous studies demonstrated that macrolides at subinhibitory concentrations inhibited bacterial motility, which contributes to biofilm formation, by affecting the gene expression responsible for producing flagella and preventing proper assembly of type IV pili on the surfaces of bacteria (30, 83), thereby causing a reduction of bacterial motility, including twitching, swarming, and swimming. Here, we demonstrated that encapsulation of clarithromycin into liposomes resulted in improving the efficacy of clarithromycin in inhibiting P. aeruginosa motility. The enhanced activity of liposomal formulations might be attributed to fusion of liposomes to the bacterial cell wall (66) so that a high concentration of antibiotic can be delivered directly to the bacterial cytoplasm, allowing inhibition of flagellar and type IV pilus activities (30, 35). Furthermore, we have noted improved efficacy of positively charged liposomal formulations, which could be explained by the interaction of the positively charged liposome surface with the negatively charged bacterial cell wall, which attracts a high concentration of liposome-loaded clarithromycin to fuse with the bacterial cell membrane (79).
A subinhibitory concentration of macrolides might reduce the production of virulence factors and host tissue damage (84, 85). A previous study demonstrated the effect of a subinhibitory concentration on reducing virulence factors using azithromycin (86). Wozniak and Keyser showed that a sub-MIC level of clarithromycin inhibited the formation of the biofilm matrix and the production of proteases (30, 87). A sub-MIC level of clarithromycin, however, is less effective than azithromycin in reducing the production of elastase and lipase (88). Considering these reports, a combination of the antitwitching property of clarithromycin, which might be enhanced when encapsulated in novel liposomal formulations, with the bactericidal properties of other drugs may prove more effective in treating chronic bacterial infections.
Exposure of A549 human lung cells to free clarithromycin reduced cell viability by 99% at 2× the MIC after 72 h of treatment. After the same period of treatment, negatively charged and uncharged liposomal clarithromycin at similar concentrations protected the cell against clarithromycin toxicity. These and other published data support our hypothesis that liposomal formulations protect host cells from toxic drugs. It was also shown in previous work that the liposomal formulation reduced the toxicity of antibiotics for cell line A549 for different formulations of liposome (DPPC–dimyristoyl glycerol-phosphoglycerol [DMPG]) (50, 51, 53). In contrast, positively charged liposomal clarithromycin at a low concentration decreased the viability of A549 cells. This phenomenon has since been observed by others for positively charged liposomes containing DDAB lipid (89). It is possible that the positively charged lipid DDAB had an adverse effect on cell proliferation (51, 90, 91).
In conclusion, these data indicate that negatively charged liposomal clarithromycin successfully reduced clarithromycin toxicity, greatly affected biofilm community members, and improved clarithromycin activity against highly resistant P. aeruginosa. Future experiments will assess the efficacy of these liposomal clarithromycin formulations in animal models.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Higher Education of the Kingdom of Saudi Arabia.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 1 April 2013
REFERENCES
- 1. Hoiby N. 2011. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uppaluri L, England SJ, Scanlin TF. 2012. Clinical evidence that V456A is a cystic fibrosis causing mutation in South Asians. J. Cyst. Fibros. 11:312–315 [DOI] [PubMed] [Google Scholar]
- 3. Sheikh HS, Tiangco ND, Harrell C, Vender RL. 2011. Severe hypercapnia in critically ill adult cystic fibrosis patients. J. Clin. Med. Res. 3:209–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munder A, Wolbeling F, Kerber-Momot T, Wedekind D, Baumann U, Gulbins E, Tummler B. 2011. Acute intratracheal Pseudomonas aeruginosa infection in cystic fibrosis mice is age-independent. Respir. Res. 12:148 doi:110.1186/1465-9921-1112-1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rogers GB, Hoffman LR, Doring G. 2011. Novel concepts in evaluating antimicrobial therapy for bacterial lung infections in patients with cystic fibrosis. J. Cyst. Fibros. 10:387–400 [DOI] [PubMed] [Google Scholar]
- 6. Ostedgaard LS, Meyerholz DK, Chen JH, Pezzulo AA, Karp PH, Rokhlina T, Ernst SE, Hanfland RA, Reznikov LR, Ludwig PS, Rogan MP, Davis GJ, Dohrn CL, Wohlford-Lenane C, Taft PJ, Rector MV, Hornick E, Nassar BS, Samuel M, Zhang Y, Richter SS, Uc A, Shilyansky J, Prather RS, McCray PB, Jr, Zabner J, Welsh MJ, Stoltz DA. 2011. The DeltaF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci. Transl. Med. 3:74ra24 doi:10.1126/scitranslmed.3001868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ntimbane T, Comte B, Mailhot G, Berthiaume Y, Poitout V, Prentki M, Rabasa-Lhoret R, Levy E. 2009. Cystic fibrosis-related diabetes: from CFTR dysfunction to oxidative stress. Clin. Biochem. Rev. 30:153–177 [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen TS, Prince A. 2012. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat. Med. 18:509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knapp JM, Wood AB, Phuan PW, Lodewyk MW, Tantillo DJ, Verkman AS, Kurth MJ. 2012. Structure-activity relationships of cyanoquinolines with corrector-potentiator activity in DeltaF508 cystic fibrosis transmembrane conductance regulator protein. J. Med. Chem. 55:1242–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clunes MT, Boucher RC. 2007. Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder. Drug Discov. Today Dis. Mech. 4:63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hauser AR, Jain M, Bar-Meir M, McColley SA. 2011. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 24:29–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Malley CA. 2009. Infection control in cystic fibrosis: cohorting, cross-contamination, and the respiratory therapist. Respir. Care 54:641–657 [DOI] [PubMed] [Google Scholar]
- 13. Sadikot RT, Blackwell TS, Christman JW, Prince AS. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171:1209–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rao J, Damron FH, Basler M, Digiandomenico A, Sherman NE, Fox JW, Mekalanos JJ, Goldberg JB. 2011. Comparisons of two proteomic analyses of non-mucoid and mucoid Pseudomonas aeruginosa clinical isolates from a cystic fibrosis patient. Front. Microbiol. 2:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kipnis E, Sawa T, Wiener-Kronish J. 2006. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med. Mal. Infect. 36:78–91 [DOI] [PubMed] [Google Scholar]
- 16. Rosenau F, Isenhardt S, Gdynia A, Tielker D, Schmidt E, Tielen P, Schobert M, Jahn D, Wilhelm S, Jaeger KE. 2010. Lipase LipC affects motility, biofilm formation and rhamnolipid production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 309:25–34 [DOI] [PubMed] [Google Scholar]
- 17. Tateda K, Ishii Y, Kimura S, Horikawa M, Miyairi S, Yamaguchi K. 2007. Suppression of Pseudomonas aeruginosa quorum-sensing systems by macrolides: a promising strategy or an oriental mystery? J. Infect. Chemother. 13:357–367 [DOI] [PubMed] [Google Scholar]
- 18. Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, Tang H, Prince A. 1998. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 66:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saiman L, Prince A. 1993. Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. J. Clin. Invest. 92:1875–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shapiro L. 1995. The bacterial flagellum: from genetic network to complex architecture. Cell 80:525–527 [DOI] [PubMed] [Google Scholar]
- 21. O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304 [DOI] [PubMed] [Google Scholar]
- 22. Caballero AR, Moreau JM, Engel LS, Marquart ME, Hill JM, O'Callaghan RJ. 2001. Pseudomonas aeruginosa protease IV enzyme assays and comparison to other Pseudomonas proteases. Anal. Biochem. 290:330–337 [DOI] [PubMed] [Google Scholar]
- 23. Kanoh S, Rubin BK. 2010. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin. Microbiol. Rev. 23:590–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ergin A, Ercis S, Hascelik G. 2006. Macrolide resistance mechanisms and in vitro susceptibility patterns of viridans group streptococci isolated from blood cultures. J. Antimicrob. Chemother. 57:139–141 [DOI] [PubMed] [Google Scholar]
- 25. Sato T, Tateda K, Kimura S, Iwata M, Ishii Y, Yamaguchi K. 2011. In vitro antibacterial activity of modithromycin, a novel 6,11-bridged bicyclolide, against respiratory pathogens, including macrolide-resistant Gram-positive cocci. Antimicrob. Agents Chemother. 55:1588–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yanagihara K, Tomono K, Imamura Y, Kaneko Y, Kuroki M, Sawai T, Miyazaki Y, Hirakata Y, Mukae H, Kadota J, Kohno S. 2002. Effect of clarithromycin on chronic respiratory infection caused by Pseudomonas aeruginosa with biofilm formation in an experimental murine model. J. Antimicrob. Chemother. 49:867–870 [DOI] [PubMed] [Google Scholar]
- 27. Moreau-Marquis S, Stanton BA, O'Toole GA. 2008. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm. Pharmacol. Ther. 21:595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makipour K, Friedenberg FK. 2011. The potential role of N-acetylcysteine for the treatment of Helicobacter pylori. J. Clin. Gastroenterol. 45:841–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parsek MR, Tolker-Nielsen T. 2008. Pattern formation in Pseudomonas aeruginosa biofilms. Curr. Opin. Microbiol. 11:560–566 [DOI] [PubMed] [Google Scholar]
- 30. Wozniak DJ, Keyser R. 2004. Effects of subinhibitory concentrations of macrolide antibiotics on Pseudomonas aeruginosa. Chest 125:62S–69S [DOI] [PubMed] [Google Scholar]
- 31. Callaghan M, McClean S. 2012. Bacterial host interactions in cystic fibrosis. Curr. Opin. Microbiol. 15:71–77 [DOI] [PubMed] [Google Scholar]
- 32. Lieleg O, Ribbeck K. 2011. Biological hydrogels as selective diffusion barriers. Trends Cell Biol. 21:543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khan W, Bernier SP, Kuchma SL, Hammond JH, Hasan F, O'Toole GA. 2010. Aminoglycoside resistance of Pseudomonas aeruginosa biofilms modulated by extracellular polysaccharide. Int. Microbiol. 13:207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mulet X, Macia MD, Mena A, Juan C, Perez JL, Oliver A. 2009. Azithromycin in Pseudomonas aeruginosa biofilms: bactericidal activity and selection of nfxB mutants. Antimicrob. Agents Chemother. 53:1552–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawamura-Sato K, Iinuma Y, Hasegawa T, Horii T, Yamashino T, Ohta M. 2000. Effect of subinhibitory concentrations of macrolides on expression of flagellin in Pseudomonas aeruginosa and Proteus mirabilis. Antimicrob. Agents Chemother. 44:2869–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mugabe C, Halwani M, Azghani AO, Lafrenie RM, Omri A. 2006. Mechanism of enhanced activity of liposome-entrapped aminoglycosides against resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50:2016–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kannan K, Mankin AS. 2011. Macrolide antibiotics in the ribosome exit tunnel: species-specific binding and action. Ann. N. Y. Acad. Sci. 1241:33–47 [DOI] [PubMed] [Google Scholar]
- 38. Wierzbowski AK, Hoban DJ, Hisanaga T, DeCorby M, Zhanel GG. 2006. The use of macrolides in treatment of upper respiratory tract infections. Curr. Allergy Asthma Rep. 6:171–181 [DOI] [PubMed] [Google Scholar]
- 39. Kohno Y, Ohno H, Miyazaki Y, Higashiyama Y, Yanagihara K, Hirakata Y, Fukushima K, Kohno S. 2007. In vitro and in vivo activities of novel fluoroquinolones alone and in combination with clarithromycin against clinically isolated Mycobacterium avium complex strains in Japan. Antimicrob. Agents Chemother. 51:4071–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sandrini A, Balter MS, Chapman KR. 2003. Diffuse panbronchiolitis in a Caucasian man in Canada. Can. Respir. J. 10:449–451 [DOI] [PubMed] [Google Scholar]
- 41. Kadota J, Mukae H, Ishii H, Nagata T, Kaida H, Tomono K, Kohno S. 2003. Long-term efficacy and safety of clarithromycin treatment in patients with diffuse panbronchiolitis. Respir. Med. 97:844–850 [DOI] [PubMed] [Google Scholar]
- 42. Kiran N, Azam S, Dhakam S. 2004. Clarithromycin induced digoxin toxicity: case report and review. J. Pak. Med. Assoc. 54:440–441 [PubMed] [Google Scholar]
- 43. Terkeltaub RA, Furst DE, Digiacinto JL, Kook KA, Davis MW. 2011. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 63:2226–2237 [DOI] [PubMed] [Google Scholar]
- 44. Viluksela M, Vainio PJ, Tuominen RK. 1996. Cytotoxicity of macrolide antibiotics in a cultured human liver cell line. J. Antimicrob. Chemother. 38:465–473 [DOI] [PubMed] [Google Scholar]
- 45. Alipour M, Suntres ZE, Omri A. 2009. Importance of DNase and alginate lyase for enhancing free and liposome encapsulated aminoglycoside activity against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 64:317–325 [DOI] [PubMed] [Google Scholar]
- 46. Yang F, Jin C, Jiang Y, Li J, Di Y, Ni Q, Fu D. 2011. Liposome based delivery systems in pancreatic cancer treatment: from bench to bedside. Cancer Treat. Rev. 37:633–642 [DOI] [PubMed] [Google Scholar]
- 47. Jia Y, Joly H, Leek DM, Demetzos C, Omri A. 2010. The effect of aminoglycoside antibiotics on the thermodynamic properties of liposomal vesicles. J. Liposome Res. 20:84–96 [DOI] [PubMed] [Google Scholar]
- 48. Muppidi K, Pumerantz AS, Wang J, Betageri G. 2012. Development and stability studies of novel liposomal vancomycin formulations. ISRN Pharm. 2012:636743 doi:636710.635402/632012/636743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frezard F, Demicheli C. 2010. New delivery strategies for the old pentavalent antimonial drugs. Expert Opin. Drug Deliv. 7:1343–1358 [DOI] [PubMed] [Google Scholar]
- 50. Halwani M, Yebio B, Suntres ZE, Alipour M, Azghani AO, Omri A. 2008. Co-encapsulation of gallium with gentamicin in liposomes enhances antimicrobial activity of gentamicin against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 62:1291–1297 [DOI] [PubMed] [Google Scholar]
- 51. Halwani M, Blomme S, Suntres ZE, Alipour M, Azghani AO, Kumar A, Omri A. 2008. Liposomal bismuth-ethanedithiol formulation enhances antimicrobial activity of tobramycin. Int. J. Pharm. 358:278–284 [DOI] [PubMed] [Google Scholar]
- 52. Halwani M, Mugabe C, Azghani AO, Lafrenie RM, Kumar A, Omri A. 2007. Bactericidal efficacy of liposomal aminoglycosides against Burkholderia cenocepacia. J. Antimicrob. Chemother. 60:760–769 [DOI] [PubMed] [Google Scholar]
- 53. Kadry AA, Al-Suwayeh SA, Abd-Allah AR, Bayomi MA. 2004. Treatment of experimental osteomyelitis by liposomal antibiotics. J. Antimicrob. Chemother. 54:1103–1108 [DOI] [PubMed] [Google Scholar]
- 54. Mugabe C, Azghani AO, Omri A. 2005. Liposome-mediated gentamicin delivery: development and activity against resistant strains of Pseudomonas aeruginosa isolated from cystic fibrosis patients. J. Antimicrob. Chemother. 55:269–271 [DOI] [PubMed] [Google Scholar]
- 55. Meers P, Neville M, Malinin V, Scotto AW, Sardaryan G, Kurumunda R, Mackinson C, James G, Fisher S, Perkins WR. 2008. Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections. J. Antimicrob. Chemother. 61:859–868 [DOI] [PubMed] [Google Scholar]
- 56. Alipour M, Halwani M, Omri A, Suntres ZE. 2008. Antimicrobial effectiveness of liposomal polymyxin B against resistant Gram-negative bacterial strains. Int. J. Pharm. 355:293–298 [DOI] [PubMed] [Google Scholar]
- 57. Marier JF, Brazier JL, Lavigne J, Ducharme MP. 2003. Liposomal tobramycin against pulmonary infections of Pseudomonas aeruginosa: a pharmacokinetic and efficacy study following single and multiple intratracheal administrations in rats. J. Antimicrob. Chemother. 52:247–252 [DOI] [PubMed] [Google Scholar]
- 58. Onyeji CO, Nightingale CH, Nicolau DP, Quintiliani R. 1994. Efficacies of liposome-encapsulated clarithromycin and ofloxacin against Mycobacterium avium intracellulare complex in human macrophages. Antimicrob. Agents Chemother. 38:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Steenwinkel JE, van Vianen W, Ten Kate MT, Verbrugh HA, van Agtmael MA, Schiffelers RM, Bakker-Woudenberg IA. 2007. Targeted drug delivery to enhance efficacy and shorten treatment duration in disseminated Mycobacterium avium infection in mice. J. Antimicrob. Chemother. 60:1064–1073 [DOI] [PubMed] [Google Scholar]
- 60. Jia Y, Joly H, Omri A. 2008. Liposomes as a carrier for gentamicin delivery: development and evaluation of the physicochemical properties. Int. J. Pharm. 359:254–263 [DOI] [PubMed] [Google Scholar]
- 61. Pumerantz A, Muppidi K, Agnihotri S, Guerra C, Venketaraman V, Wang J, Betageri G. 2011. Preparation of liposomal vancomycin and intracellular killing of meticillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 37:140–144 [DOI] [PubMed] [Google Scholar]
- 62. Alipour M, Suntres ZE, Halwani M, Azghani AO, Omri A. 2009. Activity and interactions of liposomal antibiotics in presence of polyanions and sputum of patients with cystic fibrosis. PLoS One 4:e5724 doi:10.1371/journal.pone.0005724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rukholm G, Mugabe C, Azghani AO, Omri A. 2006. Antibacterial activity of liposomal gentamicin against Pseudomonas aeruginosa: a time-kill study. Int. J. Antimicrob. Agents 27:247–252 [DOI] [PubMed] [Google Scholar]
- 64. Anderson M, Omri A. 2004. The effect of different lipid components on the in vitro stability and release kinetics of liposome formulations. Drug Deliv. 11:33–39 [DOI] [PubMed] [Google Scholar]
- 65. Halwani M, Hebert S, Suntres ZE, Lafrenie RM, Azghani AO, Omri A. 2009. Bismuth-thiol incorporation enhances biological activities of liposomal tobramycin against bacterial biofilm and quorum sensing molecules production by Pseudomonas aeruginosa. Int. J. Pharm. 373:141–146 [DOI] [PubMed] [Google Scholar]
- 66. Mugabe C, Azghani AO, Omri A. 2006. Preparation and characterization of dehydration-rehydration vesicles loaded with aminoglycoside and macrolide antibiotics. Int. J. Pharm. 307:244–250 [DOI] [PubMed] [Google Scholar]
- 67. Alipour M, Omri A, Suntres ZE. 2011. Ginseng aqueous extract attenuates the production of virulence factors, stimulates twitching and adhesion, and eradicates biofilms of Pseudomonas aeruginosa. Can. J. Physiol. Pharmacol. 89:419–427 [DOI] [PubMed] [Google Scholar]
- 68. Alipour M, Suntres ZE, Lafrenie RM, Omri A. 2010. Attenuation of Pseudomonas aeruginosa virulence factors and biofilms by co-encapsulation of bismuth-ethanedithiol with tobramycin in liposomes. J. Antimicrob. Chemother. 65:684–693 [DOI] [PubMed] [Google Scholar]
- 69. Vuong C, Gotz F, Otto M. 2000. Construction and characterization of an agr deletion mutant of Staphylococcus epidermidis. Infect. Immun. 68:1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Allan ND, Kooi C, Sokol PA, Beveridge TJ. 2003. Putative virulence factors are released in association with membrane vesicles from Burkholderia cepacia. Can. J. Microbiol. 49:613–624 [DOI] [PubMed] [Google Scholar]
- 71. Folders J, Algra J, Roelofs MS, van Loon LC, Tommassen J, Bitter W. 2001. Characterization of Pseudomonas aeruginosa chitinase, a gradually secreted protein. J. Bacteriol. 183:7044–7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Winson MK, Camara M, Latifi A, Foglino M, Chhabra SR, Daykin M, Bally M, Chapon V, Salmond GP, Bycroft BW, et al. 1995. Multiple N-acyl-L-homoserine lactone signal molecules regulate production of virulence determinants and secondary metabolites in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 92:9427–9431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kessler E, Safrin M, Olson JC, Ohman DE. 1993. Secreted LasA of Pseudomonas aeruginosa is a staphylolytic protease. J. Biol. Chem. 268:7503–7508 [PubMed] [Google Scholar]
- 74. Kong HS, Roberts DP, Patterson CD, Kuehne SA, Heeb S, Lakshman DK, Lydon J. 2012. Effect of overexpressing rsmA from Pseudomonas aeruginosa on virulence of select phytotoxin-producing strains of P. syringae. Phytopathology 102:575–587 [DOI] [PubMed] [Google Scholar]
- 75. Nombona N, Maduray K, Antunes E, Karsten A, Nyokong T. 2012. Synthesis of phthalocyanine conjugates with gold nanoparticles and liposomes for photodynamic therapy. J. Photochem. Photobiol. B 107:35–44 [DOI] [PubMed] [Google Scholar]
- 76. Lawrence SM, Alpar HO, McAllister SM, Brown MR. 1993. Liposomal (MLV) polymyxin B: physicochemical characterization and effect of surface charge and drug association. J. Drug Target. 1:303–310 [DOI] [PubMed] [Google Scholar]
- 77. Hosny KM. 2010. Ciprofloxacin as ocular liposomal hydrogel. AAPS PharmSciTech. 11:241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kadar B, Szasz M, Kristof K, Pesti N, Krizsan G, Szentandrassy J, Rokusz L, Nagy K, Szabo D. 2010. In vitro activity of clarithromycin in combination with other antimicrobial agents against biofilm-forming Pseudomonas aeruginosa strains. Acta Microbiol. Immunol. Hung. 57:235–245 [DOI] [PubMed] [Google Scholar]
- 79. Gubernator J, Drulis-Kawa Z, Dorotkiewicz-Jach A, Doroszkiewicz W, Kozubek A. 2007. In vitro antimicrobial activity of liposomes containing ciprofloxacin, meropenem and gentamicin against gram-negative clinical bacterial strains. Lett. Drug Design Discov. 4:297–304 [Google Scholar]
- 80. Yasuda H, Ajiki Y, Koga T, Kawada H, Yokota T. 1993. Interaction between biofilms formed by Pseudomonas aeruginosa and clarithromycin. Antimicrob. Agents Chemother. 37:1749–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tre-Hardy M, Vanderbist F, Traore H, Devleeschouwer MJ. 2008. In vitro activity of antibiotic combinations against Pseudomonas aeruginosa biofilm and planktonic cultures. Int. J. Antimicrob. Agents 31:329–336 [DOI] [PubMed] [Google Scholar]
- 82. Alipour M, Dorval C, Suntres ZE, Omri A. 2011. Bismuth-ethanedithiol incorporated in a liposome-loaded tobramycin formulation modulates the alginate levels in mucoid Pseudomonas aeruginosa. J. Pharm. Pharmacol. 63:999–1007 [DOI] [PubMed] [Google Scholar]
- 83. Molinari G, Paglia P, Schito GC. 1992. Inhibition of motility of Pseudomonas aeruginosa and Proteus mirabilis by subinhibitory concentrations of azithromycin. Eur. J. Clin. Microbiol. Infect. Dis. 11:469–471 [DOI] [PubMed] [Google Scholar]
- 84. Casadevall A, Pirofski LA. 2009. Virulence factors and their mechanisms of action: the view from a damage-response framework. J. Water Health 7(Suppl. 1):S2–S18 [DOI] [PubMed] [Google Scholar]
- 85. Lyczak JB, Cannon CL, Pier GB. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2:1051–1060 [DOI] [PubMed] [Google Scholar]
- 86. Skindersoe ME, Alhede M, Phipps R, Yang L, Jensen PO, Rasmussen TB, Bjarnsholt T, Tolker-Nielsen T, Hoiby N, Givskov M. 2008. Effects of antibiotics on quorum sensing in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52:3648–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Garey KW, Vo QP, Lewis RE, Saengcharoen W, LaRocco MT, Tam VH. 2009. Increased bacterial adherence and biomass in Pseudomonas aeruginosa bacteria exposed to clarithromycin. Diagn. Microbiol. Infect. Dis. 63:81–86 [DOI] [PubMed] [Google Scholar]
- 88. Molinari G, Guzman CA, Pesce A, Schito GC. 1993. Inhibition of Pseudomonas aeruginosa virulence factors by subinhibitory concentrations of azithromycin and other macrolide antibiotics. J. Antimicrob. Chemother. 31:681–688 [DOI] [PubMed] [Google Scholar]
- 89. Filion MC, Phillips NC. 1997. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta 1329:345–356 [DOI] [PubMed] [Google Scholar]
- 90. Lappalainen K, Jaaskelainen I, Syrjanen K, Urtti A, Syrjanen S. 1994. Comparison of cell proliferation and toxicity assays using two cationic liposomes. Pharm. Res. 11:1127–1131 [DOI] [PubMed] [Google Scholar]
- 91. Manosroi A, Thathang K, Werner RG, Schubert R, Peschka-Suss R, Manosroi J. 2008. Development of highly stable and low toxic cationic liposomes for gene therapy. Arzneimittelforschung 58:485–492 [DOI] [PubMed] [Google Scholar]