

Abstract
The semen-derived enhancer of viral infection (SEVI) is a positively charged amyloid fibril that is derived from a self-assembling proteolytic cleavage fragment of prostatic acid phosphatase (PAP248-286). SEVI efficiently facilitates HIV-1 infection in vitro, but its normal physiologic function remains unknown. In light of the fact that other amyloidogenic peptides have been shown to possess direct antibacterial activity, we investigated whether SEVI could inhibit bacterial growth. Neither SEVI fibrils nor the unassembled PAP248-286 peptide had significant direct antibacterial activity in vitro. However, SEVI fibrils bound to both Gram-positive (Staphylococcus aureus) and Gram-negative (Escherichia coli and Neisseria gonorrhoeae) bacteria, in a charge-dependent fashion. Furthermore, SEVI fibrils but not the monomeric PAP248-286 peptide promoted bacterial aggregation and enhanced the phagocytosis of bacteria by primary human macrophages. SEVI also enhanced binding of bacteria to macrophages and the subsequent release of bacterially induced proinflammatory cytokines (tumor necrosis factor alpha [TNF-α], interleukin-6 [IL-6], and IL-1β). Finally, SEVI fibrils inhibited murine vaginal colonization with Neisseria gonorrhoeae. These findings demonstrate that SEVI has indirect antimicrobial activity and that this activity is dependent on both the cationic charge and the fibrillar nature of SEVI.
INTRODUCTION
Human semen has recently been shown to contain cationic amyloid fibrils (1–3), the best characterized of which is the semen-derived enhancer of viral infection (SEVI) (3). SEVI is formed from a self-assembling peptide cleavage product of prostatic acid phosphatase (PAP248-286) and has been shown to facilitate HIV-1 infection of cultured cells (3). It is distinguished from other amyloids because of its strong positive charge, as well as the fact that it does not appear to be associated with disease (3–5). However, SEVI's normal physiologic function remains unknown.
Interestingly, other amyloidogenic peptides such as amyloid-β and protegrin-1 (6–8) have been shown to have bactericidal properties, which are thought to be mediated via a channel-forming mechanism (9). Amyloid-forming peptides have also been shown to mediate antimicrobial effects by binding and sequestering bacteria. For example, eosinophil cationic peptide (ECP) has been shown to form amyloid-like structures that mediate bacterial aggregation (10). The antimicrobial effects of such aggregation have been convincingly demonstrated for human α-defensin 6, which has been shown to protect the gastrointestinal mucosa of transgenic mice from infection with pathogenic bacteria, by self-assembling into amyloid-like structures (“nanonets”) that entrap bacteria (11).
We therefore reasoned that SEVI fibrils may also have antibacterial activity and that they may contribute to the well-documented antimicrobial effects of human semen (12), which contains multiple antimicrobial polypeptides, including defensins, semenogelin-derived peptides, and hCAP-18, the precursor to the human antimicrobial cathelicidin protein, LL-37 (12–16). Like LL-37, SEVI is highly cationic—a property that differentiates it from other amyloid-forming peptides. This suggested to us that SEVI and other cationic amyloid fibrils might have unique antimicrobial or immune modulatory properties and that these fibrils might bind to bacterial pathogens via electrostatic interactions. Indeed, most bacteria have an anionic surface charge at neutral pH (17), with an isoelectric point that lies in the range of pH 2 to 5, including both Gram-positive organisms (e.g., Staphylococcus aureus with a pI of 3.4, cited in reference 18) and Gram-negative organisms (e.g., Escherichia coli with a pI of 2.5, cited in reference 10, and Neisseria gonorrhoeae with a pI of 5.3, cited in reference 19).
Consistent with our prediction, we found that SEVI bound to both Gram-positive (S. aureus) and Gram-negative (E. coli and N. gonorrhoeae) bacteria. In contrast, amyloid fibrils formed from a noncationic derivative of the PAP248-286 peptide, in which all arginine and lysine residues were replaced with alanine (designated SEVIala [20]), failed to bind bacteria. SEVI fibrils also promoted bacterial aggregation and enhanced the phagocytosis of bacteria by primary human monocyte-derived macrophages (MDM), whereas the monomeric PAP248-286 peptide and the SEVIala mutant fibrils had no such effect. In addition, SEVI increased the efficiency of bacterial binding to human MDM and the subsequent release of bacterially induced proinflammatory cytokines (tumor necrosis factor alpha [TNF-α], interleukin-6 [IL-6], and IL-1β). Finally, SEVI fibrils reduced murine vaginal infection with N. gonorrhoeae. These findings demonstrate that SEVI has both immunomodulatory and indirect antimicrobial activity and that these activities are dependent on both the cationic charge and the fibrillar nature of the peptide.
MATERIALS AND METHODS
Bacterial strains.
The following bacterial strains were used in these experiments: E. coli CFT073 (K-12 [ATCC]); S. aureus UAMS1 (Mark Smeltzer, University of Arkansas Medical School), S. aureus KLA16 (UAMS1 Δspa) and its genetically complemented, protein A-positive derivative (21), and S. aureus Wood 46 (ATCC); and N. gonorrhoeae FA1090 (ATCC).
SEVI binding assay.
Fluorescent bacterial particles were used to examine bacterial binding to SEVI. For S. aureus and E. coli, we purchased fluorescein isothiocyanate (FITC)-conjugated bacterial particles (Invitrogen). No analogous reagent was available for N. gonorrhoeae, so we labeled this organism using carboxyfluorescein succinimidyl ester (CFSE; Invitrogen). Fluorescently labeled bacteria were resuspended in phosphate-buffered saline (PBS) at a concentration of 3 × 108 particles/ml and incubated at 37°C with 35 μg/ml of biotinylated SEVI fibrils (formed from a biotinylated derivative of the PAP248-286 peptide [22]). In control experiments, bacteria were incubated with biotinylated noncationic fibrils formed from a mutant derivative of the PAP248-286 peptide, in which all arginine and lysine residues were replaced with alanine (this peptide is known as SEVIala) (20). After 1 h of incubation at 37°C, antibiotin-conjugated magnetically activated cell sorting (MACS) beads (Miltenyi Biotec) in PBS containing 2.5% bovine serum albumin (BSA) were added to the mixture, which was placed on ice for 20 min to allow binding. The entire sample was then run on a magnetic separation column. The flowthrough was discarded, and the bound material was released from the column, placed into 96-well plates, and pelleted at 900 × g for 12 min prior to being resuspended in PBS for fluorescence quantification (485-nm excitation wavelength and 535-nm emission wavelength) in a Beckman Coulter DTX 880 plate reader.
Analysis of bacterial sedimentation.
SEVI-mediated aggregation of bacterial particles was assessed by sedimentation using bacterial particles labeled with the fluorescent dye pHrodo. This dye was chosen because the particles were readily visible under room light even at neutral pH, allowing us to easily visualize sedimentation. Fluorescent S. aureus and E. coli bacteria were mixed with SEVI, SEVIala (40 μg/ml), or PBS alone. After vortexing, the samples were transferred to Wintrobe tubes (Fisher Scientific), incubated at 37°C for 1 h, and then photographed to document bacterial sedimentation.
TEM.
For transmission electron microscopy (TEM) visualization, SEVI was diluted to 35 μg/ml in 15 μl of PBS. S. aureus particles (2 × 107) were then added and incubated at 37°C for 1 h. An equal volume of 2.5% phosphate-buffered glutaraldehyde was then added, and the solution was incubated for 30 min, after which 1 drop of the fixed sample was placed on a carbon film mesh nickel grid (Electron Microscopy Sciences) and air dried. Samples were stained with 2.0% phosphotungstic acid (pH 6.5) (PTA) and imaged on an H7650 Hitachi transmission electron microscope (Hitachi, Japan).
Antimicrobial assays.
Direct antimicrobial activity of SEVI fibrils and the monomeric PAP248-286 peptide was determined by microdilution MIC testing. Inocula containing ∼1× 105 mid-exponential-phase cells were dispensed into the wells of polypropylene 96-well plates (BD, San Jose, CA) containing seven 2-fold dilutions of test peptide in growth medium. Plates were then incubated overnight (12 to 18 h) at 37°C, and the MIC value was determined as the lowest concentration of peptide that inhibited bacterial growth, as measured by the unaided eye. In the case of N. gonorrhoeae, we performed a pseudo-MBC (minimal bactericidal concentration) assay. To do this, bacteria were incubated aerobically in supplemented GCP medium (23) for 2 h and then plated onto GC agar and incubated overnight at 37°C in the presence of 5% CO2. Apparent peptide MBC values were recorded as the lowest peptide concentration able to reduce cell growth at least 2-fold in the CFU assay.
Human MDM.
Peripheral blood mononuclear cells were isolated from buffy coats of healthy human donors by Ficoll density gradient centrifugation, and CD14+ monocytes were purified by positive selection using anti-CD14-conjugated magnetic beads (Miltenyi Biotec). Cells were suspended at 1.0 × 106 cells/ml in RPMI medium containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 2% glutamine, and 5 ng/ml human granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) added on day 0 and day 3. On day 5, GM-CSF-containing medium was removed, and cells were cultured until day 6 or 7 in RPMI medium containing 10% FBS.
Neutrophil isolation.
Peripheral blood was collected into plasma separator blood tubes, and polymorphonuclear leukocytes (PMNs) were purified with 1-Step Polymorphs (Accurate Chemical and Science Corp.). The PMN layer was removed, washed with Hanks balanced salt solution (HBSS) (without Ca2+ or Mg2+) plus 0.1% BSA, and red blood cells were lysed. PMNs were then counted and plated in 12-well tissue culture plates at 1 × 106 cells/well in 1 ml of PBS. After 1 h, the PMNs were used for the gentamicin protection assay as outlined below.
Phagocytosis assays using killed bacteria (flow cytometric assay).
Phagocytosis assays were conducted using pHrodo-labeled, killed bacteria (BioParticles; Invitrogen). This dye fluoresces brightly under low-pH conditions and thus allows one to conveniently differentiate phagocytosed bioparticles (which fluoresce brightly in the low-pH environment of the endosome) from bioparticles that remain bound to the cell surface (which do not fluoresce brightly). Briefly, pHrodo-labeled bacteria were resuspended in PBS at a concentration of 2 mg/ml and then mixed with SEVI, SEVIala, monomeric PAP248-286 peptides, or PBS alone. The resulting mixtures were vortexed and incubated in a water bath at 37°C for 1 h, after which 20 to 35 μl of the suspension was added to human MDM, to achieve a final concentration of between 20 and 40 μg/ml of SEVI. MDM and the fibril-bioparticle mixture were incubated for 1 h at 37°C. In control experiments, MDM were preincubated with cytochalasin-D (Sigma-Aldrich) at a concentration of 2 μg/ml for 30 min, prior to the addition of the fibril-bacterial mixture. After incubation with the fluorescent bacteria, MDM were harvested using a 1% trypsin solution and resuspended in PBS containing 2% FBS for flow cytometric analysis on a FACSCalibur (BD) cytometer.
Phagocytosis assays using live bacteria (gentamicin protection assay).
MDM were prepared as described above. Mid-log-phase bacteria were preincubated in PBS in the presence or absence of SEVI (35 μg/ml) and then added to cells. After 1 h, inocula were removed and the MDM treated with 200 μg/ml gentamicin for an additional hour. MDM were then washed extensively with PBS and lysed with 0.1% Triton X-100 in PBS (or 0.5% saponin plus 2 mM EDTA for N. gonorrhoeae). Cell lysates were then serially diluted and plated for CFU enumeration.
Analysis of cytokine release.
MDM were prepared as described above. Mid-log-phase bacteria (E. coli CFT073 and S. aureus UAMS-1) or N. gonorrhoeae swabbed into PBS from a GC agar plate were then mixed with PBS alone, monomeric PAP248-286 peptide, SEVIala fibrils, or SEVI fibrils and added to the MDMs, as described for the phagocytosis assays. For this experiment, N. gonorrhoeae and E. coli were added to cells at 5 × 105 cells/ml, and S. aureus at 5 × 107 cells/ml (different concentrations of the organisms were used because of their distinct cell wall composition and differential ability to induce cytokine release). MDM were stimulated for 1 h with bacteria (with or without peptide/fibril) and then washed thoroughly, after which they were incubated for an additional 6 h. Cytokine levels in culture supernatants were then measured by enzyme-linked immunosorbent assay (ELISA).
N. gonorrhoeae infection model.
Female BALB/c mice (>5 weeks old) were treated with water-soluble estradiol (Sigma) to promote susceptibility to N. gonorrhoeae infection as previously outlined (24, 25). To prevent commensal bacterial overgrowth, mice were given streptomycin sulfate (0.24 mg intraperitoneally twice daily); note that the infecting N. gonorrhoeae strain FA1090 is streptomycin resistant (24, 25). N. gonorrhoeae was swabbed into PBS from fresh GC agar plates and incubated for 30 min with or without SEVI (35 μg/ml). Anesthetized mice were infected intravaginally with 106 CFU with or without SEVI (in a volume of 20 μl), and infections were monitored by daily vaginal lavage with 30 μl of PBS, serial diluting, and plating on GC agar.
RESULTS
SEVI binds to both Gram-negative and Gram-positive bacteria.
To test the binding of SEVI to bacteria, we incubated fluorescently labeled E. coli, S. aureus, and N. gonorrhoeae particles with biotinylated SEVI fibrils (22) or with matched but noncationic SEVIala fibrils. After incubation with the fluorescent bacteria, we captured the biotinylated fibrils (and any associated bacterial particles) onto antibiotin-conjugated MACS beads and quantitated the magnitude of captured bacterial fluorescence. Figure 1 shows that all 3 of the bacteria bound efficiently to the cationic SEVI fibrils but bound to the noncationic SEVIala fibrils at a level equivalent to the assay background level (i.e., binding to antibiotin beads in the absence of any fibril). Binding of bacteria to SEVI was between 3- and 10-fold greater than background binding (depending on the organism). In addition, the difference in binding to SEVI versus SEVIala achieved statistical significance for all of the organisms tested (Fig. 1).
Fig 1.
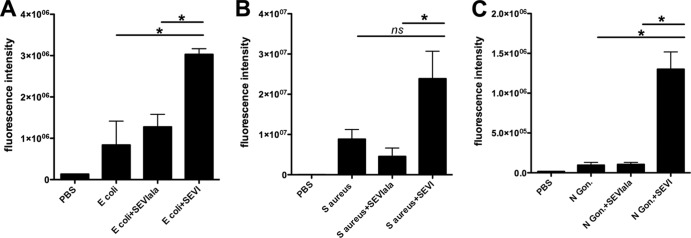
SEVI binds bacteria in a charge-dependent manner. Fluorescently labeled S. aureus (A), E. coli (B), or N. gonorrhoeae (C) cells were preincubated with 35 μg/ml of biotinylated SEVI or SEVIala fibrils. SEVI:bacteria aggregates were then captured using antibiotin magnetic beads. The fluorescence intensity of the captured aggregates is shown. Error bars represent the standard errors of the means (SEM) from three to eight independent experiments. Data were analyzed with an exact Wilcoxon rank sum test. *, P = 0.05; ns, not significant.
We next analyzed whether SEVI was capable of aggregating bacteria. Since the physiologic concentration of SEVI fibrils in semen is approximately 35 μg/ml (3, 4), we performed these and subsequent experiments using concentrations of SEVI in this range. First, we vigorously mixed fluorescent bacterial particles in transparent glass vials, in the presence or absence of SEVI or SEVIala fibrils, and then incubated the tubes on the benchtop at room temperature for 1 h. As shown in Fig. 2A, the bacteria sedimented into a thick pellet at the bottom of the tube in the presence of SEVI fibrils only. To further examine the putative complexes formed between SEVI and bound bacteria, we performed TEM using SEVI fibrils that were incubated with S. aureus. The results, presented in Fig. 2B, show that the bacteria associated closely with the SEVI fibrils.
Fig 2.

SEVI promotes bacterial aggregation. (A) Dye-labeled E. coli (left) or S. aureus (right) particles were vigorously mixed in PBS alone or in the presence of 40 μg/ml of SEVI or SEVIala fibrils. Samples were then incubated at 37°C for 1 h and photographically imaged. The arrowheads denote sedimented bacterial aggregates. (B) S. aureus was incubated with SEVI (35 μg/ml) for 1 h. Samples were fixed in 2.5% glutaraldehyde phosphate buffer, air dried, negatively stained on a carbon mesh grid, and imaged by transmission electron microscopy.
SEVI does not directly inhibit bacterial growth.
Amyloidogenic peptides such as amyloid-β and protegrin-1 have been shown to possess direct antimicrobial activity (6–9). We therefore tested whether SEVI fibrils or monomeric PAP248-286 peptide could also directly inhibit bacterial growth, using a panel of organisms known to be inhibited by the Aβ42 peptide (8). The results, shown in Table 1, reveal that neither SEVI fibrils nor the PAP248-286 peptide exhibited a potent inhibitory effect on the liquid-phase growth of a panel of bacterial organisms including E. coli, S. aureus (methicillin susceptible [MSSA] and methicillin resistant [MRSA]), N. gonorrhoeae, Staphylococcus epidermidis, Enterococcus faecalis, and Pseudomonas aeruginosa. Inhibitory activity was detected only at very high (nonphysiologic) peptide concentrations (64 or 128 μg/ml) against S. epidermidis and possibly also against E. faecalis. However, in most cases, SEVI fibrils and PAP248-286 had no measurable effect on bacterial growth—even at concentrations as high as 128 μg/ml.
Table 1.
SEVI does not have direct antimicrobial activity as determined by MICa
| Organism | Strain | MIC (μg/ml) |
|
|---|---|---|---|
| SEVI fibrils | Peptide monomer (PAP248-286) | ||
| S. aureus (MSSA) | ATCC 29213 | >128 | >128 |
| S. aureus (MRSA) | ATCC 43300 | >128 | >128 |
| S. aureus (MSSA) | UAMS-1 | >128 | >128 |
| S. epidermidis | ATCC 12228 | 128 | 128 |
| E. faecalis | ATCC 29212 | 64 | >128 |
| E. coli | ATCC 27853 | >128 | >128 |
| E. coli | CFT-073 | >128 | >128 |
| P. aeruginosa | ATCC 27853 | >128 | >128 |
| N. gonorrhoeaeb | FA1090 | >128 | >128 |
Growth inhibition was determined by visual inspection after overnight incubation and confirmed by CFU counts or by conducting CFU counts after 2 h of culturing.
Instead of an MIC assay, a pseudo-MBC assay was performed for N. gonorrhoeae.
SEVI facilitates phagocytosis of bacteria.
We next examined the effect of SEVI on bacterial phagocytosis. To do this, we used killed bacterial particles labeled with the pH-sensitive fluorescent dye pHrodo (BioParticles; Invitrogen). pHrodo-labeled E. coli and S. aureus bacteria were mixed with SEVI or PBS alone and then added to MDM. SEVI-mediated aggregation of fluorescent bacterial particles resulted in a robust enhancement of phagocytosis when measured by flow cytometry (see Fig. S1, upper panels, in the supplemental material). However, under control conditions, in which cells were pretreated with cytochalasin-D, there was significantly less fluorescence detected (see Fig. S1, lower panels, in the supplemental material), confirming that uptake of the bacterial particles was mediated by phagocytosis (26). This experiment was repeated multiple times using MDM from 3 to 5 different donors, and a summary of the results from these experiments is presented in Fig. 3. Physiological levels of SEVI increased the percentage of MDM that phagocytosed bacterial particles from 28.7% to 49.0% of the cells for E. coli and from 53.5% to 78.4% of the cells for S. aureus (P = 0.05 in both cases by the exact Wilcoxon rank sum test) (Fig. 3A and C). SEVI also increased the median fluorescent intensity (MFI) of the positive cells, 2.3-fold for S. aureus and 1.39-fold for E. coli (P = 0.05 in both cases by the exact Wilcoxon rank sum test) (Fig. 3B and D). We speculate that the greater magnitude of SEVI's effect on the number of S. aureus particles taken up per cell may be related to the smaller size of S. aureus compared to E. coli (i.e., it is easier for a cell to ingest additional S. aureus particles). As noted above, when cells were preincubated with cytochalasin-D, the uptake of the particles was abolished (Fig. 3A and C), confirming that the uptake detected was indeed phagocytic (26). Finally, experiments using the monomeric PAP248-286 peptide did not reveal any enhancement of bacterial phagocytosis (see Fig. S2 in the supplemental material), demonstrating that this effect is dependent upon fibrillization of the peptide.
Fig 3.
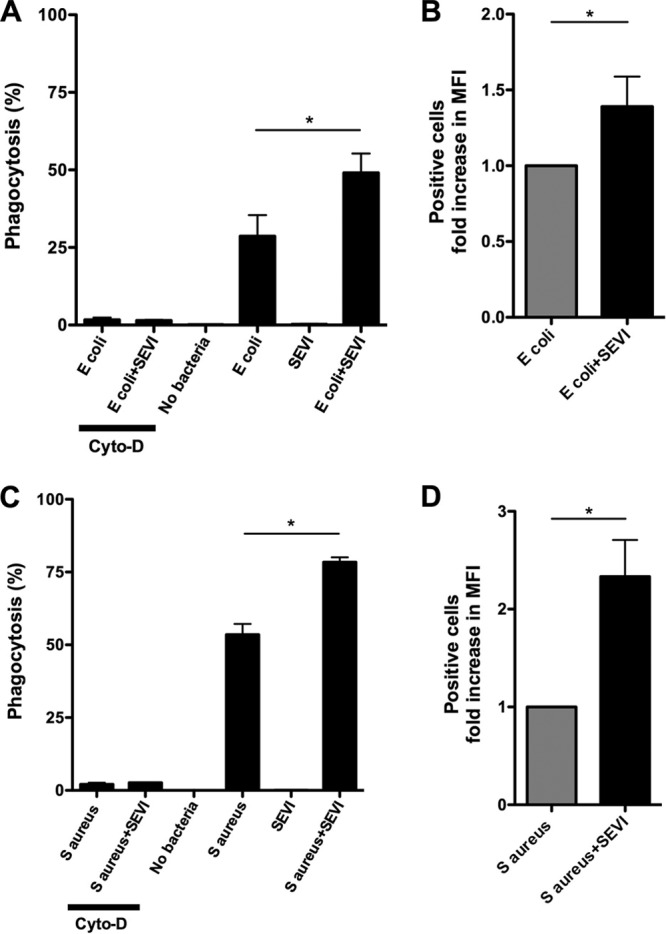
SEVI enhances phagocytic uptake of killed bacterial particles by primary monocyte-derived macrophages. Killed E. coli or S. aureus particles labeled with pHrodo (BioParticles; Invitrogen) were mixed with SEVI fibrils (SEVI) or buffer alone and then incubated with primary human monocyte-derived macrophages (MDM). After 1 h, cells were harvested and flow cytometric analysis of bacterial uptake was performed. (A, C) Percentage of MDM containing fluorescent intracellular bacterial particles for cells exposed to E. coli or S. aureus (± SEVI). (B, D) Median fluorescence intensity (MFI) of MDM containing fluorescent intracellular bacterial particles for cells exposed to E. coli or S. aureus (± SEVI). (A to D) Error bars represent the SEM from three to five independent experiments, performed with MDM from different donors in each case. Data were analyzed with an exact Wilcoxon rank sum test. *, P = 0.05.
To test whether SEVI also enhanced phagocytic uptake of live bacteria, we performed a gentamicin protection assay using the same strains of bacteria as those used to prepare the bioparticles. Mid-log-phase E. coli K-12, S. aureus Wood 46, or N. gonorrhoeae cells were preincubated with SEVI or PBS and then added to MDM or PMNs (in the case of N. gonorrhoeae). After 1 h, the inocula were removed and cells were treated with gentamicin in order to kill extracellular bacteria. MDMs or PMNs were then lysed and intracellular bacteria enumerated by serial diluting and CFU counting. SEVI enhanced the phagocytic uptake of S. aureus 13.9-fold, of N. gonorrhoeae 1.84-fold, and of E. coli 1.90-fold on MDMs (Fig. 4A to C) and of N. gonorrhoeae 17.0-fold by PMNs (Fig. 4D) (P = 0.05 in all cases by the exact Wilcoxon rank sum test).
Fig 4.
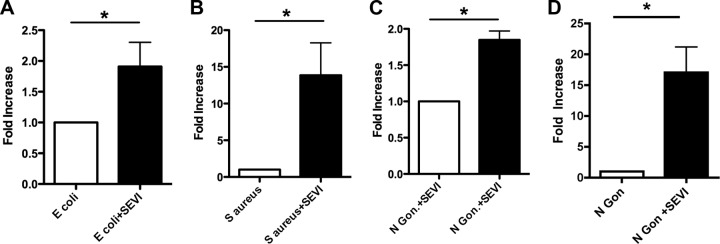
SEVI enhances phagocytic uptake of live bacteria by primary monocyte-derived macrophages and neutrophils (PMNs). Mid-log-phase bacteria were preincubated with SEVI or PBS and then added to MDM (A to C) or PMNs (D). After 1 h, inocula were removed and the MDM or PMNs were treated with gentamicin to kill extracellular bacteria. Viable intracellular bacteria were then counted by plating of serially diluted cell lysates. The results are plotted in terms of relative CFU compared to the CFU of cells exposed to bacteria alone (defined as 1). Error bars represent the SEM from three experiments, each performed with cells from different donors. Data were analyzed with a Wilcoxon signed rank test. *, P = 0.05.
Comparison of the data presented in Fig. 3 and 4 reveals that SEVI-mediated enhancement of phagocytosis was greater for live than for killed S. aureus (Fig. 3 and 4). We tentatively attribute this to the fact that live S. aureus particles readily form short chains or clusters whereas killed particles do not.
We next tested whether the well-characterized staphylococcal virulence factor, protein A, might protect S. aureus against SEVI-mediated enhancement of phagocytosis, as it does against antibody-mediated enhancement of phagocytosis (27). To do this, we took advantage of a spa-deleted mutant of the UAMS1 strain of S. aureus (KPL16) and its genetically complemented, protein A-positive counterpart (21). As shown in Fig. 5, SEVI-mediated enhancement of the phagocytosis of S. aureus was unaffected by the presence or absence of protein A.
Fig 5.
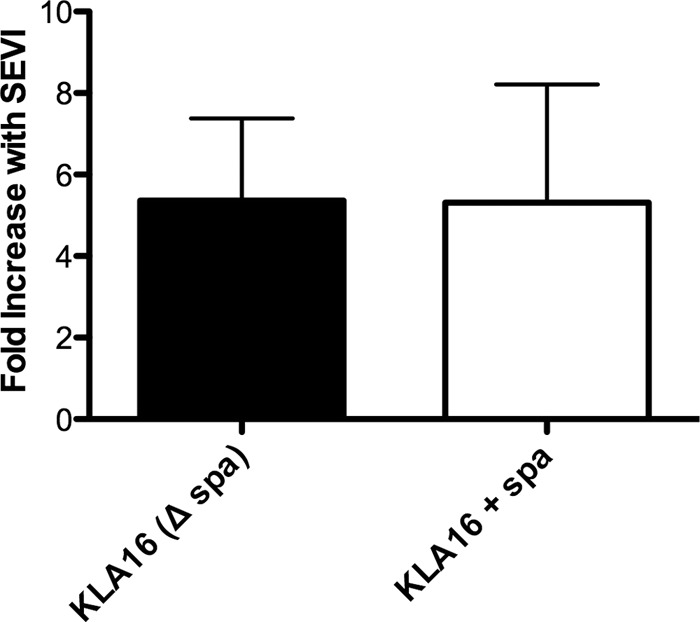
SEVI-mediated enhancement of S. aureus phagocytosis by macrophages is unaffected by staphylococcal protein A. A spa-deleted mutant of the UAMS1 strain of S. aureus (KPL16) and its genetically complemented, protein A-positive counterpart (21) were incubated with MDM in the presence or absence of SEVI, and bacterial pathogenesis was measured by gentamicin resistance assay (see Fig. 4 legend). Data are presented as the fold increase in bacterial phagocytosis in the presence of SEVI compared to that of cells exposed to bacteria alone (defined as 1). Error bars represent the SEM from three experiments, each performed with cells from different donors.
SEVI enhances binding of bacteria to the surface of MDM.
To test whether SEVI-mediated enhancement of bacterial phagocytosis might be secondary to increased binding of bacteria to MDM, we incubated FITC-labeled, killed bacterial particles with SEVI for 1 h and then added the fibril-particle mix to MDM that had been pretreated with cytochalasin-D to block phagocytosis. We found that SEVI enhanced E. coli binding to the surface of MDM over 10-fold (from an MFI of 57 to 677) and that it enhanced S. aureus binding over 5-fold (from an MFI of 1,270 to 6,480) (P = 0.05 in both cases by the exact Wilcoxon rank sum test) (Fig. 6). Control experiments showed that the monomeric PAP248-286 peptide did not enhance bacterial binding to MDM (see Fig. S3 in the supplemental material).
Fig 6.
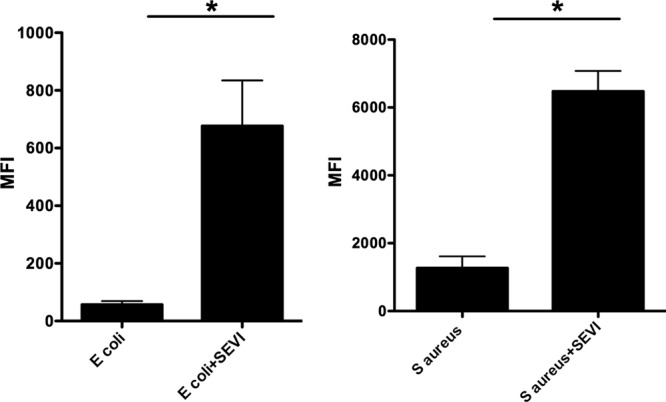
SEVI enhances binding of killed bacterial particles to human macrophages. Killed E. coli (left panels) or S. aureus (right panels) cells labeled with FITC were mixed with SEVI fibrils or buffer alone and then incubated with MDM pretreated with cytochalasin D. After 40 min, cells were harvested and flow cytometric analysis was performed. The median fluorescence intensity (MFI) of MDM incubated with fluorescent bacteria is shown. Error bars represent the SEM from three independent experiments, performed with MDM from different donors in each case. Data were analyzed with an exact Wilcoxon rank sum test. *, P = 0.05.
SEVI enhances bacterially mediated induction of cytokine release.
In light of SEVI's ability to enhance bacterial binding and phagocytosis, we examined whether SEVI could increase bacterially induced release of cytokines by MDM. To do this, mid-log-phase bacteria were incubated with SEVI for 1 h, and the fibril-particle mixture was then added to MDM. After 1 h, the cells were washed thoroughly and then incubated for an additional 6 h to allow cytokine release. As shown in Fig. 7, SEVI significantly enhanced proinflammatory cytokine release by MDM that were exposed to E. coli, S. aureus, or N. gonorrhoeae but had no effect on cytokine release in the absence of bacteria. SEVI increased TNF-α release from bacterially exposed MDM ≥2-fold for S. aureus, E. coli, and N. gonorrhoeae (from 2,970 pg/ml to 9,700 pg/ml for S. aureus, from 3,560 pg/ml to 11,900 pg/ml for E. coli, and from 5,420 pg/ml to 10,300 pg/ml for N. gonorrhoeae) (P < 0.0001 in all cases by the exact Wilcoxon rank sum test; Fig. 7, left panel). SEVI had a similar effect on IL-6 release, increasing levels of this cytokine roughly 2-fold for S. aureus, E. coli, and N. gonorrhoeae (from 5,340 pg/ml to 9,190 pg/ml for S. aureus, from 4,310 pg/ml to 9,950 pg/ml for E. coli, and from 7,230 pg/ml to 12,400 pg/ml for N. gonorrhoeae) (P < 0.01 in all cases by the exact Wilcoxon rank sum test; Fig. 7, middle panel). Finally, SEVI also stimulated IL-1β release from bacterially exposed MDM, raising levels of this cytokine by about 50% for S. aureus (from 180 to 306 pg/ml), almost 10-fold for E. coli (from 43 to 373 pg/ml), and about 5-fold for N. gonorrhoeae (from 40 to 198 pg/ml) (P = 0.05 for S. aureus and P < 0.001 for E. coli and N. gonorrhoeae by the exact Wilcoxon rank sum test; Fig. 7, right panel). The modest increase in IL-1β release from cells exposed to S. aureus (by about 50%) but strongly enhanced IL-1β release from cells exposed to N. gonorrhoeae and E. coli (5 to 10-fold) may be related to differences in the immunostimulatory activity of cell wall components of the bacteria.
Fig 7.
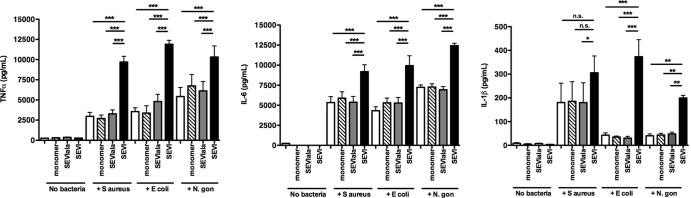
SEVI potentiates bacterially induced cytokine release from human macrophages. S. aureus, E. coli, or N. gonorrhoeae cells were mixed with the monomeric PAP248-286 peptide (monomer), SEVIala fibrils (SEVIala), SEVI fibrils (SEVI), or PBS alone and then incubated with MDM. As controls, cells were incubated in the presence or absence of the PAP248-286 monomer, SEVIala, or SEVI alone, without bacteria (lanes labeled “no bacteria”). MDM were stimulated for 1 h and then washed thoroughly and incubated for an additional 6 h, after which cytokine levels in culture supernatants were measured by ELISA. Results are shown for TNF-α (A), IL-6 (B), and IL-1β (C). Error bars represent the SEM from three independent experiments performed with MDM from different donors in each case. Data were analyzed with an exact Wilcoxon rank sum test.
SEVI inhibits vaginal colonization by N. gonorrhoeae.
To examine the effects of SEVI on bacterial infection in vivo, we used N. gonorrhoeae as a representative sexually transmissible pathogen. We further took advantage of a well-described intravaginal infection model in BALB/c mice, in which N. gonorrhoeae infection can be reliably and reproducibly established in estradiol-treated animals (24, 25). Mice were intravaginally exposed to N. gonorrhoeae, in the presence or absence of SEVI. Infections were then monitored daily by vaginally lavaging mice with PBS, followed by enumeration of colony-forming bacteria. Animals that were intravaginally exposed to N. gonorrhoeae in the presence of SEVI were found to have consistently lower levels of bacterial infection, at both 1 and 3 days after experimental inoculation (Fig. 8). Animals exposed to N. gonorrhoeae plus SEVI had mean bacterial loads of 7.2 × 104 (day 1) and 1.2 × 104 (day 3), while animals exposed to N. gonorrhoeae in the absence of SEVI had mean bacterial loads that were 40- to 90-fold higher (2.9 × 106 on day 1 and 1.1 × 106 on day 3). These differences in bacterial load were statistically different at both time points (P < 0.05 by the Mann-Whitney test), suggesting that SEVI had a protective effect in vivo against vaginal colonization by N. gonorrhoeae.
Fig 8.
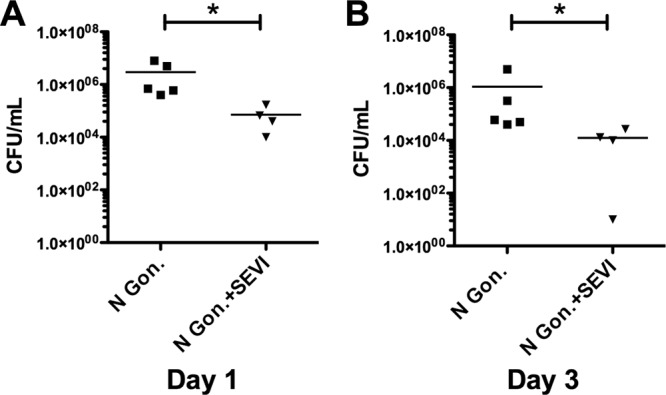
SEVI enhances cellular uptake of N. gonorrhoeae in vivo. N. gonorrhoeae was preincubated with PBS alone or with SEVI fibrils (35 μg/ml) and then injected intravaginally into BALB/c mice. After 24 and 72 h, a vaginal lavage was performed, and the number of recovered viable bacteria was measured by CFU assay. Data represent bacterial counts from individual mice from a single representative experiment. A horizontal line denotes the mean bacterial count for each group of animals. Statistical significance was determined using the Mann-Whitney test (*, P < 0.05).
DISCUSSION
While SEVI has been shown to greatly enhance HIV-1 infection in vitro, it currently has no known biological function. Based on the antimicrobial properties of other amyloidogenic peptides, we examined the antimicrobial activity of SEVI. At physiologic concentrations, SEVI had no direct inhibitory effect on bacterial growth in vitro (Table 1). However, SEVI was found to efficiently bind to both Gram-positive (S. aureus) and Gram-negative bacteria (E. coli and N. gonorrhoeae) in a charge-dependent manner (Fig. 1) and to mediate their aggregation (Fig. 2). This ability to aggregate bacteria is analogous to the recently described effects of other self-assembling host peptides, including ECP and human α-defensin 6, which can form “nanonets” that entrap bacterial pathogens (10, 11). In addition, SEVI may also promote more-efficient binding of bacteria to mammalian cells (including macrophages; Fig. 4), which is expected to be electrostatically unfavorable under normal conditions. This may result in sequestration of bacterial particles, favoring their subsequent clearance.
Overall, we attribute SEVI's ability to (i) enhance cellular uptake of bacteria, (ii) promote bacterially stimulated cytokine release, and (iii) protect against vaginal colonization by N. gonorrhoeae to its unique combination of cationic charge and fibrillar structure. Consistent with this, neither the monomeric PAP248-286 peptide nor the noncationic SEVIala fibril had any effect on bacterial phagocytosis or bacterially stimulated cytokine release.
In summary, SEVI is distinct from conventional antimicrobial peptides (28) and other bacterium-aggregating peptides, because it lacks direct antimicrobial activity and because its ability to enhance innate immune function is dependent on both its cationic charge and its fibrillar structure. We speculate that these structural properties may allow SEVI to bind to bacteria in a multivalent manner, allowing it to cross-link bacteria and to function as a “sticky mesh” capable of trapping microbial pathogens. Finally, the recent identification of other cationic amyloid fibrils in human semen (1–3) suggests that SEVI is not the only cationic amyloid fibril present in humans and that these fibrils may collectively represent a novel class of immune defense molecules. Future experiments are planned to address this hypothesis and to explore whether synthetic cationic amyloid fibrils (29) may also function as antibacterial agents and/or immune modulators.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the following grants from the National Institutes of Health (NIH): T32GM007356 (J.S.O., J.N.S.), T32DA007232 (J.S.O., F.O.), T32AI049815 (D.E.), T32GM068411 (L.R.B.), and R21AI094511.
We thank Virginia Clark and Brian Smith for advice and assistance, as well as Ann Jerse (Uniformed Sciences University, Washington, DC) for guidance and assistance with the N. gonorrhoeae infection experiments. We also thank Mark Smeltzer (University of Arkansas Medical School) for providing bacterial strains. Finally, we thank Tim Bushnell and the URMC Flow Cytometric Core facility and Karen Bentley and the URMC Electron Microscopy Core facility for technical assistance.
Footnotes
Published ahead of print 18 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02464-12.
REFERENCES
- 1. Arnold F, Schnell J, Zirafi O, Sturzel C, Meier C, Weil T, Standker L, Forssmann WG, Roan NR, Greene WC, Kirchhoff F, Munch J. 2012. Naturally occurring fragments from two distinct regions of the prostatic acid phosphatase form amyloidogenic enhancers of HIV infection. J. Virol. 86:1244–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roan NR, Muller JA, Liu H, Chu S, Arnold F, Sturzel CM, Walther P, Dong M, Witkowska HE, Kirchhoff F, Munch J, Greene WC. 2011. Peptides released by physiological cleavage of semen coagulum proteins form amyloids that enhance HIV infection. Cell Host Microbe 10:541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Munch J, Rucker E, Standker L, Adermann K, Goffinet C, Schindler M, Wildum S, Chinnadurai R, Rajan D, Specht A, Gimenez-Gallego G, Sanchez PC, Fowler DM, Koulov A, Kelly JW, Mothes W, Grivel JC, Margolis L, Keppler OT, Forssmann WG, Kirchhoff F. 2007. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 131:1059–1071 [DOI] [PubMed] [Google Scholar]
- 4. Kim KA, Yolamanova M, Zirafi O, Roan NR, Staendker L, Forssmann WG, Burgener A, Dejucq-Rainsford N, Hahn BH, Shaw GM, Greene WC, Kirchhoff F, Munch J. 2010. Semen-mediated enhancement of HIV infection is donor-dependent and correlates with the levels of SEVI. Retrovirology 7:55 doi:10.1186/1742-4690-7-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hartjen P, Frerk S, Hauber I, Matzat V, Thomssen A, Holstermann B, Hohenberg H, Schulze W, Schulze Zur Wiesch J, van Lunzen J. 2012. Assessment of the range of the HIV-1 infectivity enhancing effect of individual human semen specimen and the range of inhibition by EGCG. AIDS Res. Ther. 9:2 doi:10.1186/1742-6405-9-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kagan BL, Jang H, Capone R, Teran Arce F, Ramachandran S, Lal R, Nussinov R. 2012. Antimicrobial properties of amyloid peptides. Mol. Pharm. 9:708–717 doi:10.1021/mp200419b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jang H, Arce FT, Mustata M, Ramachandran S, Capone R, Nussinov R, Lal R. 2011. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 100:1775–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. 2010. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505 doi:10.1371/journal.pone.0009505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Capone R, Mustata M, Jang H, Arce FT, Nussinov R, Lal R. 2010. Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 98:2644–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torrent M, Pulido D, Nogues MV, Boix E. 2012. Exploring new biological functions of amyloids: bacteria cell agglutination mediated by host protein aggregation. PLoS Pathog. 8:e1003005 doi:10.1371/journal.ppat.1003005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B, Salzman NH, Underwood MA, Tsolis RM, Young GM, Lu W, Lehrer RI, Baumler AJ, Bevins CL. 2012. Human alpha-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 337:477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taylor PW, Morgan HR. 1952. Antibacterial substances in human semen and prostatic fluid. Surg. Gynecol. Obstet. 94:662–668 [PubMed] [Google Scholar]
- 13. Andersson E, Sorensen OE, Frohm B, Borregaard N, Egesten A, Malm J. 2002. Isolation of human cationic antimicrobial protein-18 from seminal plasma and its association with prostasomes. Hum. Reprod. 17:2529–2534 [DOI] [PubMed] [Google Scholar]
- 14. Malm J, Sorensen O, Persson T, Frohm-Nilsson M, Johansson B, Bjartell A, Lilja H, Stahle-Backdahl M, Borregaard N, Egesten A. 2000. The human cationic antimicrobial protein (hCAP-18) is expressed in the epithelium of human epididymis, is present in seminal plasma at high concentrations, and is attached to spermatozoa. Infect. Immun. 68:4297–4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bourgeon F, Evrard B, Brillard-Bourdet M, Colleu D, Jegou B, Pineau C. 2004. Involvement of semenogelin-derived peptides in the antibacterial activity of human seminal plasma. Biol. Reprod. 70:768–774 [DOI] [PubMed] [Google Scholar]
- 16. Com E, Bourgeon F, Evrard B, Ganz T, Colleu D, Jegou B, Pineau C. 2003. Expression of antimicrobial defensins in the male reproductive tract of rats, mice, and humans. Biol. Reprod. 68:95–104 [DOI] [PubMed] [Google Scholar]
- 17. Winslow CE, Upton MF. 1926. The electrophoretic migration of various types of vegetative cells. J. Bacteriol. 11:367–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harden VP, Harris JO. 1953. The isoelectric point of bacterial cells. J. Bacteriol. 65:198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heckels JE, Blackett B, Everson JS, Ward ME. 1976. The influence of surface charge on the attachment of Neisseria gonorrhoeae to human cells. J. Gen. Microbiol. 96:359–364 [DOI] [PubMed] [Google Scholar]
- 20. Roan NR, Munch J, Arhel N, Mothes W, Neidleman J, Kobayashi A, Smith-McCune K, Kirchhoff F, Greene WC. 2009. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J. Virol. 83:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuechenmeister LJ, Anderson KL, Morrison JM, Dunman PM. 2009. The use of molecular beacons to directly measure bacterial mRNA abundances and transcript degradation. J. Microbiol. Methods 76:146–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olsen JS, Brown C, Capule CC, Rubinshtein M, Doran TM, Srivastava RK, Feng C, Nilsson BL, Yang J, Dewhurst S. 2010. Amyloid-binding small molecules efficiently block SEVI (semen-derived enhancer of virus infection)- and semen-mediated enhancement of HIV-1 infection. J. Biol. Chem. 285:35488–35496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spence JM, Wright L, Clark VL. 2008. Laboratory maintenance of Neisseria gonorrhoeae. Curr. Protoc. Microbiol. Chapter 4:Unit 4A 1. doi:10.1002/9780471729259.mc04a01s8 [DOI] [PubMed] [Google Scholar]
- 24. Jerse AE. 1999. Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect. Immun. 67:5699–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jerse AE, Wu H, Packiam M, Vonck RA, Begum AA, Garvin LE. 2011. Estradiol-treated female mice as surrogate hosts for Neisseria gonorrhoeae genital tract infections. Front. Microbiol. 2:107 doi:10.3389/fmicb.2011.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mimura N, Asano A. 1976. Synergistic effect of colchicine and cytochalasin D on phagocytosis by peritoneal macrophages. Nature 261:319–321 [DOI] [PubMed] [Google Scholar]
- 27. Dossett JH, Kronvall G, Williams RC, Jr, Quie PG. 1969. Antiphagocytic effects of staphylococcal protein A. J. Immunol. 103:1405–1410 [PubMed] [Google Scholar]
- 28. Yang D, Liu ZH, Tewary P, Chen Q, de la Rosa G, Oppenheim JJ. 2007. Defensin participation in innate and adaptive immunity. Curr. Pharm. Des. 13:3131–3139 [DOI] [PubMed] [Google Scholar]
- 29. Easterhoff D, DiMaio JT, Doran TM, Dewhurst S, Nilsson BL. 2011. Enhancement of HIV-1 infectivity by simple, self-assembling modular peptides. Biophys. J. 100:1325–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.