

Abstract
Kaposi sarcoma-associated herpesvirus (KSHV) is the etiological agent of several human malignancies. The virus is able to modulate pro-proliferative pathways to its advantage, while simultaneously inhibiting pro-apoptotic signaling pathways. These functions are carried out by multiple viral proteins acting in concert. The overall outcome is the survival and proliferation of the infected cell. Additionally, the virus also modulates innate immune pathways to allow for prolonged survival of the infected cell following primary infection, and during viral latency. Here we review the latest advances in our knowledge of KSHV pathogenesis.
Introduction
Infection with Kaposi sarcoma-associated herpesvirus (KSHV) drives viral pathogenesis. The clinical and molecular phenotypes of these infection events are diverse and depend on the cellular environment (B cell lineage or endothelial cell lineage) as well as host cell mutations that are acquired during progressive transformation steps. KSHV pathogenesis also depends on cell extrinsic events such as co-infection with human immunodeficiency virus (HIV), degree and status of the systemic host immune response, and other host factors, which modulate the cellular environment. Currently, there is no molecular evidence that KSHV pathogenesis differs among distinct KSHV strains or clades, though SNPs in both miRNA and gene coding regions have been identified and associated with altered gene expression [1,2].
During much of its life cycle, the KSHV virus is present in a latent form. Here, only a very limited set of viral genes is expressed. They ensure maintenance and faithful segregation of the viral genome during host cell division and they ensure continued cell survival (reviewed in [3]). KSHV is able to increase the life span of infected cells and primary infection induces some features e.g. activation of pro-survival signaling pathways, that are commonly seen in transformed cells [4-6]. Individual viral genes and microRNAs augment proliferation in multiple experimental systems and also modulate autophagy and oncogene-induced senescence [3,7-9]. To ensure continued cell survival, KSHV infection also modulates cellular immunity. Recent evidence also indicates that KSHV modulates various metabolic pathways in B cells and endothelial cells to its advantage [10-12].
KSHV in endothelial cells
KSHV infects endothelial lineage cells. Clinically, KSHV infected endothelial cells constitute the bulk of Kaposi sarcoma (KS) lesions. KS lesions are composed of elongated, spindle-like, endothelial cells. All endothelial cells within a KS tumor are infected with the virus and express the KSHV latent genes, including all viral miRNAs [13,14]. Clinically, there is little evidence for long-term KSHV infected endothelial cells outside of KS lesions, or prior to KS disease manifestation. In infected endothelial lineage culture systems, KSHV is rapidly lost unless the latently infected cells undergo additional transforming events [5,6,15-17], or the virus is selected for [18]. This suggests that infection of endothelial lineage cells with KSHV places severe selection on the host cell, leading either to viral replication, viral clearance, or persistence in a transformed cell.
The many manifestations of Kaposi Sarcoma
KS (Figure 1) manifests itself in multiple forms: (i) classic KS, which is a rare disease of the elderly, (ii) endemic KS, which is a frequent disease of children in sub-Saharan Africa and was documented before the introduction of HIV, (iii) AIDS-associated KS, which marks the terminal stages of HIV infection, and (iv) iatrogenic KS, which develops in solid organ transplant recipients (reviewed in [3]). Endemic, HIV-negative KS is seen both in children and in adults with elevated incidence. In the absence of reliable pathology and a reliable clinical reporting structure, it is unclear whether the KS in these two age groups represent different diseases, whether there is a genetic predisposition that is associated with pediatric KS, or whether there exists age and environment specific environmental factors. KS incidence is also elevated in isolated parts of China [19]. AIDS-associated KS presents in multiple clinical situations as well, which may represent multiple variations in KSHV pathogenesis. Prior to the introduction of combination antiretroviral therapy (cART), AIDS KS was negatively correlated with CD4 counts. The majority of AIDS KS continues to present in persons with low CD4 counts and indicates cART failure. However, 1/3 of KS now presents in HIV+ individuals on successful long-term cART, despite undetectable HIV viral load and despite near-normal CD4 counts [20]. Lastly, KS immune reconstitution syndrome has been recognized as a complication of cART initiation, where rapid immune restoration leads to the development of KS lesions [21]. Based on limited data, KS that develops in HIV-suppressed individuals exhibits a more restricted viral gene expression profile [13].
Figure 1.
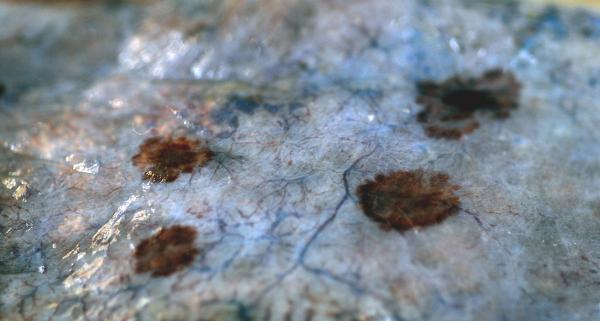
Gross pathology of internal KS lesions on the lung of an AIDS patient (with permission from Vincent J. Moylan).
The molecular drivers of KS are unclear. On the one hand, we do not know of any specific mutation that is associated with KS. On the other hand, we do know the transcriptional profiles of KS and endothelial-based model systems for KS. Profiling studies, together with the analyses of individual proteins, confirm that the tissue of origin for KS is an endothelial cell. More detailed lineage assignments are difficult to make. There exists evidence of trans- or de-differentiation of endothelial cells in response to KSHV infection and/or transformation to KS tumor cells as well as evidence for the expression of markers not exclusively associated with endothelial cells [15,22]. The most exciting recent discovery is that of the ephA2 receptor kinase as a receptor for KSHV [23,24].
KSHV-Inflammatory Cytokine Syndrome (KICS)
Primary KSHV infection does not usually result in a notable clinical phenotype. Reports of KSHV-associated mononucleosis or rash are few and far between. KSHV infection of immune competent primates mirrors the paucity of phenotypes seen in human infection. Recently, however, a new clinical entity has been proposed that describes a severe systemic infection and/or reactivation with KSHV: KSHV-Inflammatory Cytokine Syndrome (KICS) [21,25,26]. It is associated with high-level viremia, KS, and a severe cytokine storm in the absence of MCD in HIV+ patients. Whether this rare disease is the result of a viral variant or a rare genetic predisposition, analogous to X-linked predispositions to chronic viral infections, is presently unresolved. The most likely driver of the clinical phenotype seems to be the production of KSHV vIL6 and human IL6 induction by KSHV.
KSHV in B cells
KSHV establishes latency in B cells. Latency in B cells can be asymptomatic and is mostly likely restricted to a sub-set of B cells, which unlike EBV+ CD27+ memory B cells do not circulate in large numbers in peripheral blood [27]. KSHV seems to have a predilection for infecting and establishing long-term latency in only lambda expressing B cells [27,28]. KSHV infection drives primary B cells to proliferation[27]. Infection with the related MHV68 virus also induced B cell hyperplasia and resulted in fully transformed B cells [29]. Individual KSHV latent genes induce B cell proliferative phenotypes in transgenic mice. LANA transgenic mice developed splenic follicular hyperplasia, which is dependent on B cell receptor (BCR) signaling [30]. vFLIP transgenic mice develop either lymphoma or histiocydic sarcoma [31]. KSHV vIL6 transgenic mice develop hyperplasia reminiscent of MCD [32]. The viral cyclin (vCyc) gene integrates different properties of the host cyclins to foster a pro-growth and pro-virus environment [33]. Individual KSHV miRNAs also have transforming properties in in vivo B cell models, particularly the KSHV mir-155 ortholog [33,34]. Viral genes from the related MHV68 virus display similar phenotypes (reviewed in [35]). B cell specific expression of multiple viral latent KSHV proteins as well as the viral miRNAs leads to sustained hyperplasia, lymphoma, and hyper-responsiveness to antigen stimulation [36].
A putative model of KSHV B cell pathogenesis would start with the primary infection event, which drives the infected cell into an activated state. This holds true even for non-permissively infected cells. In non-permissive cell sub-types, such as IgM+ kappa B cells, or T cells [37], the virus is rapidly lost or the cell dies. In cells that are competent for the establishment of latency, the virus persists and now confers a survival advantage to the infected cell. The molecular nature of this advantage, and the exact complement of viral genes that confer this advantage are not well defined. It may very well be that genes, other than those consistently detected in KSHV tumor cells may contribute to this phenotype. This model would be analogous perhaps to the establishment of EBV latency, which starts out with expressing a more extensive set of genes (latency type II) and then contracts to latency type I or even type 0.
MCD and PEL
Once established in B cells, KSHV can lead to multiple disease manifestations, as additional mutations accumulate in the host genome over time. These B lymphoproliferative diseases include primary effusion lymphoma (PEL) and the plasmablastic variant of multicentric Castleman's disease (MCD) (reviewed in [38]) and in rare instances of diffuse large B cell lymphoma not presenting as effusions. There is also the titillating observation that KSHV increases the risk of marginal zone lymphoma (MZ) [39], and the ever present, but controversial speculation that KSHV can be found in, contribute to, or survive better in multiple myeloma cells.
MCD is considered a lymphoproliferative disorder rather than a frank lymphoma. The KSHV infected plasmablasts invariably express IgM lambda. KSHV latent genes, as well as the viral IL6 homolog are expressed in a large fraction of MCD cells (reviewed in [3,38]). Unlike PEL, MCD cells do not express the CD138/syndecan-1 marker, and also do not show hyper mutation of the IgG locus, which would argue that these cells did not participate in the canonical germinal center reaction. The viral IL6 protein, as well as elevated human IL6, seems to drive much of the clinical symptomology of MCD, which provides a novel rational target for intervention, perhaps using IL6 neutralizing antibodies [40].
PEL is a diffuse large B cell lymphoma of post germinal center origin [3,38]. All PEL cells express CD138/syndecan-1, the KSHV LANA protein, the viral miRNAs and other latent genes (vCYC, vFLIP, kaposin, vIRF3/LANA-2) [41,42]. A more detailed review about KSHV gene expression is provided elsewhere in this issue. PEL cells contain many KSHV episomes, ranging from 40 to 80 copies per cell. 50% of PEL are co-infected with EBV and the EBV positive PEL exhibit a different pattern of gene expression compared to EBV-negative PEL. At present there is no evidence that EBV-positive PEL have a different clinical outcome, respond differently to treatment, or behave differently in experimental models of tumor growth. In addition to KSHV, PEL have sustained multiple genomic alterations as ascertained by CGH [43];Luan, 2010 #53}, though the total number of cases has been too small to identify a single host cell driver mutation with a high degree of statistical significance.
The relatively consistent growth of PEL cell lines in culture and the fact that many PEL can be induced to release infectious KSHV have made them the workhorse for molecular studies in KSHV pathogenesis. There is no evidence that the virus trapped in PEL cells is defective (though the small number of cell lines which can be propagated in culture means that our knowledge about PEL biology may be heavily biased). PEL represent the extreme end of KSHV pathogenesis: a fully transformed cell, which maintains the KSHV genome in high copy numbers, has acquired additional mutations, and which easily establishes tumors in immune deficient mice (regardless of site of injection). Many PEL cell lines were derived after, and thus selected to survive, multiple rounds of chemotherapy. Importantly, all PEL cell lines depend on the presence of KSHV and the continued expression of KSHV genes for survival (reviewed in [3,6,38]).
Modulation of the innate immune response by KSHV
Similar to other herpesviruses, KSHV has to escape the host immune response during initial infection, during sustained latency, and during reactivation. The clinical experience is that (i) KSHV successfully establishes latency and thus overcomes the initial host barriers to infection, and (ii) that KS lesions spontaneously regress after immune reconstitution. MCD also wanes with immune reconstitution and MCD symptoms have a cyclical pattern. PEL never regresses but grows relentlessly in an immune compromised host. The survival time of PEL in the absence of cytotoxic (and thus immune suppressive) therapy is less than the typical time of immune reconstitution after cART initiation.
The host uses two levels of defense to counter microbial infection; the innate immune system (initial recognition of the incoming pathogen) and the adaptive immune system (systemic recognition and memory response to the pathogen). The innate immune response initiates the adaptive response, which may eventually lead to pathogen clearance. KSHV has evolved multiple molecular mechanisms to evade host immunity. These are described elsewhere in this volume. Here we focus on one aspect to the virus-host interaction, which is the interface of KSHV with the innate immune system.
Innate immunity is mediated by pattern recognition receptors (PRRs), which include the Toll-like receptors (TLRs), RIG-I like receptors (RLRs) and nucleotide-binding oligomerization domain, leucine rich repeat receptors (NLRs). TLRs, NLRs and RLRs differ in localization, regulation, stimulus, and function. Most TLRs are found on the cell surface and within endosomes, whereas the RLRs and NLRs are found within the cytosol. TLRs recognize and bind to pathogen associated molecular patterns on the incoming pathogen. In particular, TLR3, 7, 8 and 9 are all expressed in the endosome (reviewed in [44]). TLR3 recognizes double stranded RNA, while TLR7/8 can recognize single stranded RNA. Both ligands (ssRNA and dsRNA) are common intermediates in virus replication. TLR9 activation is induced upon exposure to CpG DNA in response to DNA viral infection [44]. TLR stimulation universally results in the upregulation of a large number of cytokines and chemokines, most notably type 1 interferon (IFN).
RIG-I like receptors (RLRs) include RIG-I, MDA-5 and LGP2, which are cytosolic proteins capable of sensing viral infection by binding double stranded and 5’-triphosphate RNAs. RNA-bound RIG-I becomes activated and signals through mitochondrial antiviral signaling protein (MAVS), a central node in intracellular PRR transduction pathways (reviewed in [45]).
The nucleotide-binding oligomerization domain receptor (NLR) family of innate immune sensors has been implicated in many human diseases (reviewed in [46]). NLRs sense pathogen associated molecular patterns (PAMPs), danger associated molecular patterns (DAMPs), as well as environmental irritants. Some NLR family members regulate innate immunity through the formation of large molecular complexes called inflammasomes, which lead to the upregulation of IL-1β and IL-18. However, other NLRs can inhibit NF-kB and type I IFN signaling, suggesting that this pathway has evolved mechanisms to regulate itself. Inflammasomes generally consist of oligomerization of a particular NLR, recruitment of procaspase-1 and apoptotic-associated speck-like protein (ASC). To date, several NLR family members have been shown to form inflammasomes including NLRP1, NLRP3, and NLRC4. KSHV Orf63 was shown to inhibit NLRP1- and NLRP-3 inflammasome activity and prevent the processing and activation of pro-IL-1β and pro-IL-18 to active IL-1β and IL-18 [47]
There also exist non-NLR containing inflammasomes, and KSHV has been to shown to activate interferon-inducible factor 16 (IFI16) to induce the inflammasome in KSHV-infected endothelial cells [48,49].
Primary KSHV infection of human monocytes resulted in increased levels of TLR3 and IFN-β production as well as several inflammatory cytokines [50]. In plasmacytoid dendritic cells (pDCs), TLR9 has been reported to sense KSHV leading to increased IFN-α production [51]. Furthermore, TLRs also play an important role during reactivation of KSHV from latency [52] and TLR7/8 activation led to reactivation of KSHV from latency in infected B cells. In endothelial cells, another cell type that harbors KSHV, KSHV infection resulted in the downregulation of TLR4 [53].
KSHV encodes many immune evasion genes that block the IFN response. KSHV encodes four different viral interferon regulatory factors (vIRFs) that display limited homology to cellular IRFs. The vIRFs can block the function of cellular IRFs and inhibit their transactivation of IFN-α and IFN-β (reviewed in [54]). Another KSHV viral protein, Orf45, inhibits type I interferon expression by blocking IRF7 phosphorylation and its nuclear accumulation, while ubiquitination and degradation of IRF7 and the TLR3 adaptor, TRIF, is mediated by the KSHV Rta/Orf50 protein [55-57].
Conclusions
Our insights into KSHV have significantly advanced in the recent years. A new disease entity (KICS) has been identified as being associated with acute KSHV replication. Previously recognized KSHV diseases, such as KS, continue to lead the global cancer burden in people living with HIV/AIDS. Additionally, recent advances have also been made in our understanding of how KSHV modulates autophagy, host metabolism, differentiation and innate immune responses. New tools, such as new animal model systems are on the horizon, which will allow us to investigate the life cycle of KSHV and of viral pathogenesis prior to the development of disease phenotypes. At the same time, almost 20 years after the discovery of KSHV, simple cytotoxic chemotherapy regimens remain the only clinically validated treatments. Currently, there exists no vaccine against this virus and, with the exception perhaps of mTOR inhibitors [17,58-60], no targeted agents have been added to clinical practice.
Highlights.
We review recent advances in KSHV pathogenesis
We review viral genes and microRNAs involved in KSHV pathogenesis
We review clinical manifestations of KSHV-associated diseases
Figure 2.

Different histological examples of KS. Shown is immunohistochemistry for the KSHV LANA protein in brown (panels a to d) and Hematoxilin & Eosin (H&E) stain of lesions (panel f. – i.) . Panels e and j represent non-KS control. The samples were part of the first KS tissue microarray made available through the AIDS Cancer Specimen Resource (ACSR). Magnification 40x.
Acknowledgements
BD and DD are supported by NIH grants CA019014 and CA163217 to BD and DD, and CA096500 to BD. We thank Vincent J. Moylan for providing the gross pathology image. We acknowledge the AIDS Cancer and Specimen Resource (ACSR) for the KS tissue microarray. We sincerely apologize to our colleagues who we could not cite in this article, as references were restricted to only those published in the last five years.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. Journal of virology. 2010;84:695–703. doi: 10.1128/JVI.02013-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ray A, Marshall V, Uldrick T, Leighty R, Labo N, Wyvill K, Aleman K, Polizzotto MN, Little RF, Yarchoan R, et al. Sequence analysis of Kaposi sarcoma-associated herpesvirus (KSHV) microRNAs in patients with multicentric Castleman disease and KSHV-associated inflammatory cytokine syndrome. The Journal of infectious diseases. 2012;205:1665–1676. doi: 10.1093/infdis/jis249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damania B, Cesarman E. Kaposi's Sarcoma–Associated Herpesvirus, Chapter 65. Fields Virology, Lippincott Williams & Wilkins. 2013:2080–2128. [Google Scholar]
- 4.Chandran B. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. Journal of virology. 2010;84:2188–2199. doi: 10.1128/JVI.01334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mesri EA, Cesarman E, Boshoff C. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatt AP, Damania B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front Immunol. 2012;3:401. doi: 10.3389/fimmu.2012.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7**.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [This is the first report of vFLIP modulating autophagosome biogenesis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8**.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12:193–199. doi: 10.1038/ncb2019. [This study describes an important role for KSHV miRNAs in regulating viral latency and lytic replication.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell host & microbe. 2012;11:167–180. doi: 10.1016/j.chom.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 10**.Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, Damania B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11818–11823. doi: 10.1073/pnas.1205995109. [This is the first report of KSHV-infected lymphoma cells displaying upregulated fatty acid synthesis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Delgado T, Carroll PA, Punjabi AS, Margineantu D, Hockenbery DM, Lagunoff M. Induction of the Warburg effect by Kaposi's sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10696–10701. doi: 10.1073/pnas.1004882107. [This is the first report of KSHV upregulating glycolysis in infected cells.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delgado T, Sanchez EL, Camarda R, Lagunoff M. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS pathogens. 2012;8:e1002866. doi: 10.1371/journal.ppat.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dittmer DP. Restricted Kaposi's sarcoma (KS) herpesvirus transcription in KS lesions from patients on successful antiretroviral therapy. mBio. 2011;2:e00138–00111. doi: 10.1128/mBio.00138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Hara AJ, Chugh P, Wang L, Netto EM, Luz E, Harrington WJ, Dezube BJ, Damania B, Dittmer DP. Pre-micro RNA signatures delineate stages of endothelial cell transformation in Kaposi sarcoma. PLoS pathogens. 2009;5:e1000389. doi: 10.1371/journal.ppat.1000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15**.Cheng F, Pekkonen P, Laurinavicius S, Sugiyama N, Henderson S, Gunther T, Rantanen V, Kaivanto E, Aavikko M, Sarek G, et al. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell host & microbe. 2011;10:577–590. doi: 10.1016/j.chom.2011.10.011. [This paper shows that KSHV induces endothelial-to-mesenchymal transition.] [DOI] [PubMed] [Google Scholar]
- 16.Jones T, Ye F, Bedolla R, Huang Y, Meng J, Qian L, Pan H, Zhou F, Moody R, Wagner B, et al. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. The Journal of clinical investigation. 2012;122:1076–1081. doi: 10.1172/JCI58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy D, Sin SH, Lucas AS, Venkataramanan R, Wang L, Eason A, Chavakula V, Tamburro KM, Hilton IB, Damania B, et al. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer research. 2013 doi: 10.1158/0008-5472.CAN-12-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Damania B. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008;68:4640–4648. doi: 10.1158/0008-5472.CAN-07-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu B, Sun F, Li B, Yang L, Zeng Y, Sun X, Xu F, Rayner S, Guadalupe M, Gao SJ, et al. Seroprevalence of Kaposi's sarcoma-associated herpesvirus and risk factors in Xinjiang, China. Journal of medical virology. 2009;81:1422–1431. doi: 10.1002/jmv.21550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krown SE, Lee JY, Dittmer DP. More on HIV-associated Kaposi's sarcoma. The New England journal of medicine. 2008;358:535–536. doi: 10.1056/NEJMc072994. author reply 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polizzotto MN, Uldrick TS, Hu D, Yarchoan R. Clinical Manifestations of Kaposi Sarcoma Herpesvirus Lytic Activation: Multicentric Castleman Disease (KSHV-MCD) and the KSHV Inflammatory Cytokine Syndrome. Front Microbiol. 2012;3:73. doi: 10.3389/fmicb.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scehnet JS, Ley EJ, Krasnoperov V, Liu R, Manchanda PK, Sjoberg E, Kostecke AP, Gupta S, Kumar SR, Gill PS. The role of Ephs, Ephrins, and growth factors in Kaposi sarcoma and implications of EphrinB2 blockade. Blood. 2009;113:254–263. doi: 10.1182/blood-2008-02-140020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23**.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nature medicine. 2012;18:961–966. doi: 10.1038/nm.2805. [This paper identified a new cellular receptor, EphrinA2, for KSHV.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24**.Chakraborty S, Veettil MV, Bottero V, Chandran B. Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1163–1172. doi: 10.1073/pnas.1119592109. [This paper shows that EphrinA2 is important for KSHV entry.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamburro KM, Yang D, Poisson J, Fedoriw Y, Roy D, Lucas A, Sin SH, Malouf N, Moylan V, Damania B, et al. Vironome of Kaposi sarcoma associated herpesvirus-inflammatory cytokine syndrome in an AIDS patient reveals co-infection of human herpesvirus 8 and human herpesvirus 6A. Virology. 2012;433:220–225. doi: 10.1016/j.virol.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uldrick TS, Wang V, O'Mahony D, Aleman K, Wyvill KM, Marshall V, Steinberg SM, Pittaluga S, Maric I, Whitby D, et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2010;51:350–358. doi: 10.1086/654798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hassman LM, Ellison TJ, Kedes DH. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. The Journal of clinical investigation. 2011;121:752–768. doi: 10.1172/JCI44185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chadburn A, Hyjek EM, Tam W, Liu Y, Rengifo T, Cesarman E, Knowles DM. Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV; HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology. 2008;53:513–524. doi: 10.1111/j.1365-2559.2008.03144.x. [DOI] [PubMed] [Google Scholar]
- 29.Liang X, Paden CR, Morales FM, Powers RP, Jacob J, Speck SH. Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLoS pathogens. 2011;7:e1002220. doi: 10.1371/journal.ppat.1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sin SH, Fakhari FD, Dittmer DP. The viral latency-associated nuclear antigen augments the B-cell response to antigen in vivo. Journal of virology. 2010;84:10653–10660. doi: 10.1128/JVI.00848-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballon G, Chen K, Perez R, Tam W, Cesarman E. Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice. The Journal of clinical investigation. 2011;121:1141–1153. doi: 10.1172/JCI44417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suthaus J, Stuhlmann-Laeisz C, Tompkins VS, Rosean TR, Klapper W, Tosato G, Janz S, Scheller J, Rose-John S. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood. 2012;119:5173–5181. doi: 10.1182/blood-2011-09-377705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee KS, Suarez AL, Claypool DJ, Armstrong TK, Buckingham EM, van Dyk LF. Viral cyclins mediate separate phases of infection by integrating functions of distinct mammalian cyclins. PLoS pathogens. 2012;8:e1002496. doi: 10.1371/journal.ppat.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boss IW, Nadeau PE, Abbott JR, Yang Y, Mergia A, Renne R. A Kaposi's sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. Journal of virology. 2011;85:9877–9886. doi: 10.1128/JVI.05558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol. 2011;29:351–397. doi: 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- 36.Sin SH, Dittmer DP. Viral latency locus augments B cell response in vivo to induce chronic marginal zone enlargement, plasma cell hyperplasia and lymphoma. Blood. 2013 doi: 10.1182/blood-2012-03-415620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myoung J, Ganem D. Infection of primary human tonsillar lymphoid cells by KSHV reveals frequent but abortive infection of T cells. Virology. 2011;413:1–11. doi: 10.1016/j.virol.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cesarman E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011;305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benavente Y, Mbisa G, Labo N, Casabonne D, Becker N, Maynadie M, Foretova L, Cocco PL, Nieters A, Staines A, et al. Antibodies against lytic and latent Kaposi's sarcoma-associated herpes virus antigens and lymphoma in the European EpiLymph case-control study. British journal of cancer. 2011;105:1768–1771. doi: 10.1038/bjc.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riva G, Luppi M, Barozzi P, Forghieri F, Potenza L. How I treat HHV8/KSHV-related diseases in posttransplant patients. Blood. 2012;120:4150–4159. doi: 10.1182/blood-2012-04-421412. [DOI] [PubMed] [Google Scholar]
- 41**.Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R. Ago HITS-CLIP expands understanding of Kaposi's sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS pathogens. 2012;8:e1002884. doi: 10.1371/journal.ppat.1002884. [This paper highlights both cellular and viral targets of KSHV miRNAs.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell host & microbe. 2011;10:515–526. doi: 10.1016/j.chom.2011.09.012. [This paper highlights both cellular and viral targets of KSHV miRNAs.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy D, Sin SH, Damania B, Dittmer DP. Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines. Blood. 2011;118:e32–39. doi: 10.1182/blood-2010-12-323659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 45.Ireton RC, Gale M., Jr. RIG-I like receptors in antiviral immunity and therapeutic applications. Viruses. 2011;3:906–919. doi: 10.3390/v3060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47**.Gregory SM, Damania B. Inhibition of the inflammasome response by a viral protein that interacts with NLRs. Commun Integr Biol. 2011;4:416–418. doi: 10.4161/cib.4.4.15252. [This paper was the first to report a KSHV viral protein that can inhibit NLR-containing inflammasome function.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48**.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011;9:363–375. doi: 10.1016/j.chom.2011.04.008. [This paper was the first to highlight nuclear IFI16 as a sensor of KSHV infection.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh VV, Kerur N, Bottero V, Dutta S, Chakraborty S, Ansari MA, Paudel N, Chikoti L, Chandran B. Kaposi's Sarcoma-Associated Herpesvirus Latency in Endothelial and B cells Activates Interferon Gamma-Inducible Protein 16 (IFI16) Mediated Inflammasomes. Journal of virology. 2013 doi: 10.1128/JVI.03282-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West J, Damania B. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J Virol. 2008;82:5440–5449. doi: 10.1128/JVI.02590-07. Epub 2008 Mar 5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.West JA, Gregory SM, Sivaramanan V, Su L, Damania B. Activation of Plasmacytoid Dendritic Cells by KSHV. J Virol. 2010 doi: 10.1128/JVI.01007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc Natl Acad Sci U S A. 2009;106:11725–11730. doi: 10.1073/pnas.0905316106. Epub 12009 Jun 11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagos D, Vart RJ, Gratrix F, Westrop SJ, Emuss V, Wong PP, Robey R, Imami N, Bower M, Gotch F, et al. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe. 2008;4:470–483. doi: 10.1016/j.chom.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacobs SR, Damania B. NLRs, inflammasomes, and viral infection. J Leukoc Biol. 2012;92:469–477. doi: 10.1189/jlb.0312132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu Y, Hayward GS. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity. 2010;33:863–877. doi: 10.1016/j.immuni.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, Zhang L. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing beta-interferon (TRIF). J Biol Chem. 2011;286:7865–7872. doi: 10.1074/jbc.M110.191452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu FX, Sathish N, Yuan Y. Antagonism of host antiviral responses by Kaposi's sarcoma-associated herpesvirus tegument protein ORF45. PLoS One. 2010;5:e10573. doi: 10.1371/journal.pone.0010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sin SH, Roy D, Wang L, Staudt MR, Fakhari FD, Patel DD, Henry D, Harrington WJ, Jr., Damania BA, Dittmer DP. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood. 2007;109:2165–2173. doi: 10.1182/blood-2006-06-028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59**.Krown SE, Roy D, Lee JY, Dezube BJ, Reid EG, Venkataramanan R, Han K, Cesarman E, Dittmer DP. Rapamycin with antiretroviral therapy in AIDS-associated Kaposi sarcoma: an AIDS Malignancy Consortium study. J Acquir Immune Defic Syndr. 2012;59:447–454. doi: 10.1097/QAI.0b013e31823e7884. [This paper reports the first clinical trial of rapamycin in AIDS-associated Kaposi sarcoma.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhatt AP, Bhende PM, Sin SH, Roy D, Dittmer DP, Damania B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood. 2010;115:4455–4463. doi: 10.1182/blood-2009-10-251082. [DOI] [PMC free article] [PubMed] [Google Scholar]