

Abstract
Exposure to sparsely ionising gamma- or X-ray irradiation is known to increase the risk of leukaemia in humans. However, heavy ion radiotherapy and extended space exploration will expose humans to densely ionising high linear energy transfer (LET) radiation for which there is currently no understanding of leukaemia risk. Murine models have implicated chromosomal deletion that includes the hematopoietic transcription factor gene, PU.1 (Sfpi1), and point mutation of the second PU.1 allele as the primary cause of low-LET radiation-induced murine acute myeloid leukaemia (rAML). Using array comparative genomic hybridisation, fluorescence in situ hybridisation and high resolution melt analysis, we have confirmed that biallelic PU.1 mutations are common in low-LET rAML, occurring in 88% of samples. Biallelic PU.1 mutations were also detected in the majority of high-LET rAML samples. Microsatellite instability was identified in 42% of all rAML samples, and 89% of samples carried increased microsatellite mutant frequencies at the single-cell level, indicative of ongoing instability. Instability was also observed cytogenetically as a 2-fold increase in chromatid-type aberrations. These data highlight the similarities in molecular characteristics of high-LET and low-LET rAML and confirm the presence of ongoing chromosomal and microsatellite instability in murine rAML.
Introduction
Radiation exposure is known to cause DNA damage and increase the risk of cancer, including acute myeloid leukaemia (AML). In long-term studies of atomic bomb survivors, leukaemia carried the greatest excess morbidity risk from acute, primarily low-linear energy transfer (LET) radiation exposure (1). Analysis of nuclear industry workers exposed to largely protracted and/or low dose, low-LET radiation also demonstrated increased leukaemia risk, as have a number of patient populations treated with radiotherapy (2–4). Radiation therapy- and chemotherapy-related AML (tAML) presents with a median latency of approximately 5 years and has a poor prognosis compared with de novo AML, with a median survival of only 8 months (5).
Leukaemia risk is unknown for high-LET radiation exposures that occur with extended space flight and newer heavy ion radiation therapies. Lack of human epidemiological data for high-LET radiation necessitates the use of animal models and a comparison of molecular mechanisms to allow extrapolation to humans. CBA strain mice have a low spontaneous AML incidence (<1%), but up to 25% of animals develop AML following gamma- or X-ray irradiation (6). Like its human counterpart, low-LET radiation-induced murine AML is rapidly fatal, and induction is dose dependent. Murine studies of high-LET radiation associated with neutron exposures have demonstrated similar pathology and linear dose dependence up to 3 Gy for AML induction, with a relative biological effectiveness (RBE) of three (7). We reported previously that high-LET 56Fe ion irradiation also induces rAML in CBA mice, with an RBE of approximately one (8).
Previous studies have implicated biallelic PU.1/Sfpi1 mutation as the primary cause of low-LET radiation-induced murine AML. Murine bone marrow cells have an increased incidence of chromosome 2 aberrations within 24 hours of irradiation, and more than 90% of rAMLs have clonal deletions spanning PU.1 on one copy of chromosome 2 (7,9–13). In most low-LET radiation-induced AML samples, the second PU.1 allele carries a somatic point mutation within the DNA-binding domain that likely abrogates binding, suggesting PU.1 as a candidate tumour suppressor gene (14–16). Indeed, conditional homozygous loss or dramatically reduced expression of PU.1 causes AML with at least 95% penetrance (17,18). High-LET associated radiation exposures have also been shown to induce chromosome 2 aberrations, and inactivation of the second PU.1 allele has recently been reported following neutron irradiation (7,9,13,19). The involvement of PU.1 in human AML is controversial since mutations are infrequent (20). However, several papers have reported decreased PU.1 levels in a subset of AML, and two common mutations—FLT3 internal tandem duplications and the AML1:ETO translocation—have been reported to decrease PU.1 expression (21–23). Thus the molecular targets may differ between the species though it appears that a common pathway may be affected.
It has been proposed that cancer progression requires acquisition of a mutator phenotype to account for the high number of mutations seen in tumours, with chromosomal and microsatellite instability (MSI) pathways implicated (24).While fewer mutations may be necessary to drive hematopoietic malignancies than solid tumours, it is none-the-less suspected that genetic instability is involved. Clonal chromosomal aberrations clearly play a critical role in both mouse and human AML, in which recurrent deletions and translocations are common. Less certain is the association between clonal MSI and AML. Findings from different laboratories vary significantly, as do the markers, criteria, methods of analysis and control samples employed. However, the prevailing evidence suggests that MSI is rare in de novo AML, while approximately half of therapy-related AML (including radiotherapy) exhibit some level of MSI (25–33). Studies by Haines et al. (34) and Fennelly et al. (35) have reported MSI in di- and penta-nucleotide repeat motifs in murine rAML as well. It is important to note that the majority of reports assess only clonally expanded mutations and thus provide evidence of an early instability event rather than ongoing instability.
We investigated the molecular characteristics of radiation-induced AML from both low-LET 137Cs gamma ray irradiation and high-LET 56Fe ion exposure. Using these and previously described neutron-induced AML samples, we demonstrate that the majority of high-LET rAML cases show biallelic PU.1 loss through the same combination of chromosome 2 deletion and PU.1 point mutation found in gamma- and X-ray-induced AML. MSI was present in 42% of rAML samples, including iron ion-induced AML, indicating that MSI is a common event in tumour development. Additionally, single-cell analyses of chromosome instability and microsatellite mutant frequency demonstrated statistically significant 2-fold increases in murine rAML. Overall, these data demonstrate that PU.1 is a common target for both high- and low-LET AML and support a role for ongoing instability through multiple pathways in AML.
Materials and methods
AML samples
Male CBA/CaJ mice were obtained from the Jackson Laboratory and irradiated at 8–14 weeks of age with 1 GeV/nucleon 56Fe ions (0.1, 0.2, 0.4 or 1.0 Gy) at the NASA Space Radiation Laboratory, Brookhaven National Laboratory or with 137Cs gamma rays (1.0, 2.0 or 3.0 Gy) at Colorado State University (CSU). After irradiation, the mice were maintained at CSU and monitored for enlarged spleens until moribund or until 800 days of age, then necropsied for histological evaluation and molecular analyses. Diagnosis of AML was based on histopathologic examination of spleen, liver and bone marrow stained with haemotoxylin and eosin using criteria previously published (36). The majority of AMLs were diagnosed as myeloid leukaemia with maturation. By the time AML was detected, spleens were composed predominately of leukaemic cells and were used without further isolation of blasts. Age-matched unirradiated and irradiated normal controls were taken from the same cohort. Mice, irradiation conditions and health monitoring protocols have been described (9). Animal use was approved by the Institutional Animal Care and Use Committees under protocols 03-148A (CSU) and 277 (BNL).
Fluorescence in situ hybridisation and chromosome instability assays
Spleens were minced and incubated overnight at 37°C and 5% CO2 in RPMI 1640 with 20% fetal bovine serum, 100U/ml penicillin, 100mg/ml streptomycin, 0.5 μg/ml fungizone, 2mM glutamine or ×1 Glutamax (Gibco/Life Technologies, Grand Island, NY), and 5 μg/ml Concanavalin A (Sigma–Aldrich Corp., St. Louis, MO). Colcemid was added at a final concentration of 0.1 µg/ml, and cells were further incubated for 3–4 h. Cells were collected by centrifugation, incubated in 0.75M KCl, fixed in 3:1 methanol and acetic acid and dropped directly onto slides. For detection of chromosome 2 deletions, fluorescence in situ hybridisation (FISH) was performed using a PU.1/Sfpi1-containing BAC probe as previously described (9). Images were obtained at room temperature using an Olympus Provis AX70 fluorescence microscope (×100 oil immersion objective) with a SenSys CCD camera (Photometrics, Tuscon, AZ) and processed with MacProbe imaging software (Applied Imaging, Grand Rapids, MI). A minimum of 100 metaphases were scored for each sample except AML 3269 (55 metaphases). For detection of chromosomal, chromatid and telomere aberrations, FISH was performed using a telomere-specific G-rich peptide nucleic acid probe labelled with Cy-3 (DAKO), with 4',6-diamidino-2-phenylindole (DAPI) counterstaining. In brief, slides were denatured in 70% formamide, ×2 SSC at 75°C. The probe was hybridised at 37°C for 2 hours in the dark, then slides were washed in 70% formamide, ×2 SSC for 15–20 min at 32°C in a shaking water bath and mounted with Vectashield plus DAPI (Vector Laboratories, Burlingame, CA). Slides were viewed at room temperature using an Axioplan 2ie MOT Fluorescence microscope (Carl Zeiss, Oberkochen, Germany) with ×100 oil objective. Images were obtained with a CV-m4+CL CCD camera (JAI Inc., San Jose, CA), running ISIS Fluorescence Imaging System software (MetaSystems, Altlussheim, Germany). All scorable metaphases were analysed for telomere instability events (telomere associations, duplications, deletions, double minutes and interstitial telomere signals), chromosome instability events (interstitial deletions, terminal deletions, dicentrics, Robertsonian translocations and fragments) and chromatid instability events (gaps, breaks, iso-deletions, chromatid fusions and chromatid exchanges). A minimum of 10 metaphases were scored per sample (median 33, range 10–81). Aberration frequencies were expressed for each sample as the total number of aberrations divided by the total number of metaphases analysed, and significant differences between AML and control groups were determined using the Mann–Whitney rank sum test.
Array comparative genomic hybridization
DNA quality and concentration were determined on a NanoDrop spectrophotometer. Labelling reactions were prepared using the Roche NimbleGen Labeling Protocol for array comparative genomic hybridisation (aCGH) (Version 7.0) with 1 µg total DNA input, according to manufacturer' s instructions. In brief, 1 µg of test DNA and reference DNA were labelled with Cyanine 5- and Cyanine 3-Random Nonamers, respectively. Labelled DNA was purified by isopropanol precipitation and labelling efficiency determined on the NanoDrop spectrophotometer. Next, 31 µg of labelled DNA was hybridised to a mouse 720K whole genome array (Roche NimbleGen Inc., Madison, WI) at 42°C for ~72 hours using a Maui hybridisation system. Arrays were washed and scanned at 2 µM resolution on a NimbleGen MS 200 High Resolution Scanner. Data were processed through NimbleScan v.2.6 using the mouse 100718_MM9_WG_CGH design file. The processed data were used to determine chromosome regions that have DNA copy number gains or losses using NEXUS 6.0 software (BioDiscovery Inc., El Segundo, CA). The SNP-FASST2 algorithm was used for segmentation with a significance threshold of 1 × 10–7, maximum contiguous probe spacing of 1000bp and three probes per segment minimum. The copy number gain threshold was 0.1 and copy number loss threshold was −0.09. The data set is available through the NCBI GEO database (#GSE37233).
High resolution melt analysis and sequencing
Genomic DNA was extracted from spleens of AML bearing mice or bone marrow cells of unaffected mice using the DNeasy Tissue and Blood Kit (Qiagen, Hilden, Germany) per manufacturer' s instructions. For high resolution melt analysis, a 73bp fragment including PU.1 R235 was amplified from 2.5ng spleen DNA with GoTaq® qPCR MasterMix (Promega Corp., Madison, WI) and 250nM primers (5'-GCAACCGCAAGAAGATGACCT-3' and 5'-TTCACCTCGCCTGTCTTGCC-3'). Amplification and melt were performed in duplicate on a CFX96 Real-Time Detection System (Bio-Rad) with the following conditions: 95°C (2 min); 40 cycles of 95°C (10 s), 60°C (1min); melt at 0.1°C intervals. Precision Melt Analysis Software (Bio-Rad, Hercules, CA) was used to cluster melt profiles with default settings and a minimum 95% confidence score. Mouse Genomic DNA (Promega) and rAML cell line 8016 carrying a PU.1 R235C mutation were used for wildtype and mutant controls, respectively (37). For sequencing, a portion of exon 5 was amplified from 4ng DNA with GoTaq® Hot Start Colorless Master Mix (Promega) and 250nM primers (CTACCATGGCTGGGAGGAG, CTTGGAGCGGGCTAGAGTC). Products were purified and sequenced using nested primers (GGGTCCCCATAAAATCCTTG, GGAATGTCCTCCCTGTGTCC). Bone marrow DNAs for bisulfite sequencing were treated with the EpiTect Bisulfite kit (Qiagen) as per manufacturer’s instructions. The DNA sequence of interest was amplified (GGTGGGTGGATAAGGATAAAGGT, CCCACAACCCARCACCTCACCACTAAACT), gel purified and sequenced with the forward primer.
MSI and small-pool PCR
All murine polyadenine microsatellites reported to be 50 bases or longer in the NCBI mouse genome were screened for length in CBA/CaJ mice. Fluorescently labelled primers for six of the longest amplifiable loci were multiplexed: mBAT-56a (GTGTGTATGCTGATTTTATATCCT, GCAAAAATATCATTCGGTATG), mBAT-56h (GCCAAACTAAAGTTCATCTGATA, GAAAATTTTCCAACTA CATGACTC), mBAT-57 (TGAAATCCTTGATGTTCCTCTACTAGGT, GGT CATCCTGTTGTTTCTAAATGATTGT), mBAT-58 (CCCCTAAAACTTTC CTTGCTATT, ATTTTCTGAGTTCCAGGGCAGTCTG), mBAT-59 (GGTCT TGCCCTGAGGCAAGTAAT, AACCATCCGTAACAAGATCTGACGT) and mBAT-66 (TGGGGTGTCTAAAGACAGCTAAG, GCCCACTTCATGCGTAA CAG). The same loci were tested for MSI by traditional PCR and, to measure mutant frequency at the single-cell level, by small-pool PCR (SP-PCR). For detection of MSI, 1ng spleen DNA was amplified in duplicate with ×1 CBA Multiplex, ×1 Gold ST*R Buffer (Promega) and 0.2U GoTaq Hot Start Polymerase (Promega) in a GeneAmp PCR System 9700 Thermocycler (Applied Biosystems, Foster City, CA) with the following amplification conditions: 95°C (2 min); 10 cycles of 94°C (30 s), 58°C (30 s, 29% ramp), 65°C (1 min, 28% ramp); 20 cycles of 90°C (30 s), 58°C (30 s, 29% ramp), 65°C (1 min, 28% ramp); 60°C (30 min). For analysis of mutant frequency using SP-PCR, DNA was diluted to approximately 7 pg (1 genome equivalent) per reaction in a minimum of 352 replicate reactions and amplified as for standard PCR above, with an increase from 20 to 27 cycles in the third amplification step. Products were denatured in deionised formamide with Internal Lane Standard 600 (Promega) for allele sizing and analyzed on a 3130xl Genetic Analyzer using GeneMapper 4.0 Software (Applied Biosystems) and internally developed software for automated statistical analysis. For MSI analysis, samples with one unstable locus were considered MSI-low, while samples with two or more unstable loci were considered MSI-high (≥30% of the six loci tested).
Calculation of SP-PCR mutant frequencies and statistics
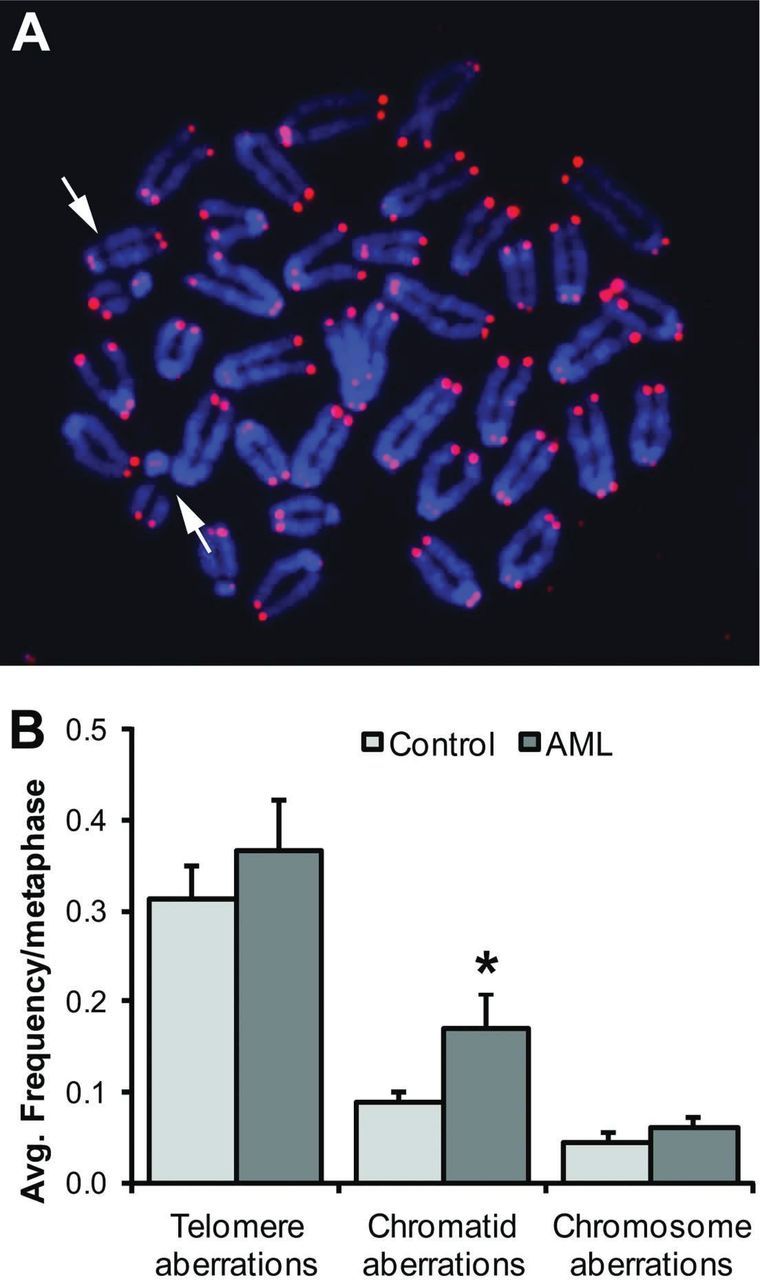
The average number of amplifiable alleles per well of SP-PCR, λ, was estimated for each locus based on the percentage of replicate wells that contained no amplifiable alleles, D. A Poisson distribution estimates λ as –ln(D). A maximum of λ = 2 (one genomic equivalent) was allowed to enable detection of rare mutant alleles that are more than 1.5bp from the average wildtype size. All clonal mutants detected by MSI PCR were excluded such that mutant frequency represents the likelihood of mutation other than the clonal MSI event. A bootstrap estimate of the mutant frequency and standard error for each locus was determined using the SP-PCR Program, Version 1.0 (Barry W. Brown, https://biostatistics.mdanderson.org/SoftwareDownload/). Mutant frequencies given in Table I are the means of six loci with a standard error of ±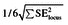 (for individual animals) or the standard error of the mean (for control groups). Mutant frequencies for all age-matched control animals were normally distributed, so the control mean mutant frequency plus 2 SD was used to establish a threshold for comparison of AML samples.
(for individual animals) or the standard error of the mean (for control groups). Mutant frequencies for all age-matched control animals were normally distributed, so the control mean mutant frequency plus 2 SD was used to establish a threshold for comparison of AML samples.
Table I.
Molecular characterisation of rAML induced by 1 GeV/n 56Fe ion or 137Cs gamma ray irradiation
| Animal # | Radiation quality | Dose, Gy | Latency, days | PU.1 deletion | PU.1 R235 mutation | Chromatid aberrationsb | MSI, loci unstable (%) | Mutant frequency | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FISH (%)a | aCGH | HRM | Sequence | MF ± SE | Increasedc | ||||||
| 2704 | 56Fe | 0.2 | 458 | nd | Normal | WT | Normal | nd | 0 | 0.024±0.003 | No |
| 4228 | 56Fe | 0.4 | 653 | 98 | Deleted | Mut | C > A | 0.23 (39) | 17 | 0.073±0.005 | Yes |
| 2816 | 56Fe | 1.0 | 452 | 85 | Deleted | Mut | C > A | 0.03 (30) | 17 | 0.051±0.004 | Yes |
| 3416 | Gamma | 1.0 | 481 | 91 | Deleted | Mut | C > A | 0.10 (49) | 17 | 0.043±0.004 | Yes |
| 3777 | Gamma | 1.0 | 636 | 89 | Deleted | nd | nd | nd | nd | nd | nd |
| 3861 | Gamma | 1.0 | 643 | 97 | Deleted | nd | nd | nd | nd | nd | nd |
| 3901 | Gamma | 1.0 | 696 | 91 | nd | nd | nd | nd | nd | nd | nd |
| 3029 | Gamma | 2.0 | 454 | 95 | nd | Mut | C > T | 0.21 (97) | 0 | 0.076±0.006 | Yes |
| 3208 | Gamma | 2.0 | 454 | 92 | Deleted | Mut | C > T | 0.05 (64) | 17 | 0.056±0.004 | Yes |
| 3229 | Gamma | 2.0 | 130 | nd | nd | WT | Normal | nd | 33 | 0.060±0.005 | Yes |
| 3250 | Gamma | 2.0 | 693 | 97 | Deletede | Mut | C > T | 0.03 (30) | 0 | 0.080±0.005 | Yes |
| 3321 | Gamma | 2.0 | 474 | 87 | nd | Mut | C > T | 0.00 (22) | 0 | 0.074±0.006 | Yes |
| 3364 | Gamma | 2.0 | 553 | 94 | Deleted | Mut | C > T | 0.19 (47) | 0 | 0.067±0.005 | Yes |
| 3576 | Gamma | 2.0 | 547 | 96 | nd | Mut | C > T | 0.24 (29) | 0 | 0.062±0.005 | Yes |
| 3999 | Gamma | 2.0 | 182 | 91 | Deleted | Mut | C > T | 0.14 (36) | 0 | 0.121±0.007 | Yes |
| 5342 | Gamma | 2.0 | 610 | 81 | Deleted | nd | nd | nd | nd | nd | nd |
| 3048 | Gamma | 3.0 | 391 | 4 | Deletede | Mut | C > T | 0.07 (56) | 0 | 0.032±0.004 | No |
| 3173 | Gamma | 3.0 | 740 | 94 | Deleted | nd | nd | nd | nd | nd | nd |
| 3189 | Gamma | 3.0 | 707 | 94 | Deleted | Mut | C > T | 0.19 (100) | 0 | 0.057±0.003 | Yes |
| 3192 | Gamma | 3.0 | 137 | nd | Deleted | Mut | C > T | nd | 17 | 0.061±0.005 | Yes |
| 3269 | Gamma | 3.0 | 290 | 89 | Deleted | Mut | C > T | 0.19 (47) | 0 | 0.146±0.008 | Yes |
| 3272 | Gamma | 3.0 | 546 | 93 | Deleted | Mut | C > T | 0.23 (22) | 0 | 0.085±0.005 | Yes |
| 3286 | Gamma | 3.0 | 464 | 98 | Deleted | Mut | C > T | 0.65 (40) | 17 | 0.055±0.005 | Yes |
| 3512 | Gamma | 3.0 | 288 | 97 | Deleted | WT | Normal | 0.17 (24) | 17 | 0.060±0.005 | Yes |
| Controlsd | 56Fe | WT | 0.08 (359) | 0 | 0.029±0.003 | ||||||
| Controlsd | Gamma | WT | 0.11 (171) | 0 | 0.031±0.005 | ||||||
| Controlsd | Sham | WT | 0.09 (130) | 0 | 0.031±0.002 | ||||||
HRM, high resolution melt; nd, not determined; Mut, mutant; WT, wildtype; MF, mutation frequency.
aPercent of metaphases with hemizygous deletion.
bFrequency of chromatid aberrations per metaphase (# of metaphases).
cMutant frequencies greater than the mean of all age-matched controls + 2 SD (0.0426) are marked ‘yes’.
dControls are the average ± SE of the group. Different animals were used for chromatid aberrations (iron, n = 10; gamma, n = 5; sham, n = 4) and mutant frequency (iron, n = 6; gamma, n = 4; sham, n = 5).
eDeletions are discontinuous for AML 3250 and small (<130kb) for AML 3048.
Results
Animals from the radiation leukaemogenesis-sensitive CBA mouse strain were exposed to acute doses of 1 GeV/n 56Fe ion radiation or 137Cs gamma rays to induce AML. As was previously reported, 1 Gy of iron ions and gamma rays resulted in AML incidences of 2.08±0.01% and 2.31±0.12%, respectively (8). The RBE of iron ion leukaemogenesis was approximately one. Tissues were collected for molecular analysis from 21 gamma ray- and 3 iron ion-irradiated mice with AML, as well as from unaffected, irradiated and sham-treated animals from the same cohort (Table I). There was no significant difference in histopathologic characteristics or tumour latency (median 458 days versus 481 days) between gamma ray- and iron ion-induced AML.
It has been demonstrated that radiation-induced AML in mice is correlated with deletion of PU.1/Sfpi1 on chromosome 2 (7,12,13). To identify PU.1 deletions in this study, we performed FISH analysis and aCGH using leukaemic cells from the spleens of affected mice. PU.1 deletions were detected in all gamma ray-induced AML samples tested (n = 20), and two of the three iron ion-induced cases (Figure 1, Table I). Only one sample, AML 3048, showed disagreement between FISH and aCGH results, likely due to the small size of the deletion (~130kb as predicted by aCGH) and the differing resolutions of the two techniques. All samples had normal ploidy except AML 3576 and AML 3173, which demonstrated >3n ploidy in 50% and 16% of metaphases, respectively.
Fig. 1.
Both high- and low-LET radiation-induced rAML show hemizygous deletion of chromosome 2 including the PU.1 gene. (A) Deletion of chromosome 2 in rAML spleen cells was assessed by FISH with a PU.1-containing BAC probe (pink) and DNA was counterstained with DAPI (blue). Cells were scored as having a hemizygous deletion if signals were absent from both chromatids of one chromosome 2 homolog. Representative metaphases are shown with 10 um scale bars. (B) Array CGH was used to identify deletions on chromosome 2. The location of PU.1 is indicated with a vertical line. Chromosomal gains (blue) and losses (red) are depicted for individual AML samples, demonstrating single copy PU.1 loss in all cases except AML 2704.
During AML development the non-deleted PU.1 allele is frequently disrupted by a missense point mutation at codon 235. We developed a rapid and sensitive high resolution melt assay to detect the most common PU.1 mutations. A 73bp amplicon containing the R235 coding sequence was amplified from spleen genomic DNA and analysed by high resolution melt (Table I, Figure 2A). The assay robustly detected R235 mutations in 84% of AML samples (Table I). Specificity and sensitivity were both 100%, as verified by sequencing (Figure 2B). Gamma ray- and iron ion-induced AML did not have significant differences in R235 mutation incidence (88 and 66%, respectively), and R235 mutations were identified only in samples with a chromosome 2 deletion. No alternative mutations were detected in PU.1 exons or splice junctions of the remaining samples, nor did we find Flt3 internal tandem duplication mutations (data not shown) as demonstrated in a recent study by Finnon et al.(19)
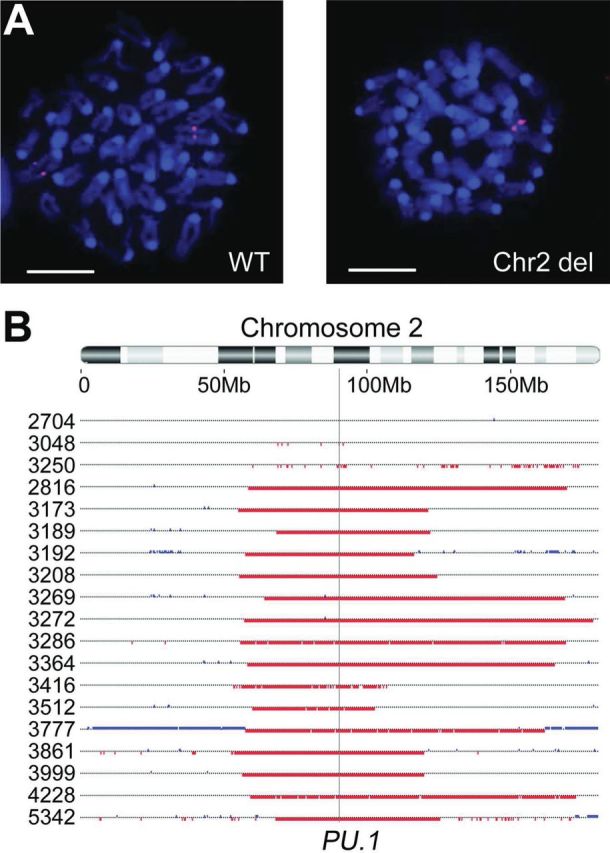
Fig. 2.
Characterisation of mutations at the undeleted PU.1 allele. (A) High resolution melt analysis was capable of genotyping samples with mutations in PU.1 R235. A 73bp amplicon containing the R235 coding sequence was amplified for each sample and a difference curve was used to compare melt profiles to reference DNA with wildtype PU.1. (Red = reference DNA and rAML samples with wildtype R235 allele; Green = rAML samples with mutant R235 allele). (B) Sequencing validated high resolution melt results for all samples. All mutations were detected at the first cytosine (asterisk) of PU.1 codon R235 (nucleotide 855 of NM_011355.1). Representative wildtype (top) and mutant (bottom) chromatograms are shown. (C) Bisulfite sequencing of bulk bone marrow demonstrated methylation of the target cytosine (asterisk). The sequencing chromatogram is shown, where methylated cytosines (indicated by blue circles) are unaffected by bisulfite conversion while unmethylated cytosines (red squares) are converted and amplified as thymine.
All but one of the gamma ray-induced AML samples with biallelic PU.1 disruption (92%) carried C to T transition mutations of the target cytosine (Table I), similar to previous reports in both gamma ray- and X-ray-induced rAML.(14,15,37) Bisulfite sequencing of bulk bone marrow identified methylation of this cytosine (Figure 2C), deamination of which would result in the common transition mutation. In contrast, both of the iron ion–induced AML samples with biallelic PU.1 mutations are C to A transversion mutations. Nine additional AML samples induced by neutron irradiation (which generates high-LET recoil protons) were kindly provided by Drs. Bouffler, Haines and Meijne, and were screened for R235 mutations by high resolution melt analysis. Of these, 56% contained PU.1 R235 mutations, all of which proved to be C to T transition mutations in common with low-LET rAML. While it is statistically unlikely that both iron-induced AML cases would be transversions merely by chance (Fisher exact, P = 0.014), the number of samples is currently too low to determine if the molecular alterations of PU.1 R235 are truly distinct between radiation qualities.
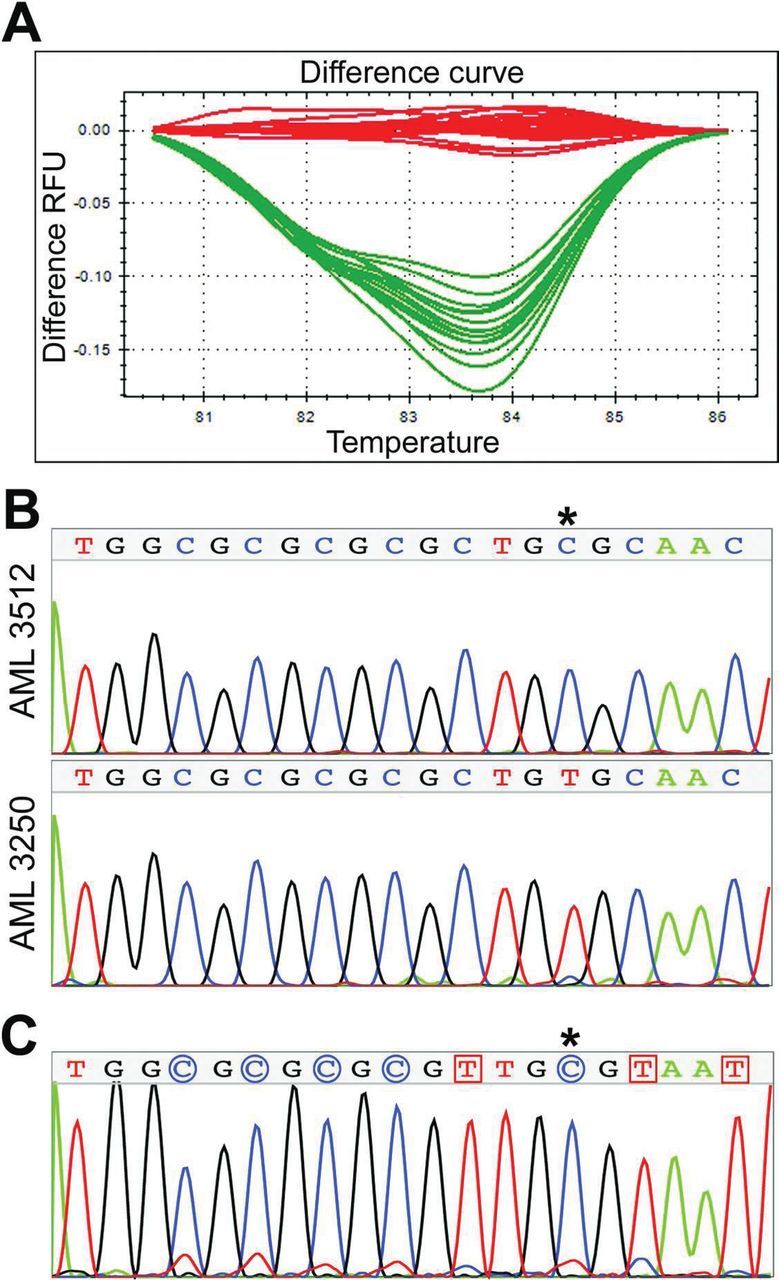
Chromosome aberrations—specifically interstitial deletions on chromosome 2—are clearly early events in AML induction. To determine if chromosomal instability is ongoing in rAML, metaphase preparations from leukemic and control splenocytes were analysed for chromatid-type aberrations (chromatid gaps, breaks, iso-deletions and chromatid-type exchanges; Figure 3), which arise in the cell cycle of collection, many generations after radiation exposure, and so are indicative of instability. No significant difference in chromatid-type aberration frequencies between age-matched unirradiated control mice (no IR; non-AML) and unaffected iron ion- and gamma ray-irradiated control mice (IR; non-AML) was seen, so they were combined. In contrast, a significant 2-fold increase in chromatid-type aberrations—primarily chromatid-type gaps and breaks—was observed in AML samples induced both by iron ion and gamma ray irradiation (Table 1, Figure 3B). Neither chromosome-type aberrations nor telomere instability endpoints were elevated within the resolution of this study.
Fig. 3.
Chromatid-type aberrations are significantly increased in rAML, indicative of ongoing instability. (A) Representative rAML mouse metaphase hybridised with peptide nucleic acid G-rich telomere probe (pink) and counterstained with DAPI (blue). Examples of chromatid breaks are shown with arrows. Original magnification x100. (B) A significant increase in chromatid-type aberrations was seen in rAML (*, P = 0.033). Total telomere aberration frequencies and total chromosome aberration frequencies were not significantly different between AML and control spleens. The control group includes age-matched animals from unirradiated (n = 4), iron irradiated (n = 10) and gamma irradiated (n = 5) cohorts that did not develop AML. Error bars indicate standard error.
MSI is an alternate mutator mechanism for cancer progression. Analysis of six non-coding mononucleotide microsatellites demonstrated that 42% of AML samples showed MSI in at least one locus, including two iron ion-induced cases (Table I). Gamma ray-induced AML 3229 was the only MSI-high rAML, with instability in two loci. Because ‘normal’ tissue was not available for genotyping (due to blast infiltration of tissues), 10 irradiated, non-leukaemic control mice and 35 unirradiated mice from the same study were also analysed. Non-standard allele sizes were detected in only two controls and are consistent with a breeding colony polymorphism due to their shared size. These data strongly support MSI as a common event in radiation-induced acute myeloid leukaemogenesis (Chi-squared test, P < 0.001).
To determine if MSI is ongoing in radiation-induced AML, we applied a variation of the traditional MSI assay, using DNA diluted to the equivalent of one genome or less per amplification reaction. This technique, called small-pool PCR (SP-PCR), allows quantification of rare mutant alleles that would otherwise be masked by an excess of wildtype alleles. Analysis of spleens from unirradiated control animals demonstrates that all animals have a low background mutation rate that is age dependent (Figure 4A). Thus we compared mutant frequency from age-matched unirradiated and irradiated normal controls with rAML samples to determine whether radiation and/or AML status could additionally increase the mutant frequency. Interestingly, neither iron ion irradiation (n = 6) nor gamma ray irradiation (n = 4) appears to have a lasting effect on mutant frequency in otherwise normal spleen cells (Figure 4B). However, it is clear that the mutant frequency in leukaemic cells is significantly increased by more than 2-fold even after removal of clonal mutants detected by the traditional MSI assay. Overall, 89% of AML samples individually have a significant increase in mutant frequency compared with controls (Table I). Microsatellite mutant frequency was not correlated with latency of disease, radiation dose or chromatid aberration frequency. Interestingly, the two AML samples with low mutant frequency lack large chromosome 2 deletions as well. These data suggest that an increase in ongoing microsatellite-type instability is prevalent in radiation-induced AML.
Fig. 4.
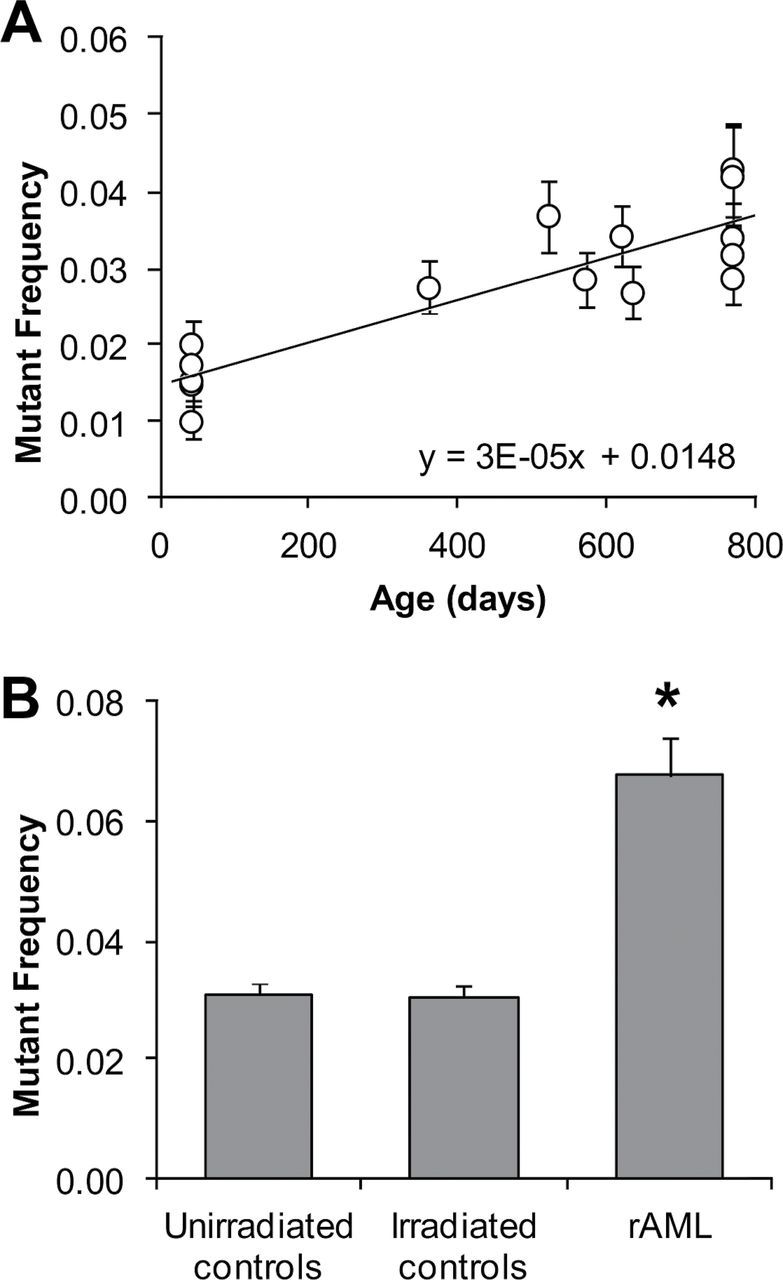
Mutant frequency at microsatellite repeats is significantly increased in rAML. (A) Spontaneous mutant frequency measured at six non-coding polyadenine microsatellites increases with age in spleen cells of unirradiated animals (Pearson’s, P < 0.0001). A linear fit is shown. (B) Non-clonal mutant frequency is increased greater than 2-fold in AML cells compared to control spleen cells from age-matched animals (*, t test P < 0.001). No persistent increase in mutant frequency was detected in normal spleen cells of irradiated animals compared with those from unirradiated age-matched controls. Error bars indicate standard error.
Discussion
To our knowledge, this is the first demonstration that high-LET iron ion radiation exposure can result in murine AML with biallelic PU.1/Sfpi1 alterations. Of the three 56Fe ion-induced AML samples available, two carried both PU.1 deletions and point mutations. Analysis of nine neutron-induced AML samples identified five additional high-LET-induced AMLs with mutations of the PU.1 R235 codon. Two of these have previously been shown to carry chromosome 2 deletions spanning PU.1; because 94% of neutron-induced AMLs carry chromosome 2 deletions, it is assumed that the remaining mutants were also biallelic (7). The difference in frequency of biallelic mutations was not significantly different between low-LET radiation-induced AML (88%) and high-LET radiation-induced AML (58%), although the limited sample sizes preclude statistically significant detection of small differences. These rates are comparable to those reported in the literature, which range from 67% to 88% (14,15,19,37).
These data show that biallelic loss of PU.1 does occur in murine rAML induction, regardless of radiation quality. However, it should be noted that a portion of the dose in high-LET 56Fe ion irradiation is due to secondary low-LET delta rays, which could contribute to AML induction. We also identified possible differences in the type of R235 mutations that occur between high- and low-LET rAML. C to T transition mutations accounted for 93% and 100% of all PU.1 R235 point mutations in gamma- and neutron-induced AML, respectively. These data are in agreement with previous reports, where 77–90% of PU.1 R235 mutations are transitions (14,15,37). Bisulfite sequencing of bone marrow DNA suggests that the target cytosine is methylated and could be mutated through deamination, though we have not specifically assayed methylation in the as-yet unknown target cell population. In contrast, C to A transversion mutations were identified in both the iron ion-induced rAML cases with R235 mutations and might suggest a different mechanism for mutation induction. It is currently unclear if radiation exposure or loss of other genes within the chromosome 2 commonly deleted region affects the incidence of PU.1 mutation. If the type of R235 mutation is indeed different between radiation qualities, these data would support a role for radiation effects on mutation rate since no difference in the extent of chromosome 2 deletion has been noted between radiation qualities.
Defects in the mismatch repair (MMR) pathway have been causally implicated in Lynch Syndrome, a cancer predisposition syndrome characterised by widespread MSI, leading to increased risk of colon and other solid cancers. Recent reports have also shown that rare patients with homozygous defects in MMR genes carry an increased risk of AML and other haematological cancers (38). However, studies of MSI within the context of human AML have been contradictory, with reports ranging from 0% to 94% of cases reported as MSI positive. None-the-less, the emerging consensus is that de novo AML is rarely characterised by MSI, while MSI is frequently present in therapy-associated AML. We re-examined the literature where MSI status was reported in sufficient detail to reassess patients with tAML secondary to chemotherapy and/or radiation therapy. The combined data suggests that more than 49% of analysed tAML cases show instability in at least one microsatellite tested, with 40% characterised as MSI-high, using a threshold of instability in ≥30% of tested loci (25–33). These data are an estimate, as the studies included use a wide variety of markers and techniques with different resolutions, employ patient populations that may be selected or artificially skewed and use various control tissues—including hematopoietic lineages—to establish constitutional genotypes. In addition, a large study by Rimsza et al. (39) was excluded because statistics for tAML and sAML (secondary to myelodysplastic syndrome) were inseparable. Regardless, the average incidence of MSI in tAML is still higher than other subsets of AML and may correlate with disease induction mechanism or patient susceptibility. In addition, a small number of MSI-positive tAML patients also show loss or hypermethylation of MMR genes and/or frameshift mutations in coding repeats of putative tumour suppressors, characteristic of MMR defects (26,33,40,41). While radiotherapy- and chemotherapy-associated AML cases are rarely reported separately, at least one study of atomic bomb survivors suggests that radiation alone is also associated with increased incidence of MSI-positive AML (25).
We identified MSI in 42% of murine rAML tumours, suggesting that this type of genomic instability is commonly elevated in AML induced by both high- and low-LET radiation. Interestingly, MSI incidence was non-significantly elevated in high-LET induced AML compared with low-LET induced samples (66% versus 38%), in keeping with reports by Haines et al. (34) assessing MSI incidence in X ray- and neutron-induced rAML using dinucleotide repeats. However, only one AML was found to be MSI-high using the standard cut-off. Haines et al. (34) also detected a predominance of MSI-low phenotypes in murine rAML. We cannot formally exclude the possibility that MSI in rAML is a chance consequence of spontaneous or radiation-induced mutations in the leukaemic stem cell, though the low single-cell mutation frequencies measured in this study are unlikely to account for such a high number of samples with MSI (Figure 4B). Further, the absence of MSI in radiation-induced tumours of several other tissues argues against MSI-low as a non-specific by-product of radiation tumorigenesis (34). Instead, it is likely that the MSI-low phenotype is due to both species and tumour type differences: Mouse tumours with known MMR defects do not exhibit MSI in as many loci as their human counterparts, and extracolonic tumours in Lynch syndrome patients are also frequently MSI low (42,43). Given that the MSI multiplex used in this study assays only a tiny fraction of the mouse genome, detection of mutations in 42% of tumours is remarkable.
To detect ongoing MSI, we also looked at non-clonal mutant frequency at the same microsatellite loci. Analysis of sham-irradiated animals demonstrates an age-dependent increase in mutant frequency in the spleen (Figure 4A), similar to levels detected in peripheral blood (J.Bacher and J.Bourdeau, in preparation). Interestingly, we find that while irradiation may increase microsatellite mutation incidence in the short term (44), phenotypically normal animals assayed more than 1 year post-irradiation show no significant increase in mutant frequency (Figure 4B). In contrast, 89% of rAML samples showed significantly elevated mutant frequencies even after removal of clonal mutants detected in the MSI assay. Thus, instability is likely ongoing during tumour proliferation. Only two samples lack significant elevation of non-clonal mutant frequency. Both of these—AML 2704 and AML 3048—also lack MSI and are the only two samples without large deletions on chromosome 2. Thus it is possible that loss of additional genes in the commonly deleted region is responsible for the incidence of MSI and/or the increase in ongoing microsatellite mutation.
The timing, target cell and mechanism of PU.1 point mutation in rAML are currently unknown. Analysis of clonal MSI events in this study suggest that microsatellite mutation is frequent and present early in the clonal history of the leukaemia since the clonal mutations account for a high percentage of observed alleles in single-cell assays (median 37%, range 14–44%). We also demonstrated that increased microsatellite mutation is ongoing in rAML. It is not yet clear if MSI is indicative of a causative mechanism or is merely correlative, but we hypothesise that defective MMR function could contribute to increased incidence of the R235 point mutation. The MMR protein, MSH6, is known to participate in correction of T:G mismatches, which would be generated by deamination of methylcytosine at R235. In addition, studies of gastrointestinal tumours in Msh6 null mouse models demonstrate a strong shift in preference for C to T transition mutations in the Apc tumour suppressor gene with the primary hot spot immediately downstream of a short dinucleotide repeat (45). PU.1 codon R235 is similarly located downstream of a dinucleotide repeat and is primarily targeted by C to T transition mutations. Thus, we are currently studying how radiation and the loss of genes on chromosome 2 influence MMR and incidence of MSI to determine if MMR is involved in PU.1 R235 mutation.
Evidence of instability can also be observed at the chromosomal level; thus, we performed cytogenetic analysis on rAML samples with particular focus on chromatid-type aberrations as they provide a signature of ongoing instability. Instability has been shown in murine bone marrow cells in response to both low-LET and high-LET irradiation in vivo (46,47). Instability has also been detected in human bone marrow cells exposed to high-LET radiation ex vivo, and it has been suggested that such a mutator phenotype may account for progression to rAML (48,49). Indeed, Bouffler et al. (7) identified chromatid-type instability in seven of 17 neutron-induced rAML cases though the instability did not consistently persist through serial passage. To our knowledge, the current study is the first report of elevated ongoing chromosomal instability in iron ion- and gamma ray-induced rAML. Inter-animal variation in the frequency of instability is consistent with previous reports, and no significant difference was detected between radiation qualities, similar to the findings of Watson et al.(7,46). These data differ from reports by Kadhim et al. (50) and may reflect differences between in vivo and ex vivo irradiations in mice. It is not clear if chromatid-type instability in iron ion- and gamma ray-induced AML would persist through serial passage, or if it plays a causal role in AML development. However, it does appear that chromatid-type instability is increased in a portion of radiation-induced AML cases in mice.
Assessment of telomere stability revealed no increase in telomere aberrations; however, mouse telomeres are maintained by active telomerase, which may mask telomere involvement (51).We also did not detect an increase in chromosome-type aberrations in this study though interstitial deletions (including known deletions of chromosome 2) are rarely within the resolution of the technique used. These data are consistent with reports that unstable chromosome-type aberrations occur at lower frequency than chromatid-type aberrations long after acute radiation exposure (46,47). Array CGH data detailing chromosomal gains and losses in finer detail will be presented elsewhere.
In conclusion, the data show that high-LET 56Fe ion radiation can induce AML with biallelic PU.1 defects, similar to results from low-LET and neutron rAML reported here and elsewhere. Clonal MSI, detected with traditional PCR, is apparent in almost half of murine AML and implies that mutations occurred early in tumour expansion. Chromatid-type aberrations and microsatellite mutant frequency at the single-cell level are also significantly elevated, indicative of ongoing instability through multiple pathways that may contribute to AML progression.
Funding
NSCOR grant from the National Aeronautics and Space Administration (NAG-1569); the US Department of Energy Low Dose Radiation Research Program (DE-FG02-05ER63946); and the National Aeronautics and Space Administration (NNX07AO85G to Y.P. and J.S.B.).
Acknowledgements
The authors would like to thank Simon D. Bouffler, Jackie W. Haines and Emmy Meijne who kindly provided the neutron-induced rAML samples. The 8016 rAML cell line was provided by Simon Bouffler and created by Dr Kazuko Yoshida (NIRS, Chiba, Japan). Xianan Liu performed the bisulfite sequencing, and Michael Betlach (Promega Corporation) and Stephen Stanhope (University of Chicago) consulted on the statistics.
Conflict of interest statement: The authors declare no competing financial interests.
References
- 1. Shimizu Y., Kato H., Schull W. J. (1991). Risk of cancer among atomic bomb survivors. J. Radiat. Res 32 Suppl 2, 54–63 [DOI] [PubMed] [Google Scholar]
- 2. Cardis E., Gilbert E. S., Carpenter L., et al. (1995). Effects of low doses and low dose rates of external ionizing radiation: cancer mortality among nuclear industry workers in three countries. Radiat. Res 142, 117–132 [PubMed] [Google Scholar]
- 3. Wright J. D., St Clair C. M., Deutsch I., Burke W. M., Gorrochurn P., Sun X., Herzog T. J. (2010). Pelvic radiotherapy and the risk of secondary leukemia and multiple myeloma. Cancer 116, 2486–2492 [DOI] [PubMed] [Google Scholar]
- 4. Travis L. B., Andersson M., Gospodarowicz M., et al. (2000). Treatment-associated leukemia following testicular cancer. J. Natl Cancer Inst 92, 1165–1171 [DOI] [PubMed] [Google Scholar]
- 5. Smith S. M., Le Beau M. M., Huo D., Karrison T., Sobecks R. M., Anastasi J., Vardiman J. W., Rowley J. D., Larson R. A. (2003). Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 102, 43–52 [DOI] [PubMed] [Google Scholar]
- 6. Mole R. H. (1986). Radiation-induced acute myeloid leukemia in the mouse: experimental observations in vivo with implications for hypotheses about the basis of carcinogenesis. Leuk. Res., 10, 859–865 [DOI] [PubMed] [Google Scholar]
- 7. Bouffler S. D., Meijne E. I., Huiskamp R., Cox R. (1996). Chromosomal abnormalities in neutron-induced acute myeloid leukemias in CBA/H mice. Radiat. Res 146, 349–352 [PubMed] [Google Scholar]
- 8. Weil M. M., Bedford J. S., Bielefeldt-Ohmann H., et al. (2009). Incidence of acute myeloid leukemia and hepatocellular carcinoma in mice irradiated with 1 GeV/nucleon (56)Fe ions. Radiat. Res 172, 213–219 [DOI] [PubMed] [Google Scholar]
- 9. Peng Y., Brown N., Finnon R., et al. (2009). Radiation leukemogenesis in mice: loss of PU.1 on chromosome 2 in CBA and C57BL/6 mice after irradiation with 1 GeV/nucleon 56Fe ions, X rays or gamma rays. Part I. Experimental observations. Radiat. Res., 171, 474–483 [DOI] [PubMed] [Google Scholar]
- 10. Bouffler S. D., Meijne E. I., Morris D. J., Papworth D. (1997). Chromosome 2 hypersensitivity and clonal development in murine radiation acute myeloid leukaemia. Int. J. Radiat. Biol., 72, 181–189 [DOI] [PubMed] [Google Scholar]
- 11. Rithidech K., Bond V. P., Cronkite E. P., Thompson M. H., Bullis J. E. (1995). Hypermutability of mouse chromosome 2 during the development of x-ray-induced murine myeloid leukemia. Proc. Natl Acad. Sci. USA 92, 1152–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Breckon G., Papworth D., Cox R. (1991). Murine radiation myeloid leukaemogenesis: a possible role for radiation-sensitive sites on chromosome 2. Genes Chromosomes Cancer 3, 367–375 [DOI] [PubMed] [Google Scholar]
- 13. Rithidech K. N., Bond V. P., Cronkite E. P., Thompson M. H. (1993). A specific chromosomal deletion in murine leukemic cells induced by radiation with different qualities. Exp. Hematol 21, 427–431 [PubMed] [Google Scholar]
- 14. Cook W. D., McCaw B. J., Herring C., John D. L., Foote S. J., Nutt S. L., Adams J. M. (2004). PU.1 is a suppressor of myeloid leukemia, inactivated in mice by gene deletion and mutation of its DNA binding domain. Blood 104, 3437–3444 [DOI] [PubMed] [Google Scholar]
- 15. Suraweera N., Meijne E., Moody J., et al. (2005). Mutations of the PU.1 Ets domain are specifically associated with murine radiation-induced, but not human therapy-related, acute myeloid leukaemia. Oncogene 24, 3678–3683 [DOI] [PubMed] [Google Scholar]
- 16. Liang H., Mao X., Olejniczak E. T., Nettesheim D. G., Yu L., Meadows R. P., Thompson C. B., Fesik S. W. (1994). Solution structure of the ets domain of Fli-1 when bound to DNA. Nat. Struct. Biol., 1, 871–875 [DOI] [PubMed] [Google Scholar]
- 17. Metcalf D., Dakic A., Mifsud S., Di Rago L., Wu L., Nutt S. (2006). Inactivation of PU.1 in adult mice leads to the development of myeloid leukemia. Proc. Natl Acad. Sci. USA 103, 1486–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenbauer F., Wagner K., Kutok J. L., Iwasaki H., Le Beau M. M., Okuno Y., Akashi K., Fiering S., Tenen D. G. (2004). Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet., 36, 624–630 [DOI] [PubMed] [Google Scholar]
- 19. Finnon R., Brown N., Moody J., et al. (2012). Flt3-ITD mutations in a mouse model of radiation-induced acute myeloid leukaemia. Leukemia 26, 1445–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mueller B. U., Pabst T., Osato M., et al. (2002). Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood 100, 998–1007 [DOI] [PubMed] [Google Scholar]
- 21. Iida H., Towatari M., Iida M., Tanimoto M., Kodera Y., Ford A. M., Saito H. (2000). Protein expression and constitutive phosphorylation of hematopoietic transcription factors PU.1 and C/EBP beta in acute myeloid leukemia blasts. Int. J. Hematol 71, 153–158 [PubMed] [Google Scholar]
- 22. Zheng R., Friedman A. D., Levis M., Li L., Weir E. G., Small D. (2004). Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood 103, 1883–1890 [DOI] [PubMed] [Google Scholar]
- 23. Vangala R. K., Heiss-Neumann M. S., Rangatia J. S., Singh S. M., Schoch C., Tenen D. G., Hiddemann W., Behre G. (2003). The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 101, 270–277 [DOI] [PubMed] [Google Scholar]
- 24. Loeb L. A. (2011). Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer 11, 450–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakanishi M., Tanaka K., Takahashi T., Kyo T., Dohy H., Fujiwara M., Kamada N. (2001). Microsatellite instability in acute myelocytic leukaemia developed from A-bomb survivors. Int. J. Radiat. Biol., 77, 687–694 [DOI] [PubMed] [Google Scholar]
- 26. Zhu Y. M., Das-Gupta E. P., Russell N. H. (1999). Microsatellite instability and p53 mutations are associated with abnormal expression of the MSH2 gene in adult acute leukemia. Blood 94, 733–740 [PubMed] [Google Scholar]
- 27. Ben-Yehuda D., Krichevsky S., Caspi O., et al. (1996). Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood 88, 4296–4303 [PubMed] [Google Scholar]
- 28. Sheikhha M. H., Tobal K., Liu Yin J. A. (2002). High level of microsatellite instability but not hypermethylation of mismatch repair genes in therapy-related and secondary acute myeloid leukaemia and myelodysplastic syndrome. Br. J. Haematol 117, 359–365 [DOI] [PubMed] [Google Scholar]
- 29. Worrillow L. J., Travis L. B., Smith A. G., et al. (2003). An intron splice acceptor polymorphism in hMSH2 and risk of leukemia after treatment with chemotherapeutic alkylating agents. Clin. Cancer Res 9, 3012–3020 [PubMed] [Google Scholar]
- 30. Boyer J. C., Risinger J. I., Farber R. A. (1998). Stability of microsatellites in myeloid neoplasias. Cancer Genet. Cytogenet 106, 54–61 [DOI] [PubMed] [Google Scholar]
- 31. Das-Gupta E. P., Seedhouse C. H., Russell N. H. (2001). Microsatellite instability occurs in defined subsets of patients with acute myeloblastic leukaemia. Br. J. Haematol 114, 307–312 [DOI] [PubMed] [Google Scholar]
- 32. Horiike S., Misawa S., Kaneko H., et al. (1999). Distinct genetic involvement of the TP53 gene in therapy-related leukemia and myelodysplasia with chromosomal losses of Nos 5 and/or 7 and its possible relationship to replication error phenotype. Leukemia 13, 1235–1242 [DOI] [PubMed] [Google Scholar]
- 33. Casorelli I. (2003). Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome. DNA Repair (Amst) 2, 547–559 [DOI] [PubMed] [Google Scholar]
- 34. Haines J., Bacher J., Coster M., Huiskamp R., Meijne E., Mancuso M., Pazzaglia S., Bouffler S. (2010). Microsatellite instability in radiation-induced murine tumours; influence of tumour type and radiation quality. Int. J. Radiat. Biol., 86, 555–568 [DOI] [PubMed] [Google Scholar]
- 35. Fennelly J., Wright E., Plumb M. (1997). Mini- and microsatellite mutations in radiation-induced acute myeloid leukaemia in the CBA/H mouse. Leukemia 11, 807–810 [DOI] [PubMed] [Google Scholar]
- 36. Dunn T. B. (1954). Normal and pathologic anatomy of the reticular tissue in laboratory mice, with a classification and discussion of neoplasms. J. Natl Cancer Inst 14, 1281–1433 [PubMed] [Google Scholar]
- 37. Hirouchi T., Takabatake T., Yoshida K., Nitta Y., Nakamura M., Tanaka S., Ichinohe K., Oghiso Y., Tanaka K. (2008). Upregulation of c-myc gene accompanied by PU.1 deficiency in radiation-induced acute myeloid leukemia in mice. Exp. Hematol 36, 871–885 [DOI] [PubMed] [Google Scholar]
- 38. Ripperger T., Beger C., Rahner N., Sykora K. W., Bockmeyer C. L., Lehmann U., Kreipe H. H., Schlegelberger B. (2010). Constitutional mismatch repair deficiency and childhood leukemia/lymphoma–report on a novel biallelic MSH6 mutation. Haematologica 95, 841–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rimsza L. M., Kopecky K. J., Ruschulte J., Chen I. M., Slovak M. L., Karanes C., Godwin J., List A., Willman C. L. (2000). Microsatellite instability is not a defining genetic feature of acute myeloid leukemogenesis in adults: results of a retrospective study of 132 patients and review of the literature. Leukemia 14, 1044–1051 [DOI] [PubMed] [Google Scholar]
- 40. Rund D., Krichevsky S., Bar-Cohen S., et al. (2005). Therapy-related leukemia: clinical characteristics and analysis of new molecular risk factors in 96 adult patients. Leukemia 19, 1919–1928 [DOI] [PubMed] [Google Scholar]
- 41. Offman J., Gascoigne K., Bristow F., et al. (2005). Repeated sequences in CASPASE-5 and FANCD2 but not NF1 are targets for mutation in microsatellite-unstable acute leukemia/myelodysplastic syndrome. Mol. Cancer Res., 3, 251–260 [DOI] [PubMed] [Google Scholar]
- 42. Bacher J. W., Abdel Megid W. M., Kent-First M. G., Halberg R. B. (2005). Use of mononucleotide repeat markers for detection of microsatellite instability in mouse tumors. Mol. Carcinog 44, 285–292 [DOI] [PubMed] [Google Scholar]
- 43. Gylling A. H., Nieminen T. T., Abdel-Rahman W. M., et al. (2008). Differential cancer predisposition in Lynch syndrome: insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis 29, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 44. Megid W. A., Ensenberger M. G., Halberg R. B., Stanhope S. A., Kent-First M. G., Prolla T. A., Bacher J. W. (2007). A novel method for biodosimetry. Radiat. Environ. Biophys., 46, 147–154 [DOI] [PubMed] [Google Scholar]
- 45. Kuraguchi M., Yang K., Wong E., et al. (2001). The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res 61, 7934–7942 [PubMed] [Google Scholar]
- 46. Watson G. E., Pocock D. A., Papworth D., Lorimore S. A., Wright E. G. (2001). In vivo chromosomal instability and transmissible aberrations in the progeny of haemopoietic stem cells induced by high- and low-LET radiations. Int. J. Radiat. Biol 77, 409–417 [DOI] [PubMed] [Google Scholar]
- 47. MacDonald D., Boulton E., Pocock D., Goodhead D., Kadhim M., Plumb M. (2001). Evidence of genetic instability in 3 Gy X-ray-induced mouse leukaemias and 3 Gy X-irradiated haemopoietic stem cells. Int. J. Radiat. Biol., 77, 1023–1031 [DOI] [PubMed] [Google Scholar]
- 48. Kadhim M. A., Lorimore S. A., Hepburn M. D., Goodhead D. T., Buckle V. J., Wright E. G. (1994). Alpha-particle-induced chromosomal instability in human bone marrow cells. Lancet 344, 987–988 [DOI] [PubMed] [Google Scholar]
- 49. Kadhim M. A., Marsden S. J., Goodhead D. T., Malcolmson A. M., Folkard M., Prise K. M., Michael B. D. (2001). Long-term genomic instability in human lymphocytes induced by single-particle irradiation. Radiat. Res 155, 122–126 [DOI] [PubMed] [Google Scholar]
- 50. Kadhim M. A., Macdonald D. A., Goodhead D. T., Lorimore S. A., Marsden S. J., Wright E. G. (1992). Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 355, 738–740 [DOI] [PubMed] [Google Scholar]
- 51. Chadeneau C., Siegel P., Harley C. B., Muller W. J., Bacchetti S. (1995). Telomerase activity in normal and malignant murine tissues. Oncogene 11, 893–898 [PubMed] [Google Scholar]