

Abstract
The cytosolic sulfotransferases (SULTs) catalyze the sulfate conjugation of nucleophilic substrates, and the cofactor for sulfonation, 3′-phosphoadenosine-5′-phosphosulfate (PAPS), is biosynthesized from sulfate and ATP. The phenotype of male knockout mice for the NaS1 sodium sulfate cotransporter includes hyposulfatemia and increased hepatic expression of mouse cytoplasmic sulfotransferase Sult2a and Sult3a1. Here we report that in 8-week-old female NaS1-null mice, hepatic Sult2a1 mRNA levels were ∼51-fold higher than they were in a wild-type liver but expression of no other Sult was affected. To address whether hyposulfatemia-inducible Sult2a1 expression might be due to reduced PAPS levels, we stably knocked down PAPS synthases 1 and 2 in HepG2 cells (shPAPSS1/2 cells). When a reporter plasmid containing at least 233 nucleotides (nt) of Sult2a1 5′-flanking sequence was transfected into shPAPSS1/2 cells, reporter activity was significantly increased relative to the activity that was seen for reporters containing 179 or fewer nucleotides. Mutation of an IR0 (inverted repeat of AGGTCA, with 0 intervening bases) nuclear receptor motif at nt −191 to 180 significantly attenuated the PAPSS1/2 knockdown-mediated increase. PAPSS1/2 knockdown significantly activated farnesoid X receptor (FXR), retinoid-related orphan receptor, and pregnane X receptor responsive reporters, and treatment with the FXR agonist GW4064 [3-(2,6-dichlorophenyl)-4-(3′-carboxy-2-chlorostilben-4-yl)oxymethyl-5-isopropylisoxazole] increased Sult2a1 promoter activity when the IR0 was intact. Transfection of shPAPSS1/2 cells with FXR small interfering RNA (siRNA) significantly reduced the Sult2a1 promoter activity. The impact of PAPSS1/2 knockdown on Sult2a1 promoter activity was recapitulated by knocking down endogenous SULT2A1 expression in HepG2 cells. We propose that hyposulfatemia leads to hepatic PAPS depletion, which causes loss of SULT2A1 activity and results in accumulation of nonsulfated bile acids and FXR activation.
Introduction
The cytosolic sulfotransferases (SULTs) are conjugating enzymes that catalyze the transfer of a sulfonate moiety from the physiologic sulfate donor 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to an appropriate nucleophilic substrate (Falany, 1997). Aside from its long-recognized function as a xenobiotic detoxifying enzyme, emerging evidence suggests that hepatic hydroxysteroid sulfotransferase (SULT2A) enzymes play critical roles in lipid and energy metabolism. SULT2A enzymes are transcriptionally regulated by and modulate the activities of nuclear receptors that are involved in hepatic lipid signaling. For example, we previously demonstrated that human hepatic SULT2A1 is transactivated by peroxisome proliferator-activated receptor α (PPARα) (Fang et al., 2005), a nuclear receptor that is activated by certain fatty acids (e.g., unsaturated C:18 fatty acids and arachidonic acid) and leukotriene LTB4 (Lin et al., 1999). We also have shown that human SULT2A1 catalyzes the sulfonation of 24-hydroxycholesterol to 3- and 24-sulfates and that these metabolites antagonize activation of the oxysterol receptor liver X receptor (LXR) (Cook et al., 2009). The pregnane X receptor (PXR) orchestrates the expression of gene expression networks involved in not only hepatic drug metabolism but also in lipid and energy metabolism (Bhalla et al., 2004; Kodama et al., 2007; Zhou et al., 2008; Moreau et al., 2009). PXR is activated by both xenobiotic and endogenous ligands such as steroid hormones, bile acids, fat-soluble vitamins, pharmaceuticals, and environmental contaminants such as phthalates (Kliewer et al., 1998; Lehmann et al., 1998; Staudinger et al., 2001; Landes et al., 2003; Hurst and Waxman, 2004). Through interactions with the transcription factor hepatocyte nuclear factor 4α (HNF4α), PXR negatively regulates the transcription of human hepatic SULT2A1 (Fang et al., 2007), thus influencing the ability of SULT2A1 to titrate lipid signaling and homeostasis in the liver.
PAPS production is entirely dependent on the cellular availability of sulfate. The obligate cofactor for sulfate conjugation, PAPS is biosynthesized through the action of PAPS synthases (PAPSS) 1 and 2. These bifunctional enzymes first use their sulfurylase domains to catalyze the reaction of ATP with inorganic sulfate to form adenosine 5′-phosphosulfate, and then the PAPSS enzymes use their kinase domains to catalyze the reaction of adenosine 5′-phosphosulfate with another molecule of ATP to form PAPS (Venkatachalam, 2003).
Sulfate, an essential macronutrient, is the fourth most abundant anion in human plasma and is readily filtered and reabsorbed in the kidney (Markovich, 2011a). To understand the physiologic consequences of disrupted sulfate homeostasis, a sulfate transporter knockout mouse model was engineered with impaired sulfate reabsorption in the kidney (Dawson et al., 2003). Sulfate conjugation is important for biotransformation of xenobiotic and endogenous compounds as well as for the production of structural components essential for normal physiologic function. Despite the importance of sulfate, the regulatory pathways responsible for sulfate homeostasis are not well understood. Genetically engineered mice with knocked-out NaS1 sodium sulfate cotransporter [solute carrier family 13 (sodium/sulfate symporters), member 1 (Slc13a1)] display hyposulfatemia and hypersulfaturia due to loss of renal sulfate reabsorption (Markovich, 2011a). The phenotype of NaS1-null mice underscores the widespread role of sulfate in normal physiology and includes a range of systemic impairments that affect fertility, circulating steroid hormone levels, intestinal barrier function, locomotion, cognitive ability, and oncogenesis (Markovich, 2011a). NaS1-null mice also display significant disturbances in hepatic lipid metabolism, and microarray analysis has revealed altered hepatic expression of several genes involved in lipid homeostasis as well as upregulation of Sult2a2 and Sult3a1 (Dawson et al., 2006).
The mechanism that is responsible for increased hepatic Sult2a expression under conditions of hyposulfatemia is unknown; however, given the aberrancies in hepatic lipid homeostasis that are most likely produced by the hyposulfatemia, the hypothesis is that lipid-sensing transcription factors in the liver may double as “sulfate sensors” during periods of metabolic stress. We 1) evaluated in greater detail the regulation of Sult genes in this mouse model, 2) developed a cell culture model to address the impact of depletion of the SULT cofactor PAPS on Sult2a1 transcription, and 3) investigated the mechanism(s) responsible for PAPS-depletion-mediated Sult2a1 transcriptional activation.
Materials and Methods
NaS1 Mice.
Breeder pairs of NaS1+/− mice were mated, and NaS1+/+ and NaS1−/− offspring were identified using DNA that was isolated from ear punch and/or tail-tip tissue using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). The DNA was analyzed by polymerase chain reaction (PCR) as described previously elsewhere (Dawson et al., 2003). At 8 weeks of age, female NaS1+/+ and Nas1−/− mice were anesthetized with pentobarbital sodium, and their livers were perfused briefly with phosphate-buffered saline, dissected, frozen in liquid nitrogen, and stored at −80°C.
Quantitative Reverse Transcription–Polymerase Chain Reaction Analysis of Sult Expression.
Total RNA was prepared from ∼15 mg samples of frozen liver using the RNeasy Mini Kit (Qiagen). Levels of Sult2a1 (RefSeq NM_001111296) mRNA were measured using a SYBR-green-based quantitative reverse transcription–polymerase chain reaction (qRT-PCR) assay that specifically detects this transcript, as previously described elsewhere (Kocarek et al., 2008). Levels of Sult1a1 (Assay ID Mm01132072_m1), Sult1b1 (Mm01213937_m1), Sult1c1 (Mm00450613_m1), Sult1c2 (Mm00471849_m1), Sult1d1 (Mm00502035_m1), Sult1e1 (Mm00499178_m1), Sult2b1 (Mm00450550_m1), Sult3a1 (Mm00491057_m1), Sult4a1 (Mm00489166_m1), Sult6b1 (Mm01233044_m1), PAPSS1 (Mm00442283_m1), and PAPSS2 (Mm00442295_m1) mRNA were measured using the indicated TaqMan Gene Expression Assays (Applied Biosystems/Life Technologies, Grand Island, NY). All TaqMan Gene Expression Assays were performed as described previously elsewhere (Fang et al., 2007) using undiluted (for Sult1a1) or a 1:10 dilution (for all other genes) of reverse transcription reaction as the template and a StepOnePlus Real-Time PCR System (Applied Biosystems/Life Technologies).
Reporter Plasmids.
A fragment of the Sult2a1 gene spanning from −1202 nt (nucleotide) upstream of the transcription start site (as considered by the 5′ position of the Sult2a1 RefSeq NM_001111296) to 46 nt downstream of the transcription start site (which was 8 nt upstream of the translation start site) was prepared by PCR using mouse genomic DNA as the template, the primers shown in Supplemental Table 1, and the HotStar Taq DNA Polymerase (Qiagen). After PCR, the fragment was digested with XhoI and HindIII and ligated into the promoterless pGL4.10[luc2] luciferase reporter plasmid (Promega Corporation, Madison, WI). This reporter plasmid (Sult2a1-1202/+46-pGL4) was used as the template for preparation of additional Sult2a1 PCR fragments containing 628, 324, 233, 179, or 128 nt of 5′-flanking region (primers shown in Supplemental Table 1), and these were also ligated into pGL4.10[luc2]. A plasmid containing a mutation of the IR0 (inverted repeat of AGGTCA, with 0 intervening bases) motif at nt −191 to 180 (GGGTCATGAACT to GGGTCAactAgT) was prepared using the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) and the primers shown in Supplemental Table 1. The indicated mutation introduced an SpeI site (ACTAGT) into the sequence, which facilitated the identification of clones containing the mutation. The sequences of all constructs were verified using the sequencing services of the Applied Genomics Technology Center at Wayne State University (Detroit, MI).
The XREM-CYP3A4-Luc (responsive to PXR), FXRE-Luc (responsive to FXR), and CYP4A1-PPRE-Luc (responsive to PPARα) reporter plasmids have been described elsewhere (Kocarek and Mercer-Haines, 2002). A reporter plasmid containing a 101 nt fragment of the sterol regulatory element-binding protein-1 promoter with an LXR response element (Repa et al., 2000) was prepared by PCR using mouse genomic DNA as the template and the primers shown in Supplemental Table 1. The PCR fragment was digested with MluI and BglII and ligated into the corresponding sites of the pGL3-Promoter luciferase reporter plasmid (Promega). A reporter plasmid containing three copies of the human secreted protein, acidic, cysteine-rich (SPARC) retinoid-related orphan receptor (ROR) response element (RORE) (hereafter designated RORE reporter plasmid) was prepared essentially as described by Chauvet et al. (2011). The oligonucleotides shown in Supplemental Table 1 were annealed and ligated into the XhoI and BglII sites of pGL4.27[luc2P/minP/Hygro] (Promega).
Preparation of PAPSS and SULT2A1 Knockdown HepG2 Cells.
A HepG2 cell clone engineered for stable knockdown of PAPSS1 and PAPSS2 (shPAPSS1/2 cells) was prepared using Mission shRNA Lentiviral Transduction Particles (Sigma-Aldrich, St. Louis, MO). HepG2 cells were first transduced with a recombinant lentivirus expressing PAPSS1 shRNA (TRCN0000010180), selected with 1.5 µg/ml puromycin, and cloned by limiting dilution. A clone with ∼88% reduction of PAPSS1 mRNA level (as determined using TaqMan Gene Expression Assay Hs00968937_m1) relative to the level measured in cells transduced with lentivirus expressing a nontargeting shRNA (shNT cells) was then transduced with lentivirus expressing PAPSS2 shRNA (TRCN0000045487), selected with 70 µg/ml puromycin, and cloned by limiting dilution. A clone with ∼86% reduction of PAPSS2 mRNA level (TaqMan Gene Expression Assay Hs00190682_m1) was selected for use. PAPSS1 and PAPSS2 protein knockdown in this clone was confirmed by Western blot hybridization.
Western blot analysis was performed using 40 μg whole cell lysate protein. Proteins were separated on Precise Protein Gels (4–20%) (Thermo Fisher Scientific, Rockford, IL) and transferred onto polyvinylidene difluoride membranes. PAPSS primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The PAPSS1 antibody (K-14) was an affinity-purified goat polyclonal antibody raised against a peptide mapping within an internal region of PAPSS1 and was used at a dilution of 1:500, and the PAPSS2 antibody (N-16) was an affinity-purified goat polyclonal antibody raised against a peptide mapping near the N-terminus of PAPSS2 and was used at a dilution of 1:800. A horseradish peroxidase-conjugated donkey anti-goat IgG (Santa Cruz Biotechnology) was used as the secondary antibody at a 1:5000 dilution. To demonstrate consistency of loading and transfer, the blots were reprobed with a rabbit polyclonal antibody against glyceraldehyde-3-phosphate dehydrogenase (FL-335; Santa Cruz Biotechnology) at a 1:2000 dilution, and a horseradish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) as the secondary antibody. Immunoreactive proteins were visualized by enhanced chemiluminescence.
To prepare HepG2 cells with reduced expression of SULT2A1 (shSULT2A1 cells), HepG2 cells were transduced with a recombinant lentivirus expressing SULT2A1 shRNA (TRCN0000035696), selected with 1.5 µg/ml puromycin, and cloned by limiting dilution. SULT2A1 mRNA levels were measured using the TaqMan Gene Expression Assay Hs00234219_m1, and SULT2A1 protein levels were measured by Western blot hybridization. Samples of whole cell lysate protein (25 μg) were separated on 4% stacking, 12.5% resolving acrylamide gels, transferred onto polyvinylidene difluoride membranes, and developed using a purified anti-SULT2A1 mouse monoclonal antibody (clone 4D7) from Origene (Rockville, MD) as the primary antibody at a 1:5000 dilution and horseradish peroxidase-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology) as the secondary antibody at a 1:20,000 dilution. The blot was then reprobed with a goat polyclonal antibody against β-tubulin (Abcam, Inc., Cambridge, MA) at 1:1000 dilution and a horseradish peroxidase-conjugated donkey anti-goat IgG (Santa Cruz Biotechnology) diluted 1:8000 to establish consistency of loading and transfer.
Transient Transfection Analysis.
Approximately 250,000 cells were seeded into the wells of 12-well plates and cultured in 1 ml of Dulbecco’s modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin (all purchased from Life Technologies) at 37°C under a humidified atmosphere of 95% air and 5% CO2. Forty-eight hours later, the culture medium was replaced with 1 ml of Opti-MEM I Reduced Serum Medium (Life Technologies). Then 200 µl was added of a premixed complex of 4 µl Lipofectamine 2000 Transfection Reagent (Life Technologies) and plasmid DNA consisting of selected combinations of the following: 1500 ng nuclear receptor-responsive or Sult2a1 firefly luciferase reporter plasmid, 150 ng pGL3-Promoter (Promega), 50 ng of pcDNA3.1 (Life Technologies), 50 ng RORα1/pCMV6-XL5 (Origene), 1 ng pRL-CMV or pRL-SV40 (Promega), and sufficient pBluescript II KS+ (Agilent Technologies) to keep the total amount of DNA constant among samples. The next day, the transfection medium was replaced with 1 ml of fresh supplemented DMEM alone or containing 0.1% dimethylsulfoxide (DMSO), 10 μM GW3965 [3-[3-[N-(2-chloro-3-trifluoromethylbenzyl)-(2,2-diphenylethyl)amino]propyloxy]phenylacetic acid hydrochloride] as the LXR agonist, 10 μM GW4064 [3-(2,6-dichlorophenyl)-4-(3′-carboxy-2-chlorostilben-4-yl)oxymethyl-5-isopropylisoxazole] as the FXR agonist, 10 μM rifampicin as the PXR agonist, or 100 μM ciprofibrate as the PPARα agonist. The cells were harvested the next day for measurement of firefly and Renilla luciferase activities using the Dual Luciferase Reporter Assay System (Promega) and a Promega GloMax Luminometer.
When all data comparisons were to be performed within an individual cell line (i.e., shNT, shPAPSS1/2, or shSULT2A1), firefly luciferase/Renilla luciferase ratios were used to make the comparisons. However, in comparing transfection data between the shNT and shPAPSS1/2 cell lines, we found that the firefly/Renilla luciferase ratio that was obtained after transfection with pGL3-Promoter, which expresses firefly luciferase under control of the SV40 promoter, and pRL-CMV, which expresses Renilla luciferase under control of the CMV promoter, varied among the cell lines. For each cell line, we therefore further normalized the firefly/Renilla ratios to the ratio obtained for the pGL3-Promoter and pRL-CMV and compared these normalized values among the cell lines.
RNA Interference.
The shPAPSS1/2 cells were cultured as described earlier. Forty-eight hours after seeding, the culture medium was replaced with 1 ml of Opti-MEM I and 200 µl of a premixed complex of 2 µl Lipofectamine 2000 (Life Technologies), 10 pmol FXR siRNA or nontargeting siRNA (ON-TARGET plus SMART pools; Thermo Fisher Scientific), 300 ng FXR-responsive luciferase reporter plasmid or Sult2a1 (−128 or −1202) luciferase reporter plasmid, and 1 ng pRL-CMV. Sufficient pBluescript II KS+ (Agilent Technologies) was added to samples not transfected with siRNA to keep the total amount of siRNA + DNA constant. The next day, the transfection medium was replaced with 1 ml fresh DMEM containing 0.1% DMSO or 10 μM GW4064. Cells were harvested 48 hours after transfection for the measurement of firefly and Renilla luciferase activities, as described earlier.
Statistical Analysis.
Data were statistically analyzed using Student’s t test or one-way analysis of variance (ANOVA) followed by the Newman-Keuls multiple comparison test. P< 0.05 was considered statistically significant.
Results
SULT Expression in NaS1 Mouse Liver.
By microarray characterization of the changes in hepatic gene expression in NaS1-null mice relative to their wild-type counterparts, Dawson et al. (2006) reported that Sult2a expression was increased ∼2.0-fold and Sult3a1 was increased ∼5.9-fold. These measurements were performed in 4-week-old male mice. We evaluated SULT expression in the livers of 8-week-old female NaS1-null and wild-type mice because we have previously shown that the levels of most Sult2a isoforms are negligible in the livers of adult male mice (Kocarek et al., 2008). For Sult2a, we used a quantitative PCR assay that specifically detects the murine Sult2a transcript that is most abundant in adult female livers (Kocarek et al., 2008). By this approach, the mean level of Sult2a1 mRNA was ∼51-fold higher in the livers of NaS1-null mice than in NaS1 wild-type mice (Fig. 1).
Fig. 1.
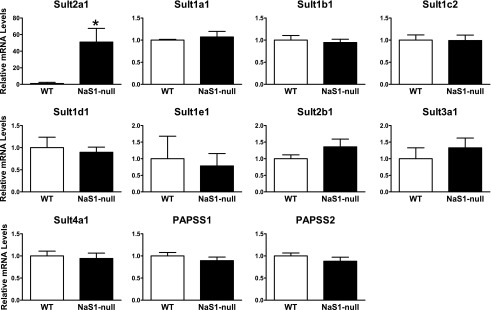
Hepatic levels of Sult mRNAs in female wild-type and NaS1-null mice. Sult mRNA levels were measured in the livers of 8-week-old female wild-type (WT) and NaS1-null mice as described in Materials and Methods. Bars represent the mean relative mRNA levels ± S.E.M. from 9 wild-type mice and 10 NaS1-null mice (except for Sult4a1: 8 wild-type and 9 NaS1-null mice). *Statistically significantly different from wild-type, P < 0.05.
TaqMan Gene Expression assays were used to measure the amounts of other murine SULTs and of PAPSS1 and PAPSS2. Using cycle threshold (Ct) values as approximations of relative transcript abundance, Sult1a1, Sult3a1, and PAPSS2 were most abundant (Ct values of ≤ 28); Sult1b1, Sult1c2, Sult1d1, and PAPSS1 were detectable in all samples (Ct values of ∼30-33); Sult1e1, Sult2b1, and Sult4a1 (reportedly a brain-specific SULT) were detectable in some but not all samples; and Sult1c1 and Sult6b1 were not detectable in most samples. The above results are generally consistent with those reported by Alnouti and Klaassen (2006) using branched DNA signal amplification assays. None of the genes were significantly differently expressed in the NaS1-null relative to the wild-type mouse livers (Fig. 1). Therefore, in the adult female mouse liver, Sult2a1 expression was selectively upregulated in response to NaS1 knockout-mediated hyposulfatemia.
Regulation of the Sult2a1 Promoter in PAPSS-Deficient HepG2 Cells.
We hypothesized that Sult2a1 upregulation is an adaptive response to diminished cellular sulfonation capacity, which occurs because less sulfate is available for the biosynthesis of PAPS, the cofactor for all sulfotransferase-catalyzed reactions. To test this possibility, we developed a hepatic cell model, based on the HepG2 cell line, in which the ability of the cell to biosynthesize PAPS was reduced by RNA interference-mediated knockdown of the PAPS synthase enzymes. Although PAPSS2 is reportedly the major PAPSS expressed in normal liver (Xu et al., 2000), we detected both PAPSS1 and PAPSS2 in HepG2 cells. Therefore, both PAPSS1 and PAPSS2 were targeted for knockdown. Simultaneous knockdown of both enzymes also eliminated the concern that knockdown of one PAPSS enzyme might result in compensatory upregulation of the other PAPSS enzyme. Figure 2 shows the levels of PAPSS1 and PAPSS2 mRNA and immunoreactive protein in HepG2 cells sequentially transduced with lentiviruses expressing shRNAs targeting PAPSS1 and PAPSS2 (shPAPSS1/2 cells) relative to the levels in cells transduced with lentivirus expressing a nontargeting shRNA (shNT cells).
Fig. 2.

Demonstration of PAPSS1 and PAPSS2 knockdown in HepG2 cells. PAPSS1 and PAPSS2 mRNA (A) and immunoreactive protein (B) levels were measured in shNT and shPAPSS1/2 cells using TaqMan Gene Expression Assays and Western blot analysis, respectively. For panel A, each bar represents the mean relative mRNA level ± S.D. from 12 wells of cells (derived by combining two independent experiments with six wells per treatment). ***Significantly different from shNT, P < 0.001.
A series of reporter constructs containing from 128 to 1202 nt of the Sult2a1 promoter was transiently transfected into shNT cells and shPAPSS1/2 cells. When the series was transfected into shNT cells, the promoter activity was not affected by the length of the promoter fragment (Fig. 3A). However, when the series was transfected into shPAPSS1/2 cells, there was a significant increase in promoter activity when the promoter length was increased from 179 to 233 nt (Fig. 3A). The 233 nt fragment contains an IR0 motif (GGGTCATGAACT at nt −191 to −180) that is known to be important for regulation of rat and mouse SULT2A gene transcription by several nuclear receptors, including PXR, FXR, constitutive androstane receptor, and vitamin D receptor (Song et al., 2001; Sonoda et al., 2002; Echchgadda et al., 2004a,b; Saini et al., 2004; Seo et al., 2007), while the 179 nt fragment begins immediately downstream of the IR0. To test the role of the IR0 in mediating Sult2a1 upregulation in response to PAPSS1/2 knockdown, the IR0 was mutated in the context of the 1,202 nt fragment (mutated IR0: GGGTCAactAgT). Figure 3B again shows that Sult2a1 promoter activity was significantly increased when the 5′-flanking region was increased from 128 to 1202 nt, and also shows that the increase was significantly attenuated when the IR0 was mutated. Mutation of the IR0 also reduced the promoter activity in the control shNT cells, indicating that the IR0 contributed to Sult2a1 promoter activity under both basal conditions and conditions of reduced PAPSS1/2 expression (Fig. 3B).
Fig. 3.

Sult2a1 promoter activity in HepG2 cells with normal and reduced PAPSS1/2 expression. (A) The shNT and shPAPSS1/2 cells were transiently transfected with luciferase reporter plasmids containing the indicated amounts of Sult2a1 5′-flanking region (n = 3 wells per group). (B) The shNT and shPAPSS1/2 cells were transiently transfected with reporter plasmid containing 128 or 1202 nt of Sult2a1 5′-flanking region or 1202 nt of Sult2a1 5′-flanking region but a mutated IR0 motif (n = 6 wells per group, derived by combining data from two independent experiments with triplicate transfections). Forty-eight hours after transfection, the cells were harvested for measurement of luciferase activities. For each cell line, each bar represents the mean ± S.D. normalized (firefly/Renilla) luciferase activity relative to the activity that was measured for the reporter containing 128 nt of Sult2a1 5′-flanking region. For shNT cells, the groups not sharing an uppercase letter are statistically significantly different from each other, P < 0.05. For shPAPSS1/2 cells, the groups not sharing a lowercase letter are statistically significantly different from each other, P < 0.05.
Because the transfection data implicated a major role for the IR0 in mediating PAPSS1/2 knockdown-inducible Sult2a1 expression, and because the SULT2A IR0 has been reported to be regulated by several nuclear receptors, we determined the impact of PAPSS1/2 knockdown on the activity of several nuclear receptor signaling pathways by transiently transfecting reporters responsive to LXR, PXR, FXR, ROR, and PPARα into shNT and shPAPSS1/2 cells. PAPSS1/2 knockdown significantly activated the FXR-, ROR-, and PXR-responsive reporters, ∼2.5-, 6.8-, and 1.6-fold, respectively (Fig. 4). We also evaluated the effects of treatments with LXR, PXR, FXR, or PPARα agonists on the reporter containing 1202 nt of the Sult2a1 5′-flanking region, with or without an intact IR0. All treatments produced strong activation of a positive control reporter but only GW4064, an FXR agonist, increased expression of the Sult2a1 reporter, and this effect was attenuated when the IR0 motif was mutated (Fig. 5). The impact of ROR on Sult2a1 promoter activity was assessed by cotransfecting shNT cells with an ROR expression plasmid. While ROR overexpression markedly activated a control ROR-responsive reporter, ROR overexpression did not activate the Sult2a1 reporter (Fig. 5). Taken together, these results suggest that PAPSS1/2 knockdown caused activation of FXR, ROR, and PXR, but only FXR activation is likely to contribute to PAPSS1/2 knockdown-mediated upregulation of Sult2a1.
Fig. 4.

Effect of PAPSS1/2 knockdown on nuclear receptor signaling pathways in HepG2 cells. shNT and shPAPSS1/2 cells were transfected with the indicated nuclear receptor-responsive reporter plasmids. Forty-eight hours after transfection, cells were harvested for measurement of luciferase activities. Each bar represents the mean ± S.D. of the normalized luciferase activities obtained in three separate experiments. For each reporter, the mean value for the shNT group is defined as 1. *Statistically significantly different from shNT cells transfected with the same reporter, P < 0.05.
Fig. 5.
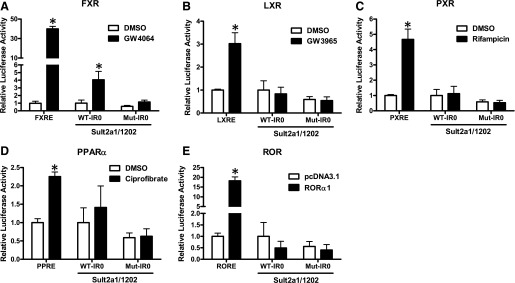
Effects of nuclear receptor activators on Sult2a1 promoter activity in shNT HepG2 cells. The shNT cells were transfected with either the indicated nuclear receptor-responsive reporter plasmid (positive control) or with a Sult2a1 reporter plasmid containing 1202 nt of 5′-flanking region and an intact (WT-IR0) or mutated (Mut-IR0) motif. (A–D) Twenty-four hours after transfection, the cells were treated for 24 hours with 0.1% DMSO or the indicated nuclear receptor agonist (all at 10 µM except ciprofibrate, which was 100 µM). (E) Together with the reporters, cells were transfected with either pcDNA3.1 (empty vector control) or RORα1 expression plasmid. Forty-eight hours after transfection, the cells were harvested for measurement of luciferase activities. Each bar represents the mean ± S.D. of normalized (firefly/Renilla) luciferase activities (n = 6 wells per group, derived by combining data from two independent experiments with triplicate transfections). For transfections with positive control reporters, the mean value for the DMSO-treated or pcDNA3.1-transfected group is defined as 1. For transfections with Sult2a1 reporters, the mean value for the DMSO-treated or pcDNA3.1-transfected group transfected with the Sult2a1 reporter with intact IR0 is defined as 1. *Statistically significantly different from the DMSO-treated or pcDNA3.1-transfected group transfected with the same reporter, P < 0.05.
To evaluate the role of FXR in PAPSS1/2 knockdown-mediated Sult2a1 upregulation specifically, RNAi was used to knock down FXR expression in shPAPSS1/2 cells. Transfection with FXR siRNA markedly reduced GW4064-mediated activation of a cotransfected FXR-responsive reporter, demonstrating effective FXR knockdown (Fig. 6A). Transfection with FXR siRNA significantly reduced Sult2a1 promoter activity in shPAPSS1/2 cells cotransfected with the PAPSS knockdown-responsive reporter containing 1202 nt of the Sult2a1 5′-flanking region but not the reporter containing 128 nt of the 5′-flanking region (Fig. 6B).
Fig. 6.

Effect of FXR knockdown on Sult2a1 promoter activity in shPAPSS1/2 HepG2 cells. (A) The shPAPSS1/2 cells were transfected with an FXR-responsive reporter plasmid. Half the cells were cotransfected with nontargeting (NT) siRNA, and half were cotransfected with siRNA targeting FXR. Twenty-four hours after transfection, the cells were treated with 0.1% DMSO or 10 μM GW4064. (B) The shPAPSS1/2 cells were transfected with reporter plasmid containing 128 or 1202 nt of the Sult2a1 5′-flanking region. Half the cells were cotransfected with NT siRNA, and half were cotransfected with FXR siRNA. Forty-eight hours after transfection, the cells were harvested for measurement of luciferase activities. Results are shown for one representative experiment; comparable results were obtained in two additional experiments. Each bar represents the mean ± S.D. of normalized (firefly/Renilla) luciferase measurements (three wells per treatment). In panel A, the mean value for the DMSO-treated, NT siRNA-transfected group is defined as 1. *Statistically significantly different from the DMSO-treated group transfected with the same siRNA, P < 0.05. In panel B, the mean value for the group transfected with the reporter containing 128 nt of Sult2a1 5′-flanking region and NT siRNA is defined as 1. *Statistically significantly different from the NT siRNA-transfected group transfected with the same reporter, P < 0.05.
PAPSS1/2 knockdown inhibits all cellular sulfonation reactions, including both those catalyzed by SULTs and those catalyzed by the membrane-bound sulfotransferases. Because Sult2a1 promoter activation in PAPSS1/2 knockdown cells was at least partially attributable to activation of the bile acid-sensing nuclear receptor FXR and because SULT2A1 is the only human SULT that catalyzes bile acid sulfonation, we hypothesized that specific knockdown of SULT2A1 might recapitulate the effect of PAPSS1/2 knockdown on Sult2a1 promoter activity. Figure 7A shows successful knockdown of SULT2A1 in a HepG2 clone stably expressing a SULT2A1-targeting shRNA. When shSULT2A1 cells were transiently transfected with the reporter containing 1202 nt of the Sult2a1 5′-flanking region, luciferase activity was significantly increased relative to the activity measured for the reporter containing 128 nt of the 5′-flanking region. When the IR0 motif was mutated, reporter activity was significantly attenuated. Therefore, knockdown of SULT2A1 had the same impact on Sult2a1 promoter activity as did PAPSS1/2 knockdown.
Fig. 7.

Sult2a1 promoter activity in HepG2 cells with normal and reduced SULT2A1 expression. (A) Demonstration of SULT2A1 knockdown in HepG2 cells. SULT2A1 mRNA and immunoreactive protein levels were measured in shNT and shSULT2A1 cells using TaqMan Gene Expression Assays and Western blot analysis, respectively. Each bar represents the mean relative mRNA level ± S.D. from 15–18 wells of cells. ***Statistically significantly different from shNT, P < 0.001. (B) The shNT and shSULT2A1 cells were transiently transfected with reporter plasmid containing 128 or 1202 nt of Sult2a1 5′-flanking region or 1202 nt of Sult2a1 5′-flanking region but a mutated IR0 motif (n = 6 wells per group, derived by combining data from two independent experiments with triplicate transfections). Forty-eight hours after transfection, cells were harvested for measurement of luciferase activities. For each cell line, each bar represents the mean ± S.D. normalized (firefly/Renilla) luciferase activity relative to the activity that was measured for the reporter containing 128 nt of Sult2a1 5′-flanking region. For shNT cells, the groups not sharing an uppercase letter are statistically significantly different from each other, P < 0.05. For shSULT2A1 cells, the groups not sharing a lowercase letter are statistically significantly different from each other, P < 0.05.
Discussion
The importance of sulfate as a determinant of metabolic homeostasis is clearly demonstrated by the impact that loss of the NaS1 transporter has on a host of physiologic processes. NaS1 plays a key role in determining whole body sulfate levels, and much of this effect is mediated in the kidney, where sulfate is freely filtered and reabsorbed in the proximal tubule. NaS1 is expressed on the brush border membrane of proximal tubular cells, and it cotransports sodium and sulfate into the cells (Markovich, 2011b). The sulfate is then transported across the basolateral membrane of the proximal tubular cells into the blood by the sulfate anion transporter (Sat1, slc26a1) (Markovich, 2011b). In mice, NaS1 is also expressed at high levels in intestine (Beck and Markovich, 2000). NaS1-null mice have been shown to exhibit the following changes: 1) reduced renal and intestinal sodium and sulfate cotransport (Dawson et al., 2003); 2) increased urinary sulfate excretion and decreased serum sulfate levels (Dawson et al., 2003); 3) retarded growth accompanied by decreased serum IGF1 levels (Dawson et al., 2003); 4) lower fertility of females (Dawson et al., 2003); 5) development of spontaneous clonic seizures (Dawson et al., 2003); 6) increased hepatic phenol sulfotransferase activity (Dawson et al., 2003); 7) increased serum levels of several bile acids (Dawson et al., 2003); 8) hepatomegaly (Dawson et al., 2003); 9) reduced serum levels of dehydroepiandrosterone and dehydroepiandrosterone sulfate (Dawson et al., 2008); 10) increased hepatic lipid levels (Dawson et al., 2006); 11) increased serum cholesterol and low-density lipoprotein levels and decreased hepatic glycogen levels (Dawson et al., 2006); 12) increased hepatic expression of several genes involved in lipid metabolism as well as two SULT genes, Sult3a1 and Sult2a2 (Dawson et al., 2006); 13) increased sensitivity to acetaminophen-induced hepatotoxicity accompanied by decreased urinary acetaminophen-sulfate to acetaminophen-glucuronide ratio and decreased hepatic glutathione levels, indicating increased oxidative metabolism to the toxic N-acetyl-p-benzoquinone imine metabolite (Lee et al., 2006); 14) altered spatial memory, olfactory function, and behaviors (Dawson et al., 2004, 2005); 15) decreased intestinal sulfomucin content, increased dextran sulfate sodium-induced colitis, and decreased intestinal barrier function (Dawson et al., 2009); 16) increased tumor growth and vascularization and decreased tumor core necrosis, collagen content, and immunoreactivity to heparan sulfate and chondroitin 4-sulfate epitopes (Dawson et al., 2010); and 17) increased life span accompanied by fewer hepatic tumors (Markovich et al., 2011).
In this study, we found that Sult2a1 mRNA was markedly and selectively upregulated in the livers of 8-week-old female NaS1−/− mice relative to NaS1+/+ mice, indicating that this SULT is distinctively sensitive to the hyposulfatemic signal(s) in hepatocytes. In the previous study characterizing hepatic gene expression in NaS1-null mice, gene expression was evaluated in 4-week-old male mice (Dawson et al., 2006). We used female mice because Sult2a expression is known to be higher in adult female relative to adult male mice, and this may explain why we observed a much larger effect on Sult2a expression than was previously observed. In addition, we used a quantitative reverse transcription–polymerase chain reaction (qRT-PCR) assay designed to measure Sult2a1 mRNA selectively relative to other Sult2a subfamily members (Kocarek et al., 2008).
The hyposulfatemia phenotype was accompanied by reduced serum levels of dehydroepiandrosterone sulfate (Dawson et al., 2008) as well as by decreased production of acetaminophen-sulfate after acetaminophen administration (Lee et al., 2006), suggesting that NaS1-null mice had decreased capacity for catalyzing sulfonation reactions. Because the sulfotransferase reaction is dependent not only on cellular sulfate levels but also the availability of the obligate cofactor PAPS, we hypothesized that the Sult2a upregulation that occurred during hyposulfatemia was an adaptive response of the hepatocyte to loss of PAPS. To test this hypothesis, we developed a HepG2 model of reduced hepatic sulfonation capacity by knocking down both of the PAPSS enzymes that catalyze formation of PAPS.
We found that Sult2a1 promoter activity was increased in shPAPSS1/2 cells when the 5′-flanking sequence contained an intact IR0 motif. This upregulation was at least partially attributable to FXR activation as indicated by 1) activation of an FXR-responsive reporter in shPAPSS1/2 cells, 2) activation of the Sult2a1 promoter by both PAPSS knockdown and an FXR agonist, and reduction of these responses when the IR0 motif was mutated, and 3) attenuation of Sult2a1 promoter activation by FXR knockdown in shPAPSS1/2 cells. The upregulation after reduction of global sulfonation capacity was also at least partially attributable to loss of SULT2A1 activity because SULT2A1 knockdown also led to activation of the Sult2a1 promoter, and the response was attenuated when the IR0 motif was mutated. Because SULT2A1 is known to catalyze bile acid sulfonation (Radominska et al., 1990) and FXR is a bile acid-activated nuclear receptor (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999), it seems likely that loss of bile acid sulfonation and accumulation of nonsulfonated bile acids causes FXR activation and Sult2a1 promoter activation in an attempt to restore bile acid homeostasis.
FXR has been shown to play a role in maintaining sulfate homeostasis in the mouse kidney and intestine. Lee et al. (2007) reported significantly higher renal NaS1 and Sult2a1 mRNA levels in mice after treatment with GW4064 or α-naphthylisothiocyanate, a hepatotoxin that is known to increase circulating bile acid levels, and identified NaS1 as a novel target gene of FXR. When FXR was knocked out, basal expression of renal and intestinal NaS1 was significantly reduced, and α-naphthylisothiocyanate treatment did not increase NaS1 expression. Renal basal expression of Sult2a1 mRNA was also significantly lower in FXR knockout mice. These data suggest that when circulating bile acids are elevated, FXR activation in the renal tubular cells causes increased reabsorption of sulfate and sulfotransferase capacity to increase the excretion of sulfonated-bile acids in the urine (Lee et al., 2007). Thus, a similar mechanism may occur in the hepatocytes to normalize bile acid levels.
Our data indicate that FXR and the IR0 play major roles in the activation of Sult2a1 promoter activity that occurs after PAPSS1/2 or SULT2A1 knockdown but also suggest that the FXR-IR0 interaction is not the sole mediator of the increase because neither knockdown of FXR nor mutation of the IR0 abolished the increase. Mutation of the IR0 also significantly reduced Sult2a1 promoter activity in the control shNT cells, which might be attributable to a small amount of FXR activation that is produced by the basal level of unconjugated bile acids in HepG2 cells.
It is noteworthy that endogenous SULT2A1 expression was not increased by PAPSS1/2 knockdown in the HepG2 model (unpublished data), demonstrating an important species difference in SULT2A regulation. SULT2A1 lacks an IR0 motif, and regulation of hepatic SULT2A by nuclear receptors that use the IR0, such as PXR and FXR, is different in human and rodent hepatic cells. For example, Song et al. (2001) reported that the potent bile acid agonist of FXR chenodeoxycholic acid activated rat SULT2A promoter activity in transfected HepG2 and Caco-2 cells through the IR0 motif, and Miyata et al. (2006) reported that treatment with chenodeoxycholic acid or GW4046 suppressed SULT2A1 expression in HepG2 cells. Also PXR activates rat and mouse SULT2A expression through the IR0 motif (Runge-Morris et al., 1999; Sonoda et al., 2002; Echchgadda et al., 2004a) but suppresses expression of human SULT2A1 in primary cultured human hepatocytes (Fang et al., 2007). In a microarray experiment to identify genes that are regulated by PAPSS1/2 knockdown, we noted that UDP-glucuronosyltransferase 2B4 (UGT2B4) expression was upregulated (Barrett et al., manuscript in preparation). Because UDP-glucuronosyltransferase 2B4 is known to glucuronidate some bile acids, activation of bile acid glucuronidation, rather than sulfonation, might be the way that human hepatic cells respond to loss of bile acid sulfonation.
PAPSS1/2 knockdown also caused activation of ROR signaling. However, increased ROR signaling, as achieved by overexpression of RORα1, did not cause Sult2a1 promoter activation. Kang et al. (2007) reported that Sult2a1 expression was markedly (43.5-fold) increased in the livers of RORα/RORγ double knockout mice relative to wild-type controls. Therefore, ROR appears to be a suppressor rather than an activator of Sult2a1 expression.
In this study, we have focused on one mechanism that could explain the increased hepatic Sult2a1 expression that occurs in NaS1-null mice and acknowledge that there might be others. For example, loss of sulfate could also be expected to impact sulfur-containing amino acids, including cysteine, a component of glutathione. Therefore, loss of glutathione could also contribute to the impact of sulfate depletion on hepatic gene expression. Studies to identify additional mechanisms by which hyposulfatemic regulates human hepatic gene expression are ongoing.
Supplementary Material
Abbreviations
- Ct
cycle threshold
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethylsulfoxide
- FXR
farnesoid X receptor
- GW3965
3-[3-[N-(2-chloro-3-trifluoromethylbenzyl)-(2,2-diphenylethyl)amino]propyloxy]phenylacetic acid hydrochloride
- GW4064
3-(2,6-dichlorophenyl)-4-(3′-carboxy-2-chlorostilben-4-yl)oxymethyl-5-isopropylisoxazole
- IR0
inverted repeat of AGGTCA, with 0 intervening bases
- LXR
liver X receptor
- nt
nucleotide
- NT
nontargeting
- PAPS
3′-phosphoadenosine-5′-phosphosulfate
- PAPSS
3′-phosphoadenosine-5′-phosphosulfate synthase
- PCR
polymerase chain reaction
- PPARα
peroxisome proliferator-activated receptor α
- PXR
pregnane X receptor
- ROR
retinoid-related orphan receptor
- RORE
retinoid-related orphan receptor response element
- siRNA
small interfering RNA
- shPAPSS1/2
knocked down PAPS synthases 1 and 2 in HepG2 cells
- SULT
cytosolic sulfotransferase
- SULT2A
hydroxysteroid sulfotransferase
- Sult2a1
mouse cytoplasmic sulfotransferase 2a1
- SULT2A1
human cytoplasmic sulfotransferase 2A1.
Authorship Contributions
Participated in research design: Barrett, Kocarek, Runge-Morris.
Conducted experiments: Barrett, Fang, Gargano.
Contributed new reagents or analytic tools: Markovich.
Performed data analysis: Barrett, Gargano, Kocarek.
Wrote or contributed to the writing of the manuscript: Barrett, Markovich, Kocarek, Runge-Morris.
Footnotes
This research was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grant R01-ES005823] (to M.R.-M.).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Alnouti Y, Klaassen CD. (2006) Tissue distribution and ontogeny of sulfotransferase enzymes in mice. Toxicol Sci 93:242–255 [DOI] [PubMed] [Google Scholar]
- Beck L, Markovich D. (2000) The mouse Na+-sulfate cotransporter gene Nas1. Cloning, tissue distribution, gene structure, chromosomal assignment, and transcriptional regulation by vitamin D. J Biol Chem 275:11880–11890 [DOI] [PubMed] [Google Scholar]
- Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. (2004) Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1α. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem 279:45139–45147 [DOI] [PubMed] [Google Scholar]
- Chauvet C, Vanhoutteghem A, Duhem C, Saint-Auret G, Bois-Joyeux B, Djian P, Staels B, Danan JL. (2011) Control of gene expression by the retinoic acid-related orphan receptor alpha in HepG2 human hepatoma cells. PLoS ONE 6:e22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook IT, Duniec-Dmuchowski Z, Kocarek TA, Runge-Morris M, Falany CN. (2009) 24-Hydroxycholesterol sulfation by human cytosolic sulfotransferases: formation of monosulfates and disulfates, molecular modeling, sulfatase sensitivity, and inhibition of liver x receptor activation. Drug Metab Dispos 37:2069–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Beck L, Markovich D. (2003) Hyposulfatemia, growth retardation, reduced fertility, and seizures in mice lacking a functional NaSi-1 gene. Proc Natl Acad Sci USA 100:13704–13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Choyce A, Chuang C, Whitelock J, Markovich D, Leggatt GR. (2010) Enhanced tumor growth in the NaS1 sulfate transporter null mouse. Cancer Sci 101:369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Gardiner B, Grimmond S, Markovich D. (2006) Transcriptional profile reveals altered hepatic lipid and cholesterol metabolism in hyposulfatemic NaS1 null mice. Physiol Genomics 26:116–124 [DOI] [PubMed] [Google Scholar]
- Dawson PA, Gardiner B, Lee S, Grimmond S, Markovich D. (2008) Kidney transcriptome reveals altered steroid homeostasis in NaS1 sulfate transporter null mice. J Steroid Biochem Mol Biol 112:55–62 [DOI] [PubMed] [Google Scholar]
- Dawson PA, Huxley S, Gardiner B, Tran T, McAuley JL, Grimmond S, McGuckin MA, Markovich D. (2009) Reduced mucin sulfonation and impaired intestinal barrier function in the hyposulfataemic NaS1 null mouse. Gut 58:910–919 [DOI] [PubMed] [Google Scholar]
- Dawson PA, Steane SE, Markovich D. (2004) Behavioural abnormalities of the hyposulphataemic Nas1 knock-out mouse. Behav Brain Res 154:457–463 [DOI] [PubMed] [Google Scholar]
- Dawson PA, Steane SE, Markovich D. (2005) Impaired memory and olfactory performance in NaSi-1 sulphate transporter deficient mice. Behav Brain Res 159:15–20 [DOI] [PubMed] [Google Scholar]
- Echchgadda I, Song CS, Oh TS, Cho SH, Rivera OJ, Chatterjee B. (2004a) Gene regulation for the senescence marker protein DHEA-sulfotransferase by the xenobiotic-activated nuclear pregnane X receptor (PXR). Mech Ageing Dev 125:733–745 [DOI] [PubMed] [Google Scholar]
- Echchgadda I, Song CS, Roy AK, Chatterjee B. (2004b) Dehydroepiandrosterone sulfotransferase is a target for transcriptional induction by the vitamin D receptor. Mol Pharmacol 65:720–729 [DOI] [PubMed] [Google Scholar]
- Falany CN. (1997) Enzymology of human cytosolic sulfotransferases. FASEB J 11:206–216 [DOI] [PubMed] [Google Scholar]
- Fang HL, Strom SC, Cai H, Falany CN, Kocarek TA, Runge-Morris M. (2005) Regulation of human hepatic hydroxysteroid sulfotransferase gene expression by the peroxisome proliferator-activated receptor alpha transcription factor. Mol Pharmacol 67:1257–1267 [DOI] [PubMed] [Google Scholar]
- Fang HL, Strom SC, Ellis E, Duanmu Z, Fu J, Duniec-Dmuchowski Z, Falany CN, Falany JL, Kocarek TA, Runge-Morris M. (2007) Positive and negative regulation of human hepatic hydroxysteroid sulfotransferase (SULT2A1) gene transcription by rifampicin: roles of hepatocyte nuclear factor 4α and pregnane X receptor. J Pharmacol Exp Ther 323:586–598 [DOI] [PubMed] [Google Scholar]
- Hurst CH, Waxman DJ. (2004) Environmental phthalate monoesters activate pregnane X receptor-mediated transcription. Toxicol Appl Pharmacol 199:266–274 [DOI] [PubMed] [Google Scholar]
- Kang HS, Angers M, Beak JY, Wu X, Gimble JM, Wada T, Xie W, Collins JB, Grissom SF, Jetten AM. (2007) Gene expression profiling reveals a regulatory role for ROR alpha and ROR gamma in phase I and phase II metabolism. Physiol Genomics 31:281–294 [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, et al. (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92:73–82 [DOI] [PubMed] [Google Scholar]
- Kocarek TA, Duanmu Z, Fang HL, Runge-Morris M. (2008) Age- and sex-dependent expression of multiple murine hepatic hydroxysteroid sulfotransferase (SULT2A) genes. Biochem Pharmacol 76:1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocarek TA, Mercer-Haines NA. (2002) Squalestatin 1-inducible expression of rat CYP2B: evidence that an endogenous isoprenoid is an activator of the constitutive androstane receptor. Mol Pharmacol 62:1177–1186 [DOI] [PubMed] [Google Scholar]
- Kodama S, Moore R, Yamamoto Y, Negishi M. (2007) Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene. Biochem J 407:373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landes N, Birringer M, Brigelius-Flohé R. (2003) Homologous metabolic and gene activating routes for vitamins E and K. Mol Aspects Med 24:337–344 [DOI] [PubMed] [Google Scholar]
- Lee H, Hubbert ML, Osborne TF, Woodford K, Zerangue N, Edwards PA. (2007) Regulation of the sodium/sulfate co-transporter by farnesoid X receptor alpha. J Biol Chem 282:21653–21661 [DOI] [PubMed] [Google Scholar]
- Lee S, Dawson PA, Hewavitharana AK, Shaw PN, Markovich D. (2006) Disruption of NaS1 sulfate transport function in mice leads to enhanced acetaminophen-induced hepatotoxicity. Hepatology 43:1241–1247 [DOI] [PubMed] [Google Scholar]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. (1998) The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest 102:1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Ruuska SE, Shaw NS, Dong D, Noy N. (1999) Ligand selectivity of the peroxisome proliferator-activated receptor alpha. Biochemistry 38:185–190 [DOI] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284:1362–1365 [DOI] [PubMed] [Google Scholar]
- Markovich D. (2011a) Physiological roles of mammalian sulfate transporters NaS1 and Sat1. Arch Immunol Ther Exp (Warsz) 59:113–116 [DOI] [PubMed] [Google Scholar]
- Markovich D. (2011b) Physiological roles of renal anion transporters NaS1 and Sat1. Am J Physiol Renal Physiol 300:F1267–F1270 [DOI] [PubMed] [Google Scholar]
- Markovich D, Ku MC, Muslim D. (2011) Increased lifespan in hyposulfatemic NaS1 null mice. Exp Gerontol 46:833–835 [DOI] [PubMed] [Google Scholar]
- Miyata M, Watase H, Hori W, Shimada M, Nagata K, Gonzalez FJ, Yamazoe Y. (2006) Role for enhanced faecal excretion of bile acid in hydroxysteroid sulfotransferase-mediated protection against lithocholic acid-induced liver toxicity. Xenobiotica 36:631–644 [DOI] [PubMed] [Google Scholar]
- Moreau A, Téruel C, Beylot M, Albalea V, Tamasi V, Umbdenstock T, Parmentier Y, Sa-Cunha A, Suc B, Fabre JM, et al. (2009) A novel pregnane X receptor and S14-mediated lipogenic pathway in human hepatocyte. Hepatology 49:2068–2079 [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284:1365–1368 [DOI] [PubMed] [Google Scholar]
- Radominska A, Comer KA, Zimniak P, Falany J, Iscan M, Falany CN. (1990) Human liver steroid sulphotransferase sulphates bile acids. Biochem J 272:597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev 14:2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge-Morris M, Wu W, Kocarek TA. (1999) Regulation of rat hepatic hydroxysteroid sulfotransferase (SULT2-40/41) gene expression by glucocorticoids: evidence for a dual mechanism of transcriptional control. Mol Pharmacol 56:1198–1206 [DOI] [PubMed] [Google Scholar]
- Saini SPS, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W. (2004) A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol Pharmacol 65:292–300 [DOI] [PubMed] [Google Scholar]
- Seo Y-K, Chung Y-T, Kim S, Echchgadda I, Song CS, Chatterjee B. (2007) Xenobiotic- and vitamin D-responsive induction of the steroid/bile acid-sulfotransferase Sult2A1 in young and old mice: the role of a gene enhancer in the liver chromatin. Gene 386:218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CS, Echchgadda I, Baek BS, Ahn SC, Oh T, Roy AK, Chatterjee B. (2001) Dehydroepiandrosterone sulfotransferase gene induction by bile acid activated farnesoid X receptor. J Biol Chem 276:42549–42556 [DOI] [PubMed] [Google Scholar]
- Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. (2002) Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR). Proc Natl Acad Sci USA 99:13801–13806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, et al. (2001) The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA 98:3369–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam KV. (2003) Human 3′-phosphoadenosine 5′-phosphosulfate (PAPS) synthase: biochemistry, molecular biology and genetic deficiency. IUBMB Life 55:1–11 [DOI] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3:543–553 [DOI] [PubMed] [Google Scholar]
- Xu ZH, Otterness DM, Freimuth RR, Carlini EJ, Wood TC, Mitchell S, Moon E, Kim UJ, Xu JP, Siciliano MJ, et al. (2000) Human 3′-phosphoadenosine 5′-phosphosulfate synthetase 1 (PAPSS1) and PAPSS2: gene cloning, characterization and chromosomal localization. Biochem Biophys Res Commun 268:437–444 [DOI] [PubMed] [Google Scholar]
- Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, et al. (2008) Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology 134:556–567 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.