

Abstract
The purpose of this study was to determine the impact of CYP3A5 expression on inhibitory potency (Ki or IC50 values) of CYP3A inhibitors, using recombinant CYP3A4 and CYP3A5 (rCYP3A4 and rCYP3A5) and CYP3A5 genotyped human liver microsomes (HLMs). IC50 ratios between rCYP3A4 and rCYP3A5 (rCYP3A5/rCYP3A4) of ketoconazole (KTZ) and itraconazole (ITZ) were 8.5 and 8.8 for midazolam (MDZ), 4.7 and 9.1 for testosterone (TST), 1.3 and 2.8 for terfenadine, and 0.6 and 1.7 for vincristine, respectively, suggesting substrate- and inhibitor-dependent selectivity of the two azoles. Due to the difference in the IC50 values for CYP3A4 and CYP3A5, nonconcordant expression of CYP3A4 and CYP3A5 protein can significantly affect the observed magnitude of CYP3A-mediated drug-drug interactions in humans. Indeed, the IC50 values of KTZ and ITZ for CYP3A-catalyzed MDZ and TST metabolism were significantly higher in HLMs with CYP3A5*1/*1 and CYP3A5*1/*3 genotypes than in HLMs with the CYP3A5*3/*3 genotype, showing CYP3A5 expression–dependent IC50 values. Moreover, when IC50 values of KTZ and ITZ for different HLMs were kinetically simulated based on CYP3A5 expression level and enzyme-specific IC50 values, a good correlation between the simulated and the experimentally measured IC50 values was observed. Further simulation analysis revealed that both the Ki ratio (for inhibitors) and Vmax/Km ratio (for substrates) between CYP3A4 and CYP3A5 were major factors for CYP3A5 expression–dependent IC50 values. In conclusion, the present study demonstrates that CYP3A5 genotype and expression level have a significant impact on inhibitory potency for CYP3A-catalyzed drug metabolism, but that the magnitude of its effect is inhibitor-substrate pair specific.
Introduction
CYP3A is an important cytochrome P450 (P450) subfamily, because its members are expressed in many tissues and metabolize more than 50% of marketed drugs. Importantly, CYP3A4 is highly expressed in human intestine and liver, and is often the locus of drug-drug interactions (DDIs) (Lown et al., 1994; Thummel and Wilkinson, 1998). A structurally related isoform, CYP3A5, is polymorphically expressed in the human intestine and liver in different populations and exhibits an overlapping substrate specificity with that of CYP3A4 (Lamba et al., 2002; Huang et al., 2004). Consequently, it has become increasingly necessary to consider the impact of “CYP3A5 expresser” status on the pharmacokinetic and DDI profiles of CYP3A substrates (Isoherranen et al., 2008; Chandel et al., 2009).
With greater access to recombinant CYP3A (rCYP3A) proteins and human liver microsome (HLM) preparations of CYP3A5-genotyped subjects, attempts have been made to compare the catalytic and inhibitory properties of CYP3A4 with those of CYP3A5 (Williams et al., 2002; Huang et al., 2004; Yamaori et al., 2004). It is accepted that the majority of CYP3A substrates favor CYP3A4 when the two P450s are incubated under similar assay conditions and one carefully considers their catalytic efficiency in light of redox partner molar ratios (Jushchyshyn et al., 2005; Christensen et al., 2011; Lee and Goldstein, 2012). To date, only drugs such as vincristine (rCYP3A5/rCYP3A4 Vmax/Km ratio ≥ 8) and tacrolimus (rCYP3A5/rCYP3A4 Vmax/Km ratio ∼2) favor rCYP3A5 (Dai et al., 2006; Dennison et al., 2006; Niwa et al., 2008). In agreement, CYP3A5 expresser status is a major determinant of vincristine M1 formation in vitro and in vivo (Dennison et al., 2007, 2008; Egbelakin et al., 2011). Likewise, the respective in vivo (oral) clearance and the HLM-scaled clearance of tacrolimus are 48 and ∼60% lower in CYP3A5 nonexpressers, compared with expressors (Dai et al., 2006; Barry and Levine, 2010).
From the standpoint of CYP3A inhibition, azole antifungal agents have been well studied (Gibbs et al., 1999; Isoherranen et al., 2008; Niwa et al., 2008). With some substrates, the IC50 ratio (rCYP3A5/rCYP3A4) has been reported to be as high as 19 and implicates CYP3A5 expresser status as an important determinant of Ki in human livers (Patki et al., 2003; Isoherranen et al., 2008; Niwa et al., 2008). Such an observation is supported by the ∼2-fold increase in fluconazole Ki with CYP3A5 expresser (versus CYP3A5 nonexpresser) HLM (Isoherranen et al., 2008). There are reports that itraconazole (ITZ) is rCYP3A4 selective with substrates such as oxybutynin and midazolam (MDZ) (Lukkari et al., 1998; Yamazaki et al., 2010). The latter authors reported a Ki ratio of ∼30 (rCYP3A5/rCYP3A4) as well as a Ki ratio of ∼2 in HLMs (CYP3A5 expressers versus nonexpressers).
Inheritance of at least one copy of the reference CYP3A5 allele (CYP3A5*1) confers a significant level of tissue CYP3A5 content, whereas inheritance of two variant alleles (e.g., CYP3A5*3, CYP3A5*6, or CYP3A5*7) leads to undetectable or very low CYP3A5 expression (Kuehl et al., 2001). Therefore, the data described earlier suggest that CYP3A5 expresser status, although not necessarily impacting the overall pharmacokinetics of a particular CYP3A substrate, may govern the inhibitor concentration ([I])/Ki value and the magnitude of a DDI. Clinically, there are at least four reports linking CYP3A5 genotype to the magnitude of CYP3A inhibition: the impact of ketoconazole (KTZ) on tacrolimus exposure (Chandel et al., 2009), effect of ITZ on the pharmacokinetics of intravenous MDZ (Yu et al., 2004), impact of ITZ on the pharmacological activity of clopidogrel (Suh et al., 2006), and the inhibition of MDZ clearance by fluconazole (Isoherranen et al., 2008). Interestingly, Isoherranen et al. (2008) also evaluated the inhibitory effect of fluconazole on erythromycin N-demethylase activity in HLM and found that its Ki was not affected by CYP3A5 expression. Clinical follow-up revealed no significant association between CYP3A5 genotype and the magnitude of effect of fluconazole on the erythromycin breath test. This raises the possibility that the impact of CYP3A5 expresser status on the magnitude of a CYP3A-mediated DDI may be substrate-dependent (Isoherranen et al., 2008).
In the present study, we aimed to determine the effect of CYP3A5 expression on the potency of inhibition of CYP3A-catalyzed drug metabolism by means of in vitro studies using rCYP3A4 and rCYP3A5 and nine CYP3A5-genotyped HLMs together with four representative CYP3A substrates [MDZ, testosterone (TST), terfenadine, and vincristine] and two inhibitors (ITZ and KTZ). It is concluded that IC50 values for inhibition of CYP3A in HLM are indeed greatly dependent on the expression levels of CYP3A5, and that the quantitative impact of CYP3A5 expression is inhibitor-substrate pair–specific.
Materials and Methods
Chemicals and Reagents
Microsomes containing human rCYP3A4 and rCYP3A5 (Supersomes) were obtained from BD Biosciences (San Jose, CA). rCYP3A4 (1.0 nmol CYP3A4/mg) had been coexpressed with cytochrome b5 (2.8 nmol/mg) and P450 reductase (2.0 µmol cytochrome c reduced/min/mg). rCYP3A5 (1.0 nmol CYP3A5/mg) had been coexpressed with cytochrome b5 (2.1 nmol/mg) and P450 reductase (3.2 µmol cytochrome c reduced/min/mg). Terfenadine, vincristine, ITZ, and KTZ were purchased from Sigma-Aldrich (St. Louis, MO). MDZ and TST were purchased from Cerilliant (Round Rock, TX). All other general reagents were of analytical grade or better and were obtained from various commercial sources. Wild-type purified human CYP3A4, mutant purified human CYP3A4 (L211F/D214E and F213W), P450 reductase, and cytochrome b5 were provided by Drs. James Halpert and Dmitri Davydov (Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, CA).
rCYP3A4- and rCYP3A5-Catalyzed Metabolism of TST, MDZ, Terfenadine, and Vincristine
Assessment of CYP3A4 and CYP3A5 Activity.
All incubations were conducted at 37°C as follows. For the determination of enzyme kinetic parameters, MDZ (0.5–25 µM) was incubated in a reaction mixture (final volume of 0.2 ml) that consisted of the following: rCYP3A4 or rCYP3A5 Supersomes (1 pmol P450/ml), potassium phosphate buffer (100 mM, pH 7.4), EDTA (1 mM), and NADPH (1 mM) for 5 minutes. The apparent product formation rate with respect to incubation time and protein concentration had previously been determined to be linear. Reactions were terminated by quenching with 1 volume of cold acetonitrile containing internal standard, 1′-hydroxy midazolam d4 (1′OH-MDZ-d4). After centrifugation at 4000g for 10 minutes, aliquots of the supernatants were subject to liquid chromatography–tandem mass spectrometry (LC/MS/MS) analysis. Analysis of 1′OH-MDZ in the incubate extracts was performed using an Accela ultra high–performance liquid chromatography (UHPLC) chromatographic system interfaced with an Orbitrap Discovery Mass Spectrometer (Thermo Fisher, Waltham, MA). Samples (10 µl) were injected via a CTC PAL autosampler (Leap Technologies, Carrboro, NC) into a BEH C18 1.7-µm 2.1 × 100–mm column (Waters Corporation, Milford, MA). The mobile phases were composed of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) at a flow rate of 0.6 ml/min. A gradient linearly increased the composition of (B) from 10 to 40% over 6 minutes, followed by an increase to 95% over the next 2.5 minutes, maintenance at 95% for 1 minute, and then a return to initial conditions. Detection of 1′OH-MDZ was achieved in the positive ion electrospray (ESI) mode, with a source voltage of 5.0 kV, a capillary voltage of 15.5 V, and a capillary temperature of 235°C. Tube voltage was set at 50 V. Data acquisition was achieved by collecting full-scan accurate mass (resolution of 30,000) mass spectra between m/z (mass-to-charge ratio) 100 and 1000. Ion chromatograms of the exact masses of the MH+ ions of 1′-OH-MDZ (m/z 342.08103) and 1′-OH-MDZ-d4 (m/z 346.10572) were extracted with a mass window of 10 ppm. Peaks in the ion chromatograms were integrated using XCalibur Quan Browser software (Thermo Fisher). The retention times of 1′-OH-MDZ and 1′-OH-MDZ-d4 were 4.8 minutes, whereas 4-hydroxy MDZ eluted at 4.5 minutes.
For TST (5–500 μM), enzyme kinetic parameters were determined after incubation with rCYP3A4 or rCYP3A5 Supersomes (10 pmol P450/ml) in potassium phosphate buffer (100 mM, pH 7.4) containing EDTA (1 mM) and NADPH (1 mM) (final volume of 0.2 ml). Incubation time was 10 minutes. Again, the apparent product formation rate with respect to incubation time and protein concentration had previously been determined to be linear. Reactions were terminated by transfer of the incubation contents into a Solvinert hydrophobic polytetrafluoroethylene filter plate (Millipore, Billerica, MA) preloaded with 3 volumes of cold methanol containing internal standard, 6β-hydroxy testosterone d3 (6βOH-TST-d3). The filter plate was stacked with a 2-ml injection plate, centrifuged, and the filtrates were subjected to LC/MS/MS analysis. Analysis of 6βOH-TST in incubate extracts was performed using an SCL-10A vp pump (Shimadzu, Columbia, MD) connected to an API4000 Qtrap (AB Sciex, Vaughan, ON, Canada). Samples (10 µl) were injected via a CTC PAL autosampler (Leap Technologies) onto a Zorbax (Agilent Technologies, Palo Alto, CA) SB-C18 5-µm, 2.1 × 150–mm column. The mobile phases were composed of 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B) at a flow rate of 0.6 ml/min. A gradient linearly increased the composition of (B) from 35 to 80% over 3.5 minutes, followed by an increase to 100% over the next 0.5 minutes, maintenance at 100% for 1 minute, and then a return to initial conditions. Detection of 6βOH-TST was achieved in the ESI multiple reaction monitoring mode by monitoring m/z transitions of 305.2 → 269.2 (6βOH-TST; retention time = 3.04 minutes) and 308.2 → 272.2 (6βOH-TST-d3; retention time = 3.03 minutes). The declustering potential was set at 66 V, the collision energy was set at 21 V, and the turbo-V source temperature was set at 400°C. Analyst software version 1.4.2 was used for data analysis (Applied Biosystems Inc., Foster City, CA).
For the determination of enzyme kinetic parameters, terfenadine was incubated for 1 minute in a mixture (final volume of 0.2 ml) consisting of the following: rCYP3A4 or rCYP3A5 Supersomes (5 pmol P450/ml), potassium phosphate buffer (100 mM, pH 7.4), EDTA (1 mM), and NADPH (1 mM). The product formation rate with respect to the incubation time and protein concentration had previously been determined to be linear. Reactions were terminated by quenching with 1 volume of cold acetonitrile containing internal standard, 1′OH-MDZ-d4. After centrifugation at 4000g for 10 minutes, aliquots of the supernatants were subject to LC/MS/MS analysis. Analysis of terfenadine alcohol in the incubate extracts was performed using an Accela UHPLC chromatographic system interfaced with an Orbitrap Discovery Mass Spectrometer (Thermo Fisher). Samples (10 µl) were injected via a CTC PAL autosampler (Leap Technologies) onto a BEH C18 1.7-µm 2.1 × 100–mm column (Waters Corporation). The mobile phases were composed of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) at a flow rate of 0.6 ml/min. A gradient linearly increased the composition of (B) from 2 to 60% over 8 minutes and up to 95% over the next 0.5 minutes, maintained it at 95% for 0.5 minutes, and then returned it to initial conditions. Detection of terfenadine alcohol was achieved in the ESI mode, with a source voltage of 5.0 kV, a capillary voltage of 15.5 V, and a capillary temperature of 235°C. Tube lens voltage was set at 50 V. Data acquisition was achieved by collection of full-scan spectrum (mass resolution 30,000) over a range of 100–1000 amu. Extracted ion chromatograms of terfenadine alcohol (MH+ 488.31665) and internal standard 1′-OH-MDZ-d4 (MH+ 346.10572) were integrated. The retention times of terfenadine alcohol and 1′-OH-MDZ-d4 were 6.3 and 5.3 minutes, respectively.
For the determination of enzyme kinetic parameters, vincristine was incubated in a reaction mixture that contained rCYP3A4 (50 pmol P450/ml) or rCYP3A5 (10 pmol P450/ml) for 10 minutes, 5 minutes, and 3 minutes, respectively. The incubation mixture (final volume of 0.2 ml) also contained potassium phosphate buffer (100 mM, pH 7.4), MgCl2 (1 mM), and NADPH (0.5 mM). The apparent reaction rate was linear with respect to incubation time and protein concentration. Reactions were terminated by the addition of 1 volume of ice-cold acetonitrile. Concentrations of vincristine metabolite (M1) were determined using UHPLC-mass spectrometry analysis (Dennison et al., 2006). Analysis of vincristine and M1 in the incubate extracts was performed using an Accela UHPLC pump connected to an LTQ Orbitrap Velos (Thermo Fisher). Samples (10 µl) were injected via a CTC PAL autosampler (Leap Technologies) onto a BEH C18 1.7-µm, 2.1 × 100–mm UPLC column (Waters Corporation). The mobile phases were composed of 0.1% formic acid in water (A) and methanol (B). A gradient linearly increased the composition of (B) from 10 to 40% over 6 minutes, followed by an increase to 95% over the next 2.5 minutes. Vincristine eluted at 6.2 minutes, and metabolite M1 at 5.7 minutes. The column eluted into the electrospray ion source, heated at 300°C and operated in the positive mode. Source voltage was set at 5.4 kV. The capillary temperature was set at 300°C. Sheath and auxiliary gases were operated at 50 and 15 arbitrary units, respectively. Quantitation of vincristine and M1 was accomplished by integration of peak areas in the respective accurate mass ion extracted chromatograms. Masses used were 413.20776 for vincristine and 406.19991 for M1, with windows of 5 ppm. These masses represent the doubly charged states of both molecules and are more abundant than the singly charged species under the conditions described.
Determination of Kinetic Parameters.
All kinetic data were analyzed by nonlinear regression analysis (KaleidaGraph 4.1; Synergy Software, Reading, PA), which involved fitting of the data to equations describing Michaelis-Menten (eq. 1) or sigmoidal (eq. 2) kinetics. In each case, the goodness of the fit was assessed based on visual inspection, assessment of χ2, and the standard error of the parameter estimates.
| (1) |
| (2) |
In eqs. 1 and 2, v is the velocity of the metabolic reaction, [S] is the substrate concentration, and n is the Hill coefficient.
Inhibition of rCYP3A4 and rCYP3A5 by ITZ and KTZ.
KTZ (0.001–5 µM) and ITZ (0.002–10 µM) were studied as inhibitors of rCYP3A4- and rCYP3A5-catalyzed metabolism of MDZ, TST, terfenadine, and vincristine. In all cases, the IC50 was generated at a [S] equivalent to the Km (S50) and at a [S] well below Km (S50) ([S]/Km ratio ∼0.05). Both azoles were dissolved in dimethylsulfoxide and the appropriate volume of the stock solution was added to the incubation mixture (final concentration of dimethylsulfoxide did not exceed 0.5%). Incubation and LC/MS/MS analysis was carried out as described for the baseline kinetic studies. Percentage of control activity was determined as the ratio of the amount of 1′OH-MDZ, 6βOH-TST, terfenadine alcohol, and vincristine M1 formed in the presence to that in the absence (solvent alone) of KTZ or ITZ. IC50 values were obtained by means of nonlinear least-squares analysis (eq. 3) using XLfit (IDBS, Guildford, UK).
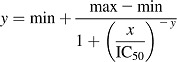 |
(3) |
In eq. 3, IC50 is the x value for the point in the curve that is midway between the max (maximum activity remaining; minimal percentage inhibition) and min (minimal activity remaining; maximal percentage inhibition). The exponent y is the slope of the curve at its midpoint. (See Supplemental Material for Determination of Ligand-Induced Binding Spectra and Reconstitution of Purified Wild-Type and Mutant CYP3A4 with P450 Reductase and Cytochrome b5.)
Inhibition of MDZ 1′-Hydroxylase and TST 6β-Hydroxylase Activity in HLM of CYP3A5-Genotyped Livers
Microsomal Preparations.
The collection and use of human tissue for research was approved by the University of Washington Human Subjects Review Board. Human liver samples were obtained from Human Tissue Bank maintained by the School of Pharmacy, University of Washington (Seattle, WA). Of the 60 available livers, three from a subset of CYP3A5*3/*3 donors (one male, two female subjects) and three from a subset of CYP3A5*1/*3 donors (three male donors) were selected randomly from each respective group for microsome preparation. Microsomes from three additional donors genotyped CYP3A5*1/*1 (one male, two female) were purchased from commercial sources to obtain microsomes from three donors of each genotype (CYP3A5*1/*1, CYP3A5*1/*3, and CYP3A5*3/*3). The procedures used for microsome isolation have been published previously (Paine et al., 1997; Lin et al., 2002; Dai et al., 2004). The CYP3A4 and CYP3A5 protein expression levels in each liver were quantified by Western blotting, and the data are summarized in Table 1. (See Supplemental Material for Assessment of CYP3A Inhibition and Determination of IC50.)
TABLE 1.
Differential expression of CYP3A4 and CYP3A5 protein in various HLM preparations
Data are presented as the mean ± S.D. (n = 3 determinations).
| Expression Level |
||||
|---|---|---|---|---|
| CYP3A5 Genotype | HLM Preparation | CYP3A4 | CYP3A5 | Percentage of CYP3A5a |
| pmol/mg | pmol/mg | % | ||
| *1/*1 | HL739 | 11.0 | 6.00 | 35.3 |
| HL867 | 110 | 17.0 | 13.4 | |
| HL47 | 62.0 | 9.30 | 13.0 | |
| Mean ± S.D. | 61.0 ± 49.5 | 10.8 ± 5.6* | 20.6 ± 12.7 | |
| *1/*3 | HL150 | 5.00 | 22.0 | 81.5 |
| HL125 | 72.0 | 58.0 | 44.6 | |
| HL167 | 19.0 | 33.0 | 63.5 | |
| Mean ± S.D. | 32.0 ± 35.3 | 37.7 ± 18.4* | 63.2 ± 18.4* | |
| *3/*3 | HL152 | 25.0 | 1.00 | 3.80 |
| HL143 | 38.0 | 1.00 | 2.60 | |
| HL131 | 28.0 | 1.00 | 3.40 | |
| Mean ± S.D. | 30.3 ± 6.8 | 1.00 ± 0.00 | 3.29 ± 0.66 | |
HL, human liver.
CYP3A5 expression represents CYP3A5 protein expression (pmol/mg) as a percentage of a total CYP3A protein expression in HLM (CYP3A4 pmol/mg + CYP3A5 pmol/mg).
P < 0.05, significantly different from expression level of CYP3A5 in HLM of a liver genotyped CYP3A5*3/*3.
Simulation of the CYP3A5 Expression-Dependent IC50 Values of KTZ and ITZ for MDZ 1′-Hydroxylation and TST 6β-Hydroxylation
IC50 values of KTZ and ITZ for MDZ 1′-hydroxylation and TST 6β-hydroxylation were simulated to model human liver microsomes with variable CYP3A4 and CYP3A5 expression. The following model was used (eq. 4), which assumes that MDZ and TST can be metabolized by CYP3A4 and CYP3A5:
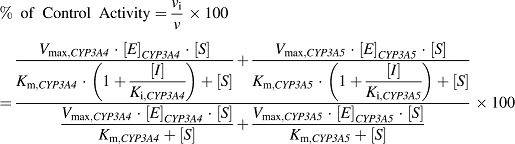 |
(4) |
where [S] is the applied concentration of substrate and [I] is the concentration of inhibitor. [E]CYP3A4 and [E]CYP3A5 are concentrations of CYP3A4 and CYP3A5, respectively, in the microsomes. v and vi are metabolite formation velocities in the absence and presence of inhibitor, respectively. By incorporating Km and Vmax values of MDZ or TST shown in Table 2, and Ki values estimated from IC50 values (Ki = IC50 based on an [S] that was greater than 10-fold lower than the Km value) of KTZ or ITZ shown in Table 3, as well as expression levels (concentrations) of CYP3A4 and CYP3A5 to eq. 4, the percentage of control activity (vi/v × 100) was predicted and the IC50 values were determined by nonlinear least-squares analysis using the MULTI program (Yamaoka et al., 1981). Leveraging the IC50 values at the low [S] circumvents the need to consider different mechanisms of inhibition (competitive versus noncompetitive versus mixed). The relationship between the CYP3A5 expression and the simulated IC50 values of KTZ and ITZ for MDZ 1′-hydroxylation and TST 6β-hydroxylation was determined. CYP3A5 expression represents the CYP3A5-specific content (pmol enzyme/mg protein) as a percentage of a total CYP3A protein content in HLM (CYP3A4 pmol/mg + CYP3A5 pmol/mg).
TABLE 2.
Kinetic parameters for the rCYP3A4- and rCYP3A5-catalyzed metabolism of MDZ, TST, terfenadine, and vincristine
Data are presented as the mean ± S.D. (n = 3 determinations), except for the CYP3A5/CYP3A4 Vmax/Km (S50) ratio.
| P450 | Substrate | Model | Km (S50) | Vmaxa | nb | Vmax/Km(S50) | Vmax/Km (S50) Ratioc |
|---|---|---|---|---|---|---|---|
| µM | |||||||
| CYP3A4 | MDZ | Hyperbolic | 1.8 ± 0.3 | 19 ± 1 | — | 11.0 | |
| TST | Sigmoidal | 78 ± 34 | 86 ± 3 | 1.2 ± 0.13 | 1.1 | ||
| Terfenadine | Hyperbolic | 3.4 ± 0.4 | 24 ± 2 | — | 7 | ||
| Vincristine | Sigmoidal | 75 ± 22 | 15 ± 4 | 1.5 ± 0.15 | 0.2 | ||
| CYP3A5 | MDZ | Hyperbolic | 3.0 ± 0.5 | 35 ± 2 | — | 11.7 | 1.1 |
| TST | Hyperbolic | 67 ± 13 | 56 ± 4 | — | 0.8 | 0.7 | |
| Terfenadine | Hyperbolic | 1.5 ± 0.2 | 18 ± 1 | — | 12.0 | 1.7 | |
| Vincristine | Sigmoidal | 47 ± 6.7 | 70 ± 7 | 1.5 ± 0.11 | 1.5 | 7.5 |
Presented as pmol/min/pmol P450, except for the formation of vincristine M1 metabolite. No attempt was made to formally quantitate M1, so data are presented as arbitrary units (see Materials and Methods).
Hill coefficient.
Ratio of CYP3A5 Vmax/Km (S50) versus CYP3A4 Vmax/Km (S50).
TABLE 3.
KTZ and ITZ as inhibitors of MDZ, TST, terfenadine, and vincristine metabolism
The data are presented as the mean ± S.D. (n = 3 determinations).
| Substrate | P450 | Substrate Conc.a | IC50b |
CYP3A5/CYP3A4 IC50 Ratio |
||
|---|---|---|---|---|---|---|
| KTZ | ITZ | KTZ | ITZ | |||
| µM | nM | nM | ||||
| MDZ | rCYP3A4 | 1.4 (0.8) | 40 ± 10 | 240 ± 50 | 5.0c | 9.6 |
| 0.1 (0.06) | 2.0 ± 0.2 | 17 ± 3 | 8.5d | 8.8 | ||
| rCYP3A5 | 2.7 (0.9) | 200 ± 4 | 2300 ± 400 | |||
| 0.2 (0.07) | 17 ± 3 | 150 ± 60 | ||||
| TST | rCYP3A4 | 78 (1.0) | 9.0 ± 1 | 40 ± 7 | 4.4 | 33 |
| 4.0 (0.05) | 17 ± 1 | 45 ± 3 | 4.7 | 9.1 | ||
| rCYP3A5 | 67 (1.0) | 40 ± 6 | 1300 ± 400 | |||
| 3.0 (0.04) | 80 ± 7 | 410 ± 180 | ||||
| Terfenadine | rCYP3A4 | 2.4 (0.7) | 90 ± 5 | 900 ± 200 | 2.2 | 2.3 |
| 0.2 (0.06) | 8.2 ± 1 | 23 ± 4 | 1.3 | 2.8 | ||
| rCYP3A5 | 1.2 (0.8) | 200 ± 5 | 2100 ± 500 | |||
| 0.1 (0.07) | 6.5 ± 2 | 64 ± 14 | ||||
| Vincristine | rCYP3A4 | 75 (1.0) | 150 ± 30 | 360 ± 50 | 0.8 | 1.9 |
| 4.0 (0.05) | 111 ± 10 | 29 ± 4 | 0.6 | 1.7 | ||
| rCYP3A5 | 45 (1.0) | 120 ± 10 | 680 ± 230 | |||
| 2.0 (0.04) | 70 ± 10 | 50 ± 5 | ||||
Conc., concentration.
[S]/Km ratio in parentheses.
IC50 was determined at a [S] close to the Km (S50) and at a [S] below Km (S50) (see Table 2).
IC50 ratio at higher [S].
IC50 ratio at lower [S].
In addition, the relationship between CYP3A5 expression and IC50 values against CYP3A-catalyzed metabolism was simulated using various Ki and Vmax/Km ratios (CYP3A5/CYP3A4). The previous model (eq. 4), which assumes that a virtual drug can be metabolized by CYP3A4 and CYP3A5, was used. Simulation of the effect of Ki ratio on CYP3A5 expression–dependent IC50 values was performed using kinetic parameters for a virtual CYP3A substrate and inhibitor set as follows: [S] = 100 nM, Vmax/Km ratio = 1, and Ki,CYP3A4 = 100 nM. The effect of Vmax/Km ratio on CYP3A5 expression–dependent IC50 values was also simulated using kinetic parameters for a virtual CYP3A substrate and inhibitor set as follows: [S] = 100 nM (< < Km) and Ki ratio = 10 (Ki,CYP3A4 = 100 nM, Ki,CYP3A5 = 1000 nM).
Statistical Analysis
Data are presented as the mean of values obtained in at least three experiments with the S.D. Statistical analyses were performed with the unpaired Student’s t test, and a probability of less than 0.05 (P < 0.05) was considered to be statistically significant.
The average fold error (afe) between the simulated and observed IC50 values was calculated using eq. 5 as described previously for evaluating accuracy of DDI predictions (Brown et al., 2005):
| (5) |
The differences between the observed and predicted IC50 values in the individual livers for each inhibitor-substrate pair were tested using a two-tailed paired t test, and a P value <0.05 was considered significant.
Results
Kinetic Parameters Describing the rCYP3A4- and rCYP3A5-Catalyzed Metabolism of MDZ, TST, Terfenadine, and Vincristine.
rCYP3A4 and rCYP3A5 (coexpressed with P450 reductase and cytochrome b5) were incubated with four different model substrates (TST, MDZ, vincristine, and terfenadine) to determine the kinetic parameters. In all cases, the rate of metabolite formation was determined under linear conditions and over a suitable substrate concentration range. Under the assay conditions chosen, rCYP3A4- and rCYP3A5-catalyzed formation of 1′OH-MDZ and terfenadine alcohol conformed to Michaelis-Menten (hyperbolic) kinetics (Table 2). On the other hand, vincristine M1 formation with both P450s was best described by sigmoidal kinetics. Only TST 6β-hydroxylation exhibited differential kinetics with rCYP3A4 (sigmoidal) and rCYP3A5 (hyperbolic).
As shown in Table 2, the kinetic parameters for rCYP3A4-catalyzed metabolism of MDZ, TST, terfenadine, and vincristine were calculated as 1.8 ± 0.3, 78 ± 34, 3.4 ± 0.4, and 75 ± 22 μM (Km or S50 values) and 19 ± 1, 86 ± 3, 24 ± 2, and 15 ± 4 pmol/min/pmol (Vmax values), respectively. The kinetic parameters for rCYP3A5-catalyzed metabolism of MTZ, TST, terfenadine, and vincristine were 3.0 ± 0.5, 67 ± 13, 1.5 ± 0.2, and 47 ± 7 μM (Km or S50 values) and 35 ± 2, 56 ± 4, 18 ± 1, and 70 ± 7 pmol/min/pmol (Vmax values), respectively. The corresponding Vmax/Km (S50) ratios (rCYP3A5/rCYP3A4) for TST, MDZ, terfenadine, and vincristine were 0.7, 1.1, 1.7, and 7.5, respectively. This permitted an assessment of CYP3A inhibition by ITZ and KTZ with a set of model substrates that spanned a 10-fold range of Vmax/Km (S50) ratios (rCYP3A5/rCYP3A4).
Inhibition of rCYP3A4 and rCYP3A5 by ITZ and KTZ.
The inhibitory effect of KTZ and ITZ on rCYP3A4- and rCYP3A5-catalyzed MDZ 1′-hydroxylation, TST 6β-hydroxylation, terfenadine alcohol formation, and vincristine M1 formation was analyzed (Supplemental Figs. 1–4; Supplemental Tables 1 and 2). In each case, IC50 values were obtained at a higher (∼Km) and lower (<Km) [S]. As expected, the IC50 decrease (≥11-fold) at the lower [S] was consistent with competitive inhibition. However, for rCYP3A4 with TST (ITZ and KTZ) and vincristine (KTZ), as well as rCYP3A5 with TST (KTZ and ITZ) and vincristine (KTZ), the IC50 change was minimal. Such a profile is more consistent with noncompetitive inhibition (Table 3).
As shown in Table 3, the IC50 ratios (rCYP3A5/rCYP3A4) of KTZ and ITZ at the higher [S] were 5.0 and 9.6 (MDZ), 4.4 and 33 (TST), 2.2 and 2.3 (terfenadine), and 0.8 and 1.9 (vincristine), respectively, suggesting substrate and/or inhibitor dependency for the difference in the IC50 values for rCYP3A4- and rCYP3A5-catalyzed drug metabolism. With the exception of the inhibition of TST 6β-hydroxylation by ITZ, similar differences in IC50 ratios were observed at the lower [S].
In the present study, rCYP3A4 exhibited sigmoidal kinetics with TST as a substrate, whereas rCYP3A5 did not (Table 2). Therefore, we hypothesized that the large rCYP3A5/rCYP3A4 IC50 ratio (33) of ITZ at the higher concentration of TST (∼S50) was largely driven by the sigmoidal kinetics exhibited by rCYP3A4 (Table 3). To test the hypothesis, ITZ was evaluated as an inhibitor of wild-type and mutant (L211F/D214E) CYP3A4. When reconstituted with P450 reductase and cytochrome b5, the former exhibits sigmoidal kinetics with TST (Hill coefficient n = 1.3). In contrast, TST 6β-hydroxylation catalyzed by the mutant form of CYP3A4 conforms to hyperbolic kinetics (Hill coefficient n = 1). Consequently, at least two amino acid residues (Leu211 and Asp214) are thought to govern the sigmoidal kinetics of CYP3A4 (Harlow and Halpert, 1998). As shown in Supplemental Table 3 and Supplemental Fig. 6, there was no major difference in the IC50 value of ITZ (wild-type versus mutant CYP3A4) when TST was incubated at two concentrations (25 and 100 µM). The higher concentration of TST approximated the S50 (∼100 µM) of both CYP3A4 forms (Harlow and Halpert, 1998).
To determine whether ITZ and KTZ bind CYP3A4 and CYP3A5 differentially in the absence of a substrate but bind within the active site of CYP3A4 and CYP3A5, ligand-induced difference spectra (binding spectra) were generated with both P450s (Supplemental Fig. 5). Both ITZ and KTZ generated typical type II binding spectra with rCYP3A4 and rCYP3A5, characteristic of nitrogen coordination to the heme and causing a heme iron high to low spin shift (Kunze et al., 2006; Locuson et al., 2007). However, the maximum change in the magnitude of the ligand-induced spectral change for ITZ was smaller with rCYP3A5 [0.010 arbitrary units (AU)] than with rCYP3A4 (0.023 AU). For KTZ, the difference in the maximum spectral change between the two P450s was smaller; the maximum absorbance difference was 0.030 AU with rCYP3A4 and 0.023 AU with rCYP3A5. With regard to ligand affinity, the Ks of ITZ was 118 nM with CYP3A4. The Ks with rCYP3A5 was 3-fold higher (352 nM). For KTZ, the determined Ks value for rCYP3A5 (460 nM) was 5-fold higher than the Ks value with CYP3A4 (86 nM).
Inhibitory Effect of ITZ and KTZ on MDZ and TST Metabolism in HLM of CYP3A5-Genotyped Livers.
The inhibitory effect of KTZ and ITZ on CYP3A-catalyzed MDZ 1′-hydroxylation and TST 6β-hydroxylation was evaluated using CYP3A5-genotyped HLM (Supplemental Figs. 7 and 8; Table 1). As summarized in Table 4, the IC50 values of KTZ and ITZ for MDZ 1′-hydroxylation and TST 6β-hydroxylation varied up to 30-fold between HLMs. Interestingly, the IC50 values of KTZ and ITZ were the lowest in the group of HLMs with CYP3A5*3/*3 genotype, followed by CYP3A5*1/*1 genotype, and the highest in CYP3A5*1/*3 genotype. Overall, the IC50 values of ITZ toward MDZ and TST were significantly higher in the CYP3A5-expressing livers than in the CYP3A5*3/*3 livers (Table 4). The IC50 values of KTZ toward MDZ were also significantly higher in the CYP3A5-expressing livers than in the CYP3A5*3/*3 livers, but no significant differences in the IC50 values of KTZ for TST metabolism were observed among CYP3A5 genotypes. Since the expression level of CYP3A5 protein in HLM with CYP3A5*1/*1 and CYP3A5*1/*3 genotypes was significantly higher than that in the HLM of livers genotyped CYP3A5*3/*3, the IC50 values overall appeared to track with the expression level of CYP3A5 (Tables 1 and 4).
TABLE 4.
Impact of CYP3A5 expression on the inhibition of MDZ and TST metabolism in HLM by KTZ and ITZ
Data are presented as the mean ± S.D. (n = 3 determinations).
| CYP3A5 Genotype | HLM Preparation | IC50 |
|||
|---|---|---|---|---|---|
| MDZa |
TSTb |
||||
| + KTZ | + ITZ | + KTZ | + ITZ | ||
| nM | nM | nM | nM | ||
| *1/*1 | HL739 | 35.8 ± 3.8 | 108 ± 24 | 31.5 ± 4.8 | 85.4 ± 10.5 |
| HL867 | 34.3 ± 3.2 | 111 ± 15 | 19.7 ± 3.8 | 120 ± 12 | |
| HL47 | 30.7 ± 4.0 | 123 ± 21 | 29.1 ± 2.7 | 111 ± 11 | |
| Mean ± S.D. | 33.6 ± 2.6* | 114 ± 8* | 26.8 ± 6.3 | 105 ± 18* | |
| *1/*3 | HL150 | 132 ± 13 | 1133 ± 782 | 308 ± 52 | 513 ± 105 |
| HL125 | 11.8 ± 1.5 | 161 ± 27 | 53.9 ± 3.8 | 158 ± 14 | |
| HL167 | 24.3 ± 2.2 | 176 ± 13 | 64.2 ± 6.6 | 195 ± 15 | |
| Mean ± S.D. | 56.1 ± 66.3 | 490 ± 557 | 142 ± 144 | 289 ± 195* | |
| *3/*3 | HL152 | 6.92 ± 1.12 | 47.3 ± 8.5 | 25.8 ± 3.8 | 20.0 ± 2.3 |
| HL143 | 12.9 ± 2.0 | 83.0 ± 5.9 | 27.1 ± 5.5 | 21.6 ± 1.7 | |
| HL131 | 11.5 ± 1.0 | 53.3 ± 7.4 | 10.2 ± 1.1 | 46.7 ± 4.2 | |
| Mean ± S.D. | 10.4 ± 3.1 | 61.2 ± 19.1 | 21.0 ± 9.4 | 29.4 ± 15.0 | |
HL, human liver.
Initial concentration of MDZ was 1 μM.
Initial concentration of TST was 10 μM.
P < 0.05, significantly different from the IC50 obtained with HLM of a liver genotyped CYP3A5*3/*3.
By incorporating the kinetic Km and Vmax values (Table 2) and Ki (IC50-derived) values of KTZ and ITZ (Table 3) for rCYP3A4- and rCYP3A5-catalyzed TST and MDZ metabolism, as well as expression levels of CYP3A4 and CYP3A5 protein in HLM (Table 1) into eq. 4, the relationship between CYP3A5 expression and IC50 values of KTZ and ITZ for CYP3A-catalyzed MDZ and TST metabolism in individual HLM was simulated (Fig. 1, solid lines). The simulated IC50 values were not significantly different from those observed (P > 0.05 for all inhibitor-substrate pairs), yet the afe analysis showed that the IC50 values of KTZ and ITZ toward TST hydroxylation (afe 0.77 and 0.999 for KTZ and ITZ, respectively) were more accurately predicted than those against MDZ (afe 0.26 and 0.40 for KTZ and ITZ, respectively). Interestingly, the relatively low prediction accuracy for the KTZ-MDZ IC50 was independent of the genotype, with the predicted IC50 values consistently lower than those observed. In contrast, the discrepancies between predicted and observed values for ITZ-MDZ interaction were mainly driven by the CYP3A5*1/*1 livers. Of the two azoles tested, the IC50 of ITZ was most sensitive to CYP3A5 expression level in HLM (Fig. 1).
Fig. 1.

Relationship between CYP3A5 expression and IC50 values of KTZ and ITZ for the CYP3A-catalyzed metabolism of MDZ and TST in HLM. Relationship between CYP3A5 expression and IC50 values of KTZ (A) and ITZ (B) for MDZ and KTZ (C) and ITZ (D) for TST metabolism. CYP3A5 expression represents CYP3A5 protein expression (pmol/mg) as a percentage of a total CYP3A protein expression in HLM (CYP3A4 pmol/mg + CYP3A5 pmol/mg). Solid and dotted lines represent simulated data and their 2-fold error ranges, respectively (see Materials and Methods); ●, ▲, and ■ represent observed data using HLMs with CYP3A5*1/*1, CYP3A5*1/*3, and CYP3A5*3/*3 genotypes, respectively. Data are presented as the mean ± S.D. (n = 3 determinations).
Simulation Analysis of the Effect of CYP3A5 Expression on Inhibition of CYP3A-Catalyzed Drug Metabolism.
To consider the effect of the different Ki values of novel CYP3A inhibitors, and Vmax/Km values between CYP3A4 and CYP3A5 of CYP3A substrates, on CYP3A5 expression–dependent IC50 values, the relationship between CYP3A5 expression and IC50 values against CYP3A-catalyzed metabolism was simulated using various Ki and Vmax/Km ratios (CYP3A5/CYP3A4) (Fig. 2). The effect of the Ki ratio on CYP3A5 expression–dependent IC50 values was simulated using kinetic parameters for a virtual CYP3A substrate and inhibitor set as follows: substrate concentration = 100 nM, Vmax/Km ratio = 1 (i.e., CYP3A4 and CYP3A5 have equal intrinsic clearances for the substrate), and Ki,CYP3A4 = 100 nM (Fig. 2A). When the Ki ratio (CYP3A5/CYP3A4) was 1.0, no effect of CYP3A5 expression on the IC50 value for CYP3A-catalyzed drug metabolism was observed. The simulated IC50 value increased with increasing relative CYP3A5 expression with an increase in the Ki ratio and conversely decreased with decreased Ki ratio.
Fig. 2.

Simulation analysis of effect of CYP3A5 expression on inhibition of CYP3A-catalyzed drug metabolism. (A) Impact of Ki ratio (CYP3A5/CYP3A4) on CYP3A5 expression–dependent IC50 values. Kinetic parameters for a virtual CYP3A substrate and inhibitor used in current simulations are set as follows: substrate concentration = 100 nM, Vmax/Km ratio = 1, and Ki,CYP3A4 = 100 nM. (B) Impact of Vmax/Km ratio (CYP3A5/CYP3A4) on CYP3A5 expression–dependent IC50 values. Kinetic parameters for a virtual CYP3A substrate and inhibitor used in current simulations are set as follows: substrate concentration = 100 nM (< < Km) and Ki ratio = 10 (Ki,CYP3A4 = 100 nM, Ki,CYP3A5 = 1000 nM). CYP3A5 expression represents the percentage of total CYP3A (CYP3A4 + CYP3A5) expression (see Materials and Methods).
Simulation of the effect of CYP3A Vmax/Km ratio on CYP3A5 expression–dependent IC50 values was also performed using kinetic parameters for a virtual CYP3A substrate and inhibitor set as follows: substrate concentration = 100 nM (< < Km) and Ki ratio = 10 (Ki,CYP3A4 = 100 nM, Ki,CYP3A5 = 1000 nM) (Fig. 2B). When the Vmax/Km ratio was higher, the IC50 value for CYP3A-catalyzed drug metabolism increased even when CYP3A5 expression was low (<20% of total CYP3A). In contrast, when the Vmax/Km ratio was lower, the IC50 value was constant or slightly changed at lower CYP3A5 levels, then increased at higher CYP3A5 levels. This simulation suggested that variability of IC50 values for CYP3A-catalyzed drug metabolism became more sensitive to CYP3A5 expression when CYP3A5 contribution to substrate clearance was greater.
Discussion
The results of this study show that CYP3A5 genotype and protein expression level have a significant impact on the inhibitory potency of ITZ and KTZ, but that the magnitude of the effect is inhibitor-substrate pair–specific. The IC50 ratios (rCYP3A5/rCYP3A4) of KTZ and ITZ for the inhibition of rCYP3A4- and rCYP3A5-catalyzed metabolism of MDZ and TST were much higher (>4) than those for terfenadine and vincristine (<3). This difference in the IC50 values for CYP3A4 and CYP3A5 may result in a complex CYP3A-mediated DDI profile in humans carrying the CYP3A5*1 allele that confers significant expression of the encoded enzyme (Kuehl et al., 2001). It is noteworthy that the range of IC50 ratios cannot be rationalized solely on the basis of higher ITZ (∼3-fold) and KTZ (∼5-fold) CYP3A4 binding affinity (Ks) in the absence of a substrate. This implies that one has to consider the binding of both substrate and inhibitor within the CYP3A active site and the effect of allosterism in substrate-dependent inhibitory potency. In agreement with what would be predicted from rP450 data, the IC50 values of KTZ and ITZ for MDZ 1′-hydroxylation and ITZ for TST 6β-hydroxylation were significantly higher in HLMs with CYP3A5*1/*1 and CYP3A5*1/*3 genotypes than in HLMs with CYP3A5*3/*3 genotype. However, no significant difference in the IC50 values of KTZ for TST metabolism was observed among the CYP3A5 genotype groups (Table 4). Based on the higher CYP3A5 protein expression level in HLMs with CYP3A5*1/*1 and CYP3A5*1/*3 genotypes compared with CYP3A5*3/*3 genotype, the greater IC50 values of KTZ and ITZ for CYP3A-catalyzed MDZ and TST metabolism are likely due to the higher expression level of CYP3A5 and lower inhibitory potency of KTZ and ITZ toward CYP3A5 compared with CYP3A4 (higher IC50 ratios) (Tables 1, 3, and 4). Alternatively, one can anticipate no significant effect of CYP3A5 expression on the IC50 values of KTZ and ITZ for CYP3A-catalyzed metabolism of terfenadine and vincristine, based on the low IC50 ratios of KTZ and ITZ shown in Table 3. Further investigation would be needed to clarify the impact of CYP3A5 expression on the inhibition of CYP3A-catalyzed metabolism of terfenadine and vincristine. Overall, the relative CYP3A5 expression level rather than genotype (heterozygous or homozygous CYP3A5*1) determined the magnitude of IC50 values of ITZ and KTZ in HLMs.
Results from our simulation of the IC50 values at different CYP3A5 expression levels with KTZ and ITZ using kinetic data from CYP3A4 and CYP3A5 Supersomes and protein expression levels of CYP3A4 and CYP3A5 in the HLMs support our conclusion that relative CYP3A5 expression determines the inhibitory potency toward a given substrate (Fig. 1). When the simulated IC50 values of KTZ and ITZ were compared with the experimentally measured IC50 values, the predicted IC50 values were not significantly different from each other, demonstrating that simulation can be used to predict the effect of CYP3A5 expression on the IC50 values for CYP3A-catalyzed drug metabolism (Fig. 1). Interestingly, the prediction of the IC50 values was more accurate for both inhibitors when TST was used as a substrate than with MDZ (afe 0.32 versus 0.87). This difference in prediction accuracy could be due to unaccounted allosteric, substrate-specific effects. Such allosteric effects were recently shown in vitro and in vivo between fluconazole and MDZ (Yang et al., 2012). However, for both MDZ and TST metabolism, the observed IC50 values were generally within the 2-fold range of the predicted values, with the exception of HL150. In this donor, the observed IC50 value was significantly higher than that predicted and that observed in the other donors. One possible explanation for this inconsistency is the potential contribution of CYP3A7 to drug metabolism in HL150 and relatively weak inhibition of CYP3A7 by KTZ and ITZ, yielding a higher IC50 value of KTZ and ITZ for MDZ and TST metabolism (Nishimura et al., 2003; Sim et al., 2005; Hata et al., 2010). Such a hypothesis is supported by the much higher (∼16-fold) IC50 of KTZ for rCYP3A7-catalyzed TST metabolism versus rCYP3A5 (630 versus 40 nM; unpublished data). Another possible explanation is that minor contributions from other P450 enzymes to MDZ and TST metabolism, such as CYP2C9 and CYP2C19, may be factors in the deviation (Choi et al., 2005).
To further evaluate the potential impact of CYP3A5 expression in CYP3A IC50 values, the effect of Ki ratios (CYP3A5/CYP3A4) of CYP3A inhibitors and Vmax/Km ratios (CYP3A5/CYP3A4) of CYP3A substrates on CYP3A5 expression-dependent IC50 values was simulated. As shown in Fig. 2A, a CYP3A5 expression–dependent change in the IC50 values for CYP3A-catalyzed drug metabolism is clearly predicted when the Ki ratio deviates from 1, whereas no effect of CYP3A5 expression on IC50 value is expected when the Ki ratio is unity. Therefore, it is concluded that the Ki ratio is one of the major determinants for CYP3A5 expression–dependent IC50 values. On the other hand, as shown in Fig. 2B, the effect of CYP3A5 expression on the IC50 value for CYP3A-catalyzed drug metabolism is also dependent on the relative catalytic efficiency of CYP3A5 when compared with CYP3A4, and is more sensitive at higher CYP3A5/CYP3A4 Vmax/Km ratios. Accordingly, if the contribution of CYP3A5 to total CYP3A-catalyzed drug metabolism is high (that is, the intrinsic clearance of the drug is higher for CYP3A5 than for CYP3A4), even very low relative CYP3A5 expression has a significant effect on the observed apparent IC50 values. In such a case, CYP3A5 expression would again become an important factor to consider when determining IC50 values for CYP3A-catalyzed drug metabolism in HLMs. However, if the contribution of CYP3A5 to total CYP3A-catalyzed drug metabolism is low, i.e., intrinsic clearance of CYP3A5 ∼0.1 (Fig. 2), the effect of an individual difference in CYP3A5 expression on the IC50 values is likely to be negligible, despite any difference in the Ki value for CYP3A4 and CYP3A5.
The present findings suggest that, when CYP3A-mediated DDI studies are planned, the inhibition potency of the new chemical entity toward CYP3A4 and CYP3A5 should be tested in vitro using the same probe as will be used in the clinical study to determine whether CYP3A5 genotype will affect interindividual variability in the magnitude of the DDI observed and significantly affect study power. The inhibitor selectivity should be considered in relation to the intrinsic clearance ratio of the substrate. The influence of CYP3A5 expression on the magnitude of DDIs could also be taken into consideration when the in vivo probe substrate is selected. On the other hand, when the new chemical entity is considered as the object drug of the DDI, it will be prudent to determine the contribution of CYP3A5 to the overall clearance of the drug (in CYP3A5 expressers) and to test the effect of CYP3A5 expression on the IC50 of KTZ or another strong CYP3A inhibitor toward that specific compound. It is important to note that, due to the allosteric behavior of CYP3A, the effect of CYP3A5 expression on in vivo IC50 values is specific for the inhibitor-substrate pair and needs to be determined individually in vitro for the combination in the study. Unfortunately, very few compounds have been studied in vivo or in vitro for their potency as CYP3A5 inhibitors, and, as such, extrapolation of these data to the overall variability of the magnitude of CYP3A-mediated DDIs can be challenging. This is emphasized by the fact that, at present, very limited data are available for CYP3A5 contribution to the clearance of the known sensitive substrates of CYP3A. More systematic studies are needed to establish the overall significance of CYP3A5 inhibition in clinical DDIs.
In conclusion, the present study demonstrates that the IC50 value for CYP3A-catalyzed drug metabolism measured using HLMs varies due to differences in CYP3A5 expression/genotype, the substrate studied, and the relative inhibitory potency toward CYP3A4 and CYP3A5. These findings imply that the HLM assay has the potential to misread the IC50 value for CYP3A-catalyzed drug metabolism. However, simulation analysis, as described in the present study, may be useful when considering variability of IC50 values for CYP3A-catalyzed drug metabolism in individual HLMs, and may enable relatively precise prediction of DDIs in vivo involving the inhibition of CYP3A.
Supplementary Material
Acknowledgments
The authors thank Drs. James Halpert and Dmitri Davydov (Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, CA) for the gifts of purified human CYP3A4 (wild-type and mutant forms), P450 reductase, and cytochrome b5.
Abbreviations
- afe
average fold error
- AU
arbitrary units
- DDI
drug-drug interaction
- ESI
positive ion electrospray
- HLM
human liver microsome
- [I]
inhibitor concentration
- ITZ
itraconazole
- Ki
inhibition constant
- Ks
spectral binding constant
- KTZ
ketoconazole
- LC/MS/MS
liquid chromatography–tandem mass spectrometry
- MDZ
midazolam
- m/z
mass-to-charge ratio
- 1′OH-MDZ
1′-hydroxy midazolam
- 6βOH-TST
6β-hydroxy testosterone
- P450
cytochrome P450
- rCYP3A4
recombinant CYP3A4
- rCYP3A5
recombinant CYP3A5
- [S]
substrate concentration
- TST
testosterone
- UHPLC
ultra high–performance liquid chromatography
Authorship Contributions
Participated in research design: Isoherranen, Thummel, Rodrigues.
Conducted experiments: Peng, Grubb, Chang, Shirasaka.
Performed data analysis: Shirasaka, Chang, Grubb, Isoherranen, Rodrigues.
Wrote or contributed to the writing of the manuscript: Shirasaka, Rodrigues, Thummel, Isoherranen, Chang, Grubb.
Footnotes
This work was supported in part by a Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science [Research Project Number H23-694]; and the National Institutes of Health National Institute of General Medical Sciences [Grant P01 GM32165].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Barry A, Levine M. (2010) A systematic review of the effect of CYP3A5 genotype on the apparent oral clearance of tacrolimus in renal transplant recipients. Ther Drug Monit 32:708–714 [DOI] [PubMed] [Google Scholar]
- Brown HS, Ito K, Galetin A, Houston JB. (2005) Prediction of in vivo drug-drug interactions from in vitro data: impact of incorporating parallel pathways of drug elimination and inhibitor absorption rate constant. Br J Clin Pharmacol 60:508–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N, Aggarwal PK, Minz M, Sakhuja V, Kohli KK, Jha V. (2009) CYP3A5*1/*3 genotype influences the blood concentration of tacrolimus in response to metabolic inhibition by ketoconazole. Pharmacogenet Genomics 19:458–463 [DOI] [PubMed] [Google Scholar]
- Choi MH, Skipper PL, Wishnok JS, Tannenbaum SR. (2005) Characterization of testosterone 11 beta-hydroxylation catalyzed by human liver microsomal cytochromes P450. Drug Metab Dispos 33:714–718 [DOI] [PubMed] [Google Scholar]
- Christensen H, Hestad AL, Molden E, Mathiesen L. (2011) CYP3A5-mediated metabolism of midazolam in recombinant systems is highly sensitive to NADPH-cytochrome P450 reductase activity. Xenobiotica 41:1–5 [DOI] [PubMed] [Google Scholar]
- Dai Y, Hebert MF, Isoherranen N, Davis CL, Marsh C, Shen DD, Thummel KE. (2006) Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab Dispos 34:836–847 [DOI] [PubMed] [Google Scholar]
- Dai Y, Iwanaga K, Lin YS, Hebert MF, Davis CL, Huang W, Kharasch ED, Thummel KE. (2004) In vitro metabolism of cyclosporine A by human kidney CYP3A5. Biochem Pharmacol 68:1889–1902 [DOI] [PubMed] [Google Scholar]
- Dennison JB, Jones DR, Renbarger JL, Hall SD. (2007) Effect of CYP3A5 expression on vincristine metabolism with human liver microsomes. J Pharmacol Exp Ther 321:553–563 [DOI] [PubMed] [Google Scholar]
- Dennison JB, Kulanthaivel P, Barbuch RJ, Renbarger JL, Ehlhardt WJ, Hall SD. (2006) Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab Dispos 34:1317–1327 [DOI] [PubMed] [Google Scholar]
- Dennison JB, Mohutsky MA, Barbuch RJ, Wrighton SA, Hall SD. (2008) Apparent high CYP3A5 expression is required for significant metabolism of vincristine by human cryopreserved hepatocytes. J Pharmacol Exp Ther 327:248–257 [DOI] [PubMed] [Google Scholar]
- Egbelakin A, Ferguson MJ, MacGill EA, Lehmann AS, Topletz AR, Quinney SK, Li L, McCammack KC, Hall SD, Renbarger JL. (2011) Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 56:361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs MA, Thummel KE, Shen DD, Kunze KL. (1999) Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos 27:180–187 [PubMed] [Google Scholar]
- Harlow GR, Halpert JR. (1998) Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc Natl Acad Sci USA 95:6636–6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata S, Miki Y, Fujishima F, Sato R, Okaue A, Abe K, Ishida K, Akahira J, Unno M, Sasano H. (2010) Cytochrome 3A and 2E1 in human liver tissue: Individual variations among normal Japanese subjects. Life Sci 86:393–401 [DOI] [PubMed] [Google Scholar]
- Huang W, Lin YS, McConn DJ, 2nd, Calamia JC, Totah RA, Isoherranen N, Glodowski M, Thummel KE. (2004) Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab Dispos 32:1434–1445 [DOI] [PubMed] [Google Scholar]
- Isoherranen N, Ludington SR, Givens RC, Lamba JK, Pusek SN, Dees EC, Blough DK, Iwanaga K, Hawke RL, Schuetz EG, et al. (2008) The influence of CYP3A5 expression on the extent of hepatic CYP3A inhibition is substrate-dependent: an in vitro-in vivo evaluation. Drug Metab Dispos 36:146–154 [DOI] [PubMed] [Google Scholar]
- Jushchyshyn MI, Hutzler JM, Schrag ML, Wienkers LC. (2005) Catalytic turnover of pyrene by CYP3A4: evidence that cytochrome b5 directly induces positive cooperativity. Arch Biochem Biophys 438:21–28 [DOI] [PubMed] [Google Scholar]
- Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, et al. (2001) Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet 27:383–391 [DOI] [PubMed] [Google Scholar]
- Kunze KL, Nelson WL, Kharasch ED, Thummel KE, Isoherranen N. (2006) Stereochemical aspects of itraconazole metabolism in vitro and in vivo. Drug Metab Dispos 34:583–590 [DOI] [PubMed] [Google Scholar]
- Lamba JK, Lin YS, Schuetz EG, Thummel KE. (2002) Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev 54:1271–1294 [DOI] [PubMed] [Google Scholar]
- Lee S-J, Goldstein JA. (2012) Comparison of CYP3A4 and CYP3A5: the effects of cytochrome b5 and NADPH-cytochrome P450 reductase on testosterone hydroxylation activities. Drug Metab Pharmacokinet 27:663–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YS, Dowling AL, Quigley SD, Farin FM, Zhang J, Lamba J, Schuetz EG, Thummel KE. (2002) Co-regulation of CYP3A4 and CYP3A5 and contribution to hepatic and intestinal midazolam metabolism. Mol Pharmacol 62:162–172 [DOI] [PubMed] [Google Scholar]
- Locuson CW, Hutzler JM, Tracy TS. (2007) Visible spectra of type II cytochrome P450-drug complexes: evidence that “incomplete” heme coordination is common. Drug Metab Dispos 35:614–622 [DOI] [PubMed] [Google Scholar]
- Lown KS, Kolars JC, Thummel KE, Barnett JL, Kunze KL, Wrighton SA, Watkins PB. (1994) Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack of prediction by the erythromycin breath test. Drug Metab Dispos 22:947–955 [PubMed] [Google Scholar]
- Lukkari E, Taavitsainen P, Juhakoski A, Pelkonen O. (1998) Cytochrome P450 specificity of metabolism and interactions of oxybutynin in human liver microsomes. Pharmacol Toxicol 82:161–166 [DOI] [PubMed] [Google Scholar]
- Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. (2003) Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi 123:369–375 [DOI] [PubMed] [Google Scholar]
- Niwa T, Murayama N, Emoto C, Yamazaki H. (2008) Comparison of kinetic parameters for drug oxidation rates and substrate inhibition potential mediated by cytochrome P450 3A4 and 3A5. Curr Drug Metab 9:20–33 [DOI] [PubMed] [Google Scholar]
- Paine MF, Khalighi M, Fisher JM, Shen DD, Kunze KL, Marsh CL, Perkins JD, Thummel KE. (1997) Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther 283:1552–1562 [PubMed] [Google Scholar]
- Patki KC, Von Moltke LL, Greenblatt DJ. (2003) In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab Dispos 31:938–944 [DOI] [PubMed] [Google Scholar]
- Sim SC, Edwards RJ, Boobis AR, Ingelman-Sundberg M. (2005) CYP3A7 protein expression is high in a fraction of adult human livers and partially associated with the CYP3A7*1C allele. Pharmacogenet Genomics 15:625–631 [DOI] [PubMed] [Google Scholar]
- Suh JW, Koo BK, Zhang SY, Park KW, Cho JY, Jang IJ, Lee DS, Sohn DW, Lee MM, Kim HS. (2006) Increased risk of atherothrombotic events associated with cytochrome P450 3A5 polymorphism in patients taking clopidogrel. CMAJ 174:1715–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel KE, Wilkinson GR. (1998) In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol 38:389–430 [DOI] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Hamman MA, Hall SD, Wrighton SA. (2002) Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30:883–891 [DOI] [PubMed] [Google Scholar]
- Yamaoka K, Tanigawara Y, Nakagawa T, Uno T. (1981) A pharmacokinetic analysis program (multi) for microcomputer. J Pharmacobiodyn 4:879–885 [DOI] [PubMed] [Google Scholar]
- Yamaori S, Yamazaki H, Iwano S, Kiyotani K, Matsumura K, Honda G, Nakagawa K, Ishizaki T, Kamataki T. (2004) CYP3A5 Contributes significantly to CYP3A-mediated drug oxidations in liver microsomes from Japanese subjects. Drug Metab Pharmacokinet 19:120–129 [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Nakamoto M, Shimizu M, Murayama N, Niwa T. (2010) Potential impact of cytochrome P450 3A5 in human liver on drug interactions with triazoles. Br J Clin Pharmacol 69:593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Atkins WM, Isoherranen N, Paine MF, Thummel KE. (2012) Evidence of CYP3A allosterism in vivo: analysis of interaction between fluconazole and midazolam. Clin Pharmacol Ther 91:442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KS, Cho JY, Jang IJ, Hong KS, Chung JY, Kim JR, Lim HS, Oh DS, Yi SY, Liu KH, et al. (2004) Effect of the CYP3A5 genotype on the pharmacokinetics of intravenous midazolam during inhibited and induced metabolic states. Clin Pharmacol Ther 76:104–112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.