

Abstract
The neuropeptides oxytocin (OT) and vasopressin (AVP) have been shown to play a central role in social behaviors; as a consequence, they have been recognized as potential drugs to treat neurodevelopmental and psychiatric disorders characterized by impaired social interactions. However, despite the basic and preclinical relevance of mouse strains carrying genetic alterations in the OT/AVP systems to basic and preclinical translational neuroscience, the pharmacological profile of mouse OT/AVP receptor subtypes has not been fully characterized. To fill in this gap, we have characterized a number of OT and AVP agonists and antagonists at three murine OT/AVP receptors expressed in the nervous system as follows: the oxytocin (mOTR) and vasopressin V1a (mV1aR) and V1b (mV1bR) subtypes. These three receptors were transiently expressed in vitro for binding and intracellular signaling assays, and then a homology model of the mOTR structure was constructed to investigate how its molecular features compare with human and rat OTR orthologs. Our data indicate that the selectivity profile of the natural ligands, OT and AVP, is conserved in humans, rats, and mice. Furthermore, we found that the synthetic peptide [Thr4Gly7]OT (TGOT) is remarkably selective for the mOTR and, like the endogenous OT ligand, activates Gq and Gi and recruits β-arrestins. Finally, we report three antagonists that exhibit remarkably high affinities and selectivities at mOTRs. These highly selective pharmacological tools will contribute to the investigation of the specific physiologic and pathologic roles of mOTR for the development of selective OT-based therapeutics.
Introduction
Central oxytocin (OT) and vasopressin (AVP) effects are mediated by three G-protein-coupled receptors (GPCRs) that are evolutionarily highly conserved and closely related, with overall homology varying from 40 to 85%: the vasopressin 1a receptor (V1aR), the vasopressin 1b receptor (V1bR), and the OT receptor (OTR) (Barberis et al., 1999; Birnbaumer, 2000; Zingg and Laporte, 2003). OT and AVP are also structurally very similar, differing by only two amino acids in most mammals (Wallis, 2012). Given this high degree of conservation in both receptors and peptides, the development of selective agonists and antagonists has proved to be a daunting task (Manning et al., 2008). Over the last decades, at least 1000 synthetic peptides have been synthesized and examined for their ability to bind to and activate the different OT/AVP receptor subtypes, an effort that has led to the identification of a number of subtype-selective analogs for human and rat subtypes (Manning et al., 2012). However, subtle differences between receptor sequences in different animal species are responsible for important changes in the selectivity profile of some ligands, and so the pharmacological data obtained in one species cannot be extrapolated tout court to others (Chini and Manning, 2007).
In addition to their well established systemic physiologic effects, OT and AVP are potent modulators of social behavior; consistently, the OT/AVP system is emerging as a relevant target for the treatment of impaired social functions associated with neurodevelopmental and psychiatric disorders (Miller, 2013). Even as OT has been recently reported to improve cognitive deficits in autistic patients (Modi and Young, 2012), its use is hampered by several of the following factors: 1) OT does not cross the blood-brain barrier and is administered intranasally, with unknown pharmacokinetics; 2) it also binds to and activates the V1aR and V1bR, which are highly expressed in the brain, where they exert different and even opposite effects (Pittman and Spencer, 2005); and 3) OT promotes the coupling of the OTR to different G proteins and β-arrestins (Busnelli et al., 2012) and, consequently, activates multiple signaling pathways whose precise roles within the brain are currently unknown. The development of more selective, potent, and therefore, longer-lasting analogs acting on brain is a priority in the field to understand how (in terms of biologic mechanisms) and where (in terms of neural specificity) OT exerts its effects.
In particular, subtype-selective analogs are crucial for defining the role of different receptor subtypes in rodent models currently used to investigate the OT/AVP system in the processing of socially relevant clues. Studies using transgenic mice genetically engineered to eliminate OT, OTR, or CD38, a protein involved in OT secretion, show that these animals lose important social behaviors (Ferguson et al., 2000; Takayanagi et al., 2005; Jin et al., 2007; Higashida et al., 2011). The deficits in the social paradigm can be fully restored by a single intracerebroventricular infusion of OT given prior to the test (Ferguson et al., 2001; Jin et al., 2007; Sala et al., 2011). Although this effect is consistent with the genetic alteration in CD38- and OT-null mice, which have a decreased level of circulating (and centrally released) OT, the efficacy of OT in restoring social recognition in the OTR-null mice is particularly intriguing. We have recently shown that the social behavioral deficits associated with the complete loss of Oxtr gene expression can be rescued by the activation of cognate vasopressin receptors, thus suggesting that the OT/AVP brain systems have overlapping and/or compensatory functions (Sala et al., 2011).
Another level of complexity in developing selective analogs derives from the finding that a single GPCR may couple to more than one G-protein, potentially activating multiple responses. Interestingly, different ligands show different degrees of intrinsic efficacy to different signaling pathways activated by the same receptor, a phenomenon referred to as “functional selectivity” (Urban et al., 2007; Kenakin, 2011). Because functional selective ligands have been recently described in the OT/AVP receptor family (in particular for the vasopressin 2 receptor (Jean-Alphonse et al., 2009), OTR (Reversi et al., 2005; Gravati et al., 2010; Busnelli et al., 2012), and V1aR (MacKinnon et al., 2009), the screening of the functional selective properties of ligands is becoming a crucial issue for the pharmacological characterization of selective ligands.
The aim of this study was to pharmacologically characterize a number of OT/AVP analogs at the OT/AVP receptor subtypes expressed in mouse brain: mOTR, mV1aR, and mV1bR. We found that [Thr4Gly7]OT (TGOT) (Lowbridge et al., 1977) has a remarkable selectivity for the mouse OTR through which, like the endogenous OT ligand, it activates Gq and Gi and recruits β-arrestins. Furthermore, we identified several antagonists that exhibit remarkable selectivity profiles at mOTR. These analogs represent valuable tools to investigate the specific role of the mOTRs in the brain.
Materials and Methods
Reagents, Constructs, and Peptides.
[3H]OT (NET-858, 30–60 Ci/mmol) and [3H]AVP (NET-800, 35–85 Ci/mmol) came from PerkinElmer Life and Analytical Sciences (Monza, Italy); coelenterazine H from Invitrogen (Milan, Italy); DeepBlueC coelenterazine 400a (CLz400) from Biotium (Hayward, CA); OT, AVP, TGOT, and atosiban from Bachem (Weil am Rhein, Germany); and SR49059 [(2S)1-[(2R,3S)-1,5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxybenzene-sulfonyl)-3-hydroxy-2,3-dihydro-1H-indole-2-carbonyl]-pyrrolidine-2-carboxamide] from sanofi-aventis (Toulouse, France). All of the other peptides used in this study were from the laboratory of M. Manning of the University of Toledo (Toledo, OH).
The mouse V1aR cDNA (mV1aR) came from OriGene (Rockville, MD); the mouse V1bR (mV1bR) cDNA was a gift from M.A. Ventura (U-567; INSERM, Paris, France).
The GFP10-Gγ2 cDNA, in which the blue-shifted variant of Aequorea victoria (GFP10) was fused to Gγ2, and the Gβ1 cDNA are described in Gales et al. (2006). The Gα subunit expression vector cDNAs came from Missouri S&T cDNA Resource Center (Rolla, MO). The expression vector of β-arrestin2 fused at its C terminus to the yellow fluorescent protein (β-arrestin2–YFP) (originally developed in M. Bouvier’s laboratory) came from Dr. J. Perroy (IGF, Montpellier, France), and the expression vector for β-arrestin1–YFP came from C. Hoffmann of the University of Wuerzburg (Wuerzburg, Germany). The mOTR C-terminally fused to Renilla luciferase (mOTR-Rluc) was generated by subcloning the entire coding region of mOTR into an Rluc vector (PerkinElmer BioSignal, Inc., Monza, Italy).
Cell Cultures.
HEK293 and COS7 cells purchased from the American Type Culture Collection (Manassas, VA) were grown in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, Milan, Italy), supplemented with 10% fetal calf serum and 1% penicillin-streptomycin (Sigma-Aldrich) in a 10% CO2 humidified atmosphere at 37°C.
Transfection.
For the ligand binding assays, the COS7 cells were transfected by means of electroporation as previously described (Chini et al., 1995). For the homogeneous time-resolved fluorescence (HTRF) and bioluminescence resonance energy transfer (BRET) assays, HEK293 cells were seeded at a density of 3,100,000 cells/well in 100-mm plates on the day before transfection. A mix containing 20 μg of DNA and 60 μg of polyethylenimine (PEI linear, MW 25000; Polysciences Europe GmbH, Eppelheim, Germany) was prepared with 1 ml of basic medium (without additives such as serum or antibiotics) and, after 15 minutes of incubation at room temperature, added directly to cells maintained in 10 ml of complete medium containing 10% fetal bovine serum. For the HTRF experiments, the cells were detached 24 hours after transfection and seeded with 100,000 cells/well in white half-area 96-well microplates (Corning Life Sciences, Amsterdam, The Netherlands). For the BRET experiments, the supplemented Dulbecco’s modified Eagle’s medium was renewed 24 hours after the transfections, and the cells were maintained in culture for an additional 24 hours before being washed twice, detached, and resuspended with phosphate-buffered saline (PBS)-MgCl2 (0.5 mM) at room temperature.
Ligand Binding Assays.
The binding assays were performed at 30°C on membranes prepared from COS7 cells as previously described (Chini et al., 1995). Compound affinities were determined by means of competition experiments in which the unlabeled compound concentrations varied from 10−11 to 10−5 M, and the concentration of the radioligand ([3H]OT for mOTR and [3H]AVP for mV1aR and mV1bR) was 2–4 × 10−9 M. Nonspecific binding was determined in the presence of unlabeled OT or AVP (10−3 M). The ligand binding data were analyzed by means of nonlinear regression using Prism version 5 (GraphPad Software, Inc., La Jolla, CA). The Ki values were calculated from the experimental IC50 values using the Cheng-Prusoff equation for a single population of competitive sites: Ki = IC50/[1 + (L/Kd)], where L is the concentration of radioligand used in each experiment and the Kd values were as previously reported: OT Kd = 0.54 nM for mOTR (Ring et al., 2010), AVP Kd = 1.3 nM for mV1aR (Oshikawa et al., 2004), and AVP Kd = 0.67 nM for mV1bR (Serradeil-Le Gal et al., 2007). All of the assays were performed in triplicate and repeated at least three times.
BRET Assay.
The interactions between mOTR and the different Gα subunits were analyzed by means of BRET2 experiments that use RLuc as the donor, the DeepBlueC coelenterazine derivative as its substrate, and GFP10 as the acceptor. HEK293 cells were cotransfected with mOTR-Rluc, GFP10-Gγ2, or Gβ1, without (-Gα) or with one of Gαq, Gαi1, Gαi2, Gαi3, Gαs, or Gαo. Cells were incubated for 2 minutes with OT, TGOT, and PBS (untreated cells) before the addition of Rluc substrate, DeepBlueC; BRET2 was measured immediately after using an Infinite F500 reader plate (Tecan, Milan, Italy) that allows the sequential integration of light signals detected with two filter settings (Rluc, 370–450 nm; GFP10 filter, 510–540 nm). The BRET2 signal was calculated as the ratio between GFP10 emission and the light emitted by Rluc. The positive changes in BRET induced by the ligands indicated the closest interaction between the donor and the acceptor and were expressed on graphs as “BRET ligand effect” using the formula: (emission GFP10 ligand/emission Rluc ligand) − (emission GFP10 PBS/emission Rluc PBS).
To analyze the kinetics of the mOTR–β-arrestin interactions, BRET1 experiments that use RLuc as the donor, coelenterazine H as its substrate, and YFP as the acceptor were performed. HEK293 cells were cotransfected with mOTR-Rluc and β-arrestin2–YFP or β-arrestin1–YFP. The transfected cells were distributed in a white 96-well microplate (100 μg of proteins per well) (Optiplate; PerkinElmer Life and Analytical Sciences) and incubated in the presence or absence of ligands. Coelenterazine H was added 8 minutes before the addition of the different ligands, and readings were made for 10 minutes using an Infinite F500 reader plate (Tecan) and filter set (Rluc filter, 370–480 nm; YFP filter, 520–570 nm). To determine the half-time (t1/2) of OT- and other ligand-induced BRET, the data were recorded as the difference between the ligand-promoted BRET signal and the average of the baseline (PBS-treated) BRET signal, and the time at which the half-BRET peak was reached was estimated.
Inositol Phosphate Measurements.
Myoinositol 1-phosphate (IP1) accumulation in HEK293 cells transiently transfected with mOTR, mV1aR, and mV1bR (100,000 cells) was determined in 96-well half-area microplates (Corning Life Sciences) using the HTRF-IP-One Kit (CisBio International, Bagnols-sur-Cèze, France) after 1 hour of stimulation with increasing concentrations of OT, AVP, and TGOT at 37°C. The time-resolved fluorescence resonance energy transfer signals were measured 50 μs after excitation at 620 and 665 nm using a Tecan Infinite F500 instrument. The IP1 concentrations were interpolated from the IP1 standard curve supplied with the kit.
Statistical Analysis.
All of the data were analyzed using GraphPad Prism software, version 5. Data from radioligand binding were evaluated by the nonlinear, least-squares curve-fitting procedure. Concentration-response IP1 curves were analyzed by means of nonlinear curve fitting using the sigmoidal dose-response equation. Parameters errors (Ki and EC50) are all expressed in %CV and calculated by simultaneous analysis of at least three different experiments performed in triplicate. Ki comparison has been performed on the basis of the F test for the extra sum of squares principle (*P < 0.05; **P < 0.01; ***P < 0.001). Ligand-induced BRET ratios are expressed as mean ± S.E.M and were analyzed with one-way analysis of variance followed by Tukey’s post hoc test to determine statistically significant differences in treatments (***P < 0.001). The BRET1 kinetics data were normalized by setting the zero time point immediately after the addition of the ligand, and the data were analyzed by means of nonlinear least-squares fitting to the one-phase exponential association equation.
Homology Modeling of the mOTR Structure.
A large number of GPCR crystal structures in different activity-state-related conformations have been published in recent years (Zhao and Wu, 2012), most of them cocrystallized with specific ligands (agonists or antagonists) (Kobilka and Schertler, 2008; Hanson and Stevens, 2009). Therefore, they serve as optimal templates for family A GPCR homology modeling (OTRs are members of family A GPCRs) with the purpose to study potential details of ligand binding or signal transduction.
Based on high sequence similarity and overlapping structural features in the transmembrane helices (TMHs), the β2-adrenergic receptor crystal structure in an active conformation was used here as a template (Protein Data Bank code 3SN6) (Rasmussen et al., 2011) for modeling of the murine OTR. The general modeling procedure (sequence alignment, crystal structure preparation for modeling, side chain substitutions) was performed as recently described (Costanzi, 2012).
In addition, extracellular loop (ECL) 2 of the β2-adrenergic receptor was partially deleted, because of significant differences in length and amino acid composition (biophysical properties) compared with the sequence of mOTR ECL2. According to advanced insights from previous GPCR modeling studies [supplemental material in Michino et al. (2009)], only the C-terminal part of the template ECL2 structure, which includes the highly conserved cysteine bridge to TMH3, was kept. Other mOTR ECL2 residues, most likely not involved in direct constitution of the ligand binding region, were manually added as spacers. The putative general OTR ligand binding region, located between the extracellular ends of the TMHs and the ECLs, was defined based on similarity to the ligand binding regions of known GPCR crystal structures (Deupi and Standfuss, 2011; Kratochwil et al., 2011; Wichard et al., 2011; Jacobson and Costanzi, 2012).
The OTR model is constituted by amino acids from positions Glu36 to Leu344 (the extracellular N terminus and intracellular C terminus are not included because of missing structural templates). For modeling procedures, the Sybyl-X 2.0 version (Tripos International, St. Louis, MO) was used. Gaps of missing residues in the loops of the template structure were closed manually by adding OTR-specific amino acids. Side chains were subjected to conjugate gradient minimizations (until converging at a termination gradient of 0.1 kcal/mol/Å) and molecular dynamics simulation (2 ns) by fixing the backbone of the TMHs. Finally, the model was minimized without constraints using the AMBER 7 forcefield. Structure images were produced using the PyMOL software (PyMOL Molecular Graphics System, version 1.3; Schrödinger, LLC, Portland, OR).
Results
Binding Affinities of Commonly Used OT/AVP Analogs at Mouse Receptors.
The ligand binding properties of commonly used OT/AVP analogs at mOTR, mV1aR, and mV1bR were determined by competition experiments; calculated Ki values ± %CV are reported in Table 1.
TABLE 1.
Amino acid sequences and affinity values (Ki) of the investigated ligands
Substitutions and/or modifications of the amino acid sequence of OT are indicated in boldface type; the superscript numbers indicate the position of the residue in the peptide sequence.
| Analog | Sequence |
Ki for: |
||
|---|---|---|---|---|
| mOTR | mV1a | mV1b | ||
| nM ± %CV | ||||
| OT | Cys1, Tyr2, Ile3, Gln4, Asn5, Cys6, Pro7, Leu8, Gly-NH29 | 0.83 ± 17 | 20.38 ± 26 | 36.32 ± 7 |
| AVP | Cys1, Tyr2, Phe3, Gln4, Asn5, Cys6, Pro7, Arg8, Gly-NH29 | 0.87 ± 8 | 1.11 ± 27 | 0.43 ± 12 |
| dLVT | dCys1, Tyr2, Ile3, Gln4, Asn5, Cys6, Pro7, Lys8, Gly-NH29 | 0.43 ± 20 | 3.39 ± 28 | 0.82 ± 7 |
| TGOT | Cys1, Tyr2, Ile3, Thr4, Asn5, Cys6, Gly7, Leu8, Gly-NH29 | 0.04 ± 32 | >1000 | >10,000 |
| Atosiban | dCys1, DTyr(Et)2, Ile3, Thr4, Asn5, Cys6, Pro7, Orn8, Gly-NH29 | 1.29 ± 46 | >1000 | >1000 |
| OTA1 | d(CH2)51, Tyr(Me)2, Ile3, Thr4, Asn5, Cys6, Pro7, Orn8, Tyr-NH29 | 0.13 ± 42 | 34.3 ± 33 | 374.9 ± 20 |
| OTA2 | desGly-NH2,d(CH2)51, Tyr(Me)2, Ile3, Thr4, Asn5, Cys6, Pro7, Orn8 | 0.27 ± 25 | 232 ± 48 | >10,000 |
| OTA3 | desGly-NH2,d(CH2)51, DTyr2, Ile3, Thr4, Asn5, Cys6, Pro7, Orn8 | 1.24 ± 36 | >10,000 | >10,000 |
| LVA | Phenylac1, DTyr(Me)2, Phe3, Gln4, Asn5, Arg6, Pro7, Arg8 | 3.90 ± 30 | 0.10 ± 19 | 9.66 ± 12 |
| Manning compound | d(CH2)51, Tyr(Me)2, Phe3, Gln4, Asn5, Cys6, Pro7, Arg8, Gly-NH29 | 42.6 ± 18 | 1.72 ± 28 | 73.87 ± 9 |
| SR49059 | 13.2 ± 19 | 0.94 ± 22 | 97.35 ± 12 | |
d, deamino; d(CH2)5, β-mercapto-β,β-penthamethylenepropionic; desGly-NH2, desglycineamide; DTyr(Et), O-ethyl-d-tyrosine; Orn, ornithine; Phenylac, phenylacetyl; Tyr(Me), O-methyltyrosine.
With regard to the peptides with agonist activity, the endogenous OT and AVP ligands had very different selectivity profiles (Fig. 1, A and B). AVP bound to the three brain-expressed OT/AVP receptors with almost identical affinity (Ki values for OTR, V1aR, and V1bR of, respectively, 0.87 nM ± 8% CV, n = 3; 1.11 nM ± 27% CV, n = 4; and 0.43 nM ± 12% CV, n = 4), whereas OT had a receptor-specific affinity range that was highest for OTR (Ki = 0.83 nM ± 17% CV, n = 4) and lower for V1aR (Ki = 20.38 ± 26% CV, n = 5) (P < 0.001 versus mOTR) and V1bR (Ki = 36.32 nM ± 7% CV, n = 4) (P < 0.001 versus mOTR). The dLVT peptide agonist binds with significantly different Ki values for OTR, V1aR, and V1bR of, respectively, 0.43 nM ± 20% CV, n = 5; 3.39 nM ± 28% CV, n = 5 (P < 0.001 versus mOTR); and 0.82 nM ± 7% CV, n = 3 (P < 0.01 versus mOTR) (Fig. 1C). However, we should mention here that a significant difference in Ki is not sufficient to define ligand selectivity. As an operational criterion, it has been proposed that, to be “selective” for a particular subtype, any ligand should display a Ki at least 2 orders of magnitude lower than that for the other receptor subtypes (Chini et al., 2008). On these premises, the only agonist provided with a good selectivity profile is TGOT, with a Ki of 0.04 nM ± 32% CV, n = 5, for OTR and Ki values of >1000 nM for V1aR and V1bR (Fig. 1D).
Fig. 1.

Binding properties of OT/AVP agonists at mOTR, mV1aR, and mV1bR. Competition binding experiments were performed using increasing concentrations (from 10−11 to 10−5 M) of the endogenous ligands OT (A) and AVP (B) and the synthetic ligands dLVT (C) and TGOT (D). Ligand binding was determined on membrane preparations of COS7 cells transiently transfected with mOTR (black circles), mV1aR (blue triangles), and mV1bR (red squares). Specific binding was determined in the presence of 2–4 × 10−9 M [3H]OT for mOTR and [3H]AVP for mV1aR and mV1bR; nonspecific binding was determined in the presence of OT or AVP (10−3 M). Each curve is the mean of triplicate determinations of a single representative experiment.
Among the Gq OTR antagonists (OTAs) that we analyzed, whose original synthesis and pharmacological properties are reviewed in Manning et al. (2008, 2012), atosiban, OTA2, and OTA3 were selective for mOTR (Fig. 2, A, C, and D), with affinities in the nanomolar range (Ki = 1.29 nM ± 46% CV, n = 4 for atosiban; Ki = 0.27 nM ± 25% CV, n = 4 for OTA2; and Ki = 1.24 nM ± 36% CV, n = 4 for OTA3), and showing Ki values for V1aR and V1bR that were >1000-fold higher (P < 0.001). The widely used OTR antagonist OTA1 also had a good selectivity profile (Fig. 2B), with highest affinity for OTR (Ki = 0.13 nM ± 42% CV, n = 5), intermediate for V1aR (Ki = 34.3 nM ± 33% CV, n = 4) (P < 0.001), and lowest for V1bR (Ki = 374 nM ± 20% CV, n = 5) (P < 0.001).
Fig. 2.

Binding properties of commonly used OTAs at mOTR, mV1aR, and mV1bR. Competition binding experiments were performed using increasing concentrations (from 10−11 to 10−5 M) of the Gq antagonists atosiban (A), OTA1 (B), OTA2 (C), and OTA3 (D). Ligand binding was determined on membrane preparations of COS7 cells transiently transfected with mOTR (black circles), mV1aR (blue triangles), and mV1bR (red squares). Specific binding was determined in the presence of 2–4 × 10−9 M [3H]OT for mOTR and [3H]AVP for mV1aR and mV1bR; nonspecific binding was determined in the presence of OT or AVP (10−3 M). Each curve is the mean of triplicate determinations of a single representative experiment.
We finally analyzed three compounds commonly used as V1aR-selective antagonists: LVA (linear vasopressin antagonist), the Manning compound, and the nonpeptidic antagonist SR49059 [reviewed in Manning et al. (2008, 2012)]. As shown in Fig. 3, A–C, they bound with different affinities to mouse OTR, V1aR, and V1bR. The three antagonists bounds with significantly different Ki values to OTR and V1aR (P < 0.001); SR49059 also had a significantly different Ki value for V1bR (P < 0.001 versus mOTR). However, none of them had a good selectivity profile for mouse OTR, V1aR, and V1bR, as their Ki affinities for the V1aR were at most 10–20 times lower than those for OTR (Table 1).
Fig. 3.

Binding properties of commonly used V1a antagonists at mOTR, mV1aR, and mV1bR. Competition binding experiments were performed using increasing concentrations (from 10−11 to 10−5 M) of three compounds commonly used as selective V1aR antagonists: LVA (A), the Manning compound (B), and SR49059 (C). Ligand binding was determined on membrane preparations of COS7 cells transiently transfected with mOTR (black circles), mV1aR (blue triangles), and mV1bR (red squares). Specific binding was determined in the presence of 2–4 × 10−9 M [3H]OT for mOTR and [3H]AVP for mV1aR and mV1bR; nonspecific binding was determined in the presence of OT or AVP (10−3 M). Each curve is the mean of triplicate determinations of a single representative experiment.
Coupling Properties of Commonly Used OT/AVP Analogs at Mouse Receptors
As mOTR, mV1aR, and mV1bR are all coupled to Gq, leading to phospholipase C activation, inositol phosphate production, and an increase in intracellular calcium, we assayed the efficacy of the agonists by drawing up concentration-response curves of IP1 production at the three receptor subtypes. Our results indicate that OT activated mOTR with an EC50 of 4.45 nM ± 31% CV, n = 3; and mV1aR and mV1bR with similar, lower, EC50 values (171 nM ± 19% CV, n = 3; and 87 nM ± 45% CV, n = 3) (Fig. 4A), displaying a good selectivity profile. On the contrary, AVP activates mV1aR, mV1bR, and mOTR with decreasing potency (Fig. 4B), with calculated EC50 values of, respectively, 0.65 nM ± 89% CV, n = 3; 6.62 nM ± 32% CV, n = 3; and 47.9 nM ± 69% CV, n = 3. There were no significant differences between OT and AVP Emax in the three receptor subtypes. In comparison with OT, the highly selective analog TGOT was characterized by a left-shifted dose-response curve (Fig. 4C), as expected on the basis of its high binding affinity. The calculated EC50 of TGOT for mOTR was 0.18 nM ± 83% CV, n = 3; whereas no IP1 production was observed for mV1aR and mV1bR also at very high peptide doses (>1000 nM), which is in accordance with its low affinity for these receptor subtypes.
Fig. 4.

Receptor/Gq coupling properties of OT/AVP analogs determined by means of IP1 inositol phosphate production. IP1 production was measured using an immune-competitive HTRF-based assay in HEK293 cells transiently transfected with mOTR (black circles), mV1aR (blue triangles), and mV1bR (red squares). A total of 100,000 cells were stimulated for 30 minutes with increasing concentrations (10−14 to 10−5 M) of OT (A), AVP (B), and TGOT (C). Each curve is the mean of triplicate determinations of a single representative experiment.
The functional selective properties of TGOT on mOTR coupling were investigated by means of a BRET2-based assay in which the energy donor RLuc is fused to the C-terminal end of mOTR cDNA and GFP10 used as the acceptor is N-terminally fused to the Gγ2 subunit (GFP10-Gγ2). As shown in Fig. 5, upon OT (10−5 M) and TGOT (10−5 M) binding, mOTR significantly (P < 0.001 versus PBS) recruits Gq, Gi1, Gi2, Gi3, and G0, but not Gs, thus confirming its coupling to the same G-protein subtypes recruited by OT (Busnelli et al., 2012).
Fig. 5.

TGOT-induced G-protein recruitment determined by means of a BRET-based assay. BRET2 was measured in HEK293 cells transiently transfected with mOTR-RLuc, GFP10-Gγ2, and Gβ1 in the absence (-Gα) or presence of the indicated Gα subunits. The data represent the differences in BRET signals between the specified BRET partners in the absence (PBS; empty bars) or presence of OT (10−5 M; black bars) and TGOT (10−5 M; red bars), and are expressed as the mean value ± S.E.M. of three independent assays performed in triplicate. One-way analysis of variance followed by Tukey’s test was used to determine the statistical differences between treatments. ***P < 0.001 vs. untreated controls (PBS).
Finally, to investigate whether TGOT induces β-arrestin recruitment after receptor activation, we used a “real-time” BRET1 assay that uses the mOTR-RLuc construct as the energy donor and β-arrestin2–YFP or β-arrestin1–YFP as the acceptor (Fig. 6). In cells coexpressing mOTR-Rluc and β-arrestin2–YFP or β-arrestin1–YFP, OT at a final concentration of 10 μM increased the BRET ratio with t1/2 values of, respectively, 89.9 ± 2.3 seconds (n = 3) and 101 ± 6.1 seconds (n = 3); similarly, TGOT at the same concentration increased the BRET ratio with t1/2 values of, respectively, 124.6 ± 4.2 seconds (n = 3) and 121.9 ± 5.1 seconds (n = 3). Moreover, the BRET ratio remained stable for at least 10 minutes (Fig. 6), thus indicating a sustained agonist-induced association between mOTR and β-arrestins.
Fig. 6.

TGOT-mediated β-arrestin1 and β-arrestin2 recruitment. BRET1 was monitored in HEK293 cells transiently transfected with mOTR-RLuc and β-arrestin1–YFP or β-arrestin2–YFP. The cells were stimulated with OT (10−5 M; black squares) or TGOT (10−5 M; red circles). Real-time BRET1 measurements were made every 20 seconds for 10 minutes. The data represent the differences in BRET signals between the specified BRET partners in the absence or presence of the OT and TGOT agonist. Each curve is the mean of triplicate determinations of a single representative experiment.
Insights into Molecular Differences between OTR Subtypes Based on a Structural Model.
To evaluate differences in amino acid composition among mouse, rat, and human OTR subtypes, we first performed an alignment of their amino acid sequences (Fig. 7). Secondly, we designed a three-dimensional homology model of a putative mOTR conformation to analyze the spatial distribution of different amino acids at corresponding positions and to study structural-functional features of the mOTR (Fig. 8). The sequence alignment shows that several not highly conserved amino acids are distributed over the entire receptor structure, in particular in the N-terminal region and C-terminal regions (Fig. 7). In a previous study, it was shown that truncation of the first 32 residues of the N terminus of the hOTR did not influence OT binding (Wesley et al., 2002), and the only residue in the N terminus found to be relevant for high-affinity OT binding was the conserved arginine at position 34 (Wesley et al., 2002). Variations in the N terminus are thus unlikely to be involved in determining TGOT high-affinity binding to the mOTR.
Fig. 7.
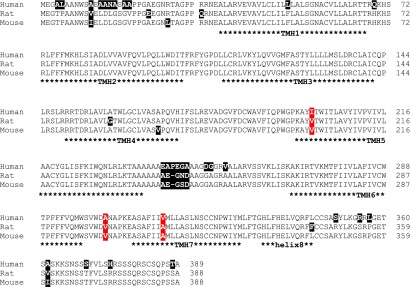
Sequence alignment of human, rat, and mouse OTR. The sequences of the human, rat, and mouse OTR orthologs are aligned to each other. Asterisks indicate potential TMHs numbered from 1 to 7 and helix 8. Positions of varying amino acids between the OTR subtypes are highlighted with black shadows, whereby the three positions of side chain variations that are in close spatial proximity to the putative ligand binding pocket are highlighted with red shadows.
Fig. 8.

Structural homology model of the monomeric mouse OTR. The structural model (active conformation) of the mOTR (backbone cartoon, white) is represented without the N-terminal extracellular part (Ntt), the intracellular tail (Ctt), and the middle portion of ICL3. The potential ligand binding region is highlighted by an inner surface (green). This pocket-like crevice is constituted by specific amino acids (green lines) located at the ECLs and the TMHs toward the extracellular site. This three-dimensional representation of the OTR is helpful to identify potential links between functional differences of the receptor subspecies with particular residue variations. Amino acid positions that are not conserved between rodent and human OTR are shown as sticks (red and brown, labeled). Amino acids Val201 (TMH5), Val301 (ECL3), and Ala313 (TMH7) (red sticks) are located in close spatial proximity to the ligand binding region, whereby only Val201 is a direct determinant of the main ligand binding pocket. Alanine at position 159 and Val169 (TMH4) as well as Phe51 (TMH1) or His69 (ICL1) are outside the putative ligand binding site, and side chain variations should not have direct influences on ligand binding properties.
The three-dimensional visualization of the mouse OTR serpentine domain (TMHs and loops) shows that Val201 in TMH5 (hOTR: isoleucine, rOTR: valine), Val301 in ECL3 (hOTR: alanine, rOTR: valine) and Ala313 in TMH7 (hOTR: valine, rOTR: alanine) are located in close spatial proximity to the putative ligand binding region (Fig. 8). Based on our model, only Val201 directly participates in the determination of the ligand pocket properties (such as the shape) and biophysical parameters. At this position, an isoleucine is located in the hOTR, which is different in bulkiness and length compared with the rodent valine.
Alanine at position 159 (hOTR: alanine; rOTR: glycine) and valine at position 169 (hOTR: alanine; rOTR: alanine), located at TMH4, are outside the ligand binding pocket. Residue Phe51 inTMH1 (hOTR: leucine, rOTR: phenylalanine) points toward the membrane without any intramolecular interaction, and His69 (hOTR: glutamine; rOTR: histidine) is located at intracellular loop 1 (ICL1). Therefore, they should not have a direct impact on ligand binding, even though indirect effects due to changes in intrinsic signaling capacity (e.g., helix movement flexibility) cannot be excluded.
Discussion
We describe the in vitro pharmacological characterization of several analogs of mouse OTR, V1a, and V1b receptors, the three OT/AVP receptor subtypes expressed in mammalian brain. Peptidic and nonpeptidic OT/AVP analogs have primarily been assayed for their agonistic and antagonistic activities in in vitro and in vivo assays based on the peripheral effects of OT/AVP receptors, with OTR activities being quantified on the basis of myometrial contractility, V1aR activities on the basis of vasoconstriction, V1bR activities on the basis of adrenocorticotropin release, and vasopressin 2 receptor activities on the basis of antidiuresis. Much fewer pharmacological data have been collected concerning the selective effects of these analogs within the brain, where their use at very high doses has often led to conflicting or inconsistent results (Engelmann et al., 1996). A systematic analysis of the affinity and efficacy of OT/AVP compounds in selected brain areas would therefore be extremely valuable in preclinical research. However, this approach is hindered by a number of technical issues. First of all, OT/AVP receptors are not highly expressed in brain, and secondly, tritiated OT and AVP radiotracers have low specific activity. Overcoming these limitations would require tissue enrichment procedures to obtain consistent and reproducible results (Elands et al., 1988), but this would involve the use of a large number of animals, increase costs, and raise ethical concerns. Consequently, we believe that the in vitro characterization of transfected receptors represents a preliminary step for the selection of candidate drugs to be tested in vivo.
Our in vitro results indicate that, as observed in other animal species, endogenous OT and AVP ligands have different selectivity profiles for mouse OT/AVP receptors. AVP binds to the three brain-expressed OT/AVP receptors with almost identical affinity, but activates the mV1aR, mV1bR, and mOTR with decreasing potency over 2 orders of magnitude. On the contrary, OT has a receptor-specific affinity range that is highest for OTR and lower for V1aR and V1bR, and this correlates with its potency at the sites of the three receptor subtypes. Depending on the dose and site of administration, some of the actions of AVP may be mediated via the OTR, and OT can bind and activate V1aR and V1bR expressed in the brain, albeit with lower affinity than AVP itself, which means that exogenously administered OT agonists can elicit substantial responses by binding to AVP receptors. On the other hand, a low AVP dose may also act as a “competitive antagonist” at the mOTR, and particularly the Gq-mediated pathway, which is activated with a high EC50. Moreover, the fact that AVP is devoid of agonist activity upon OTR-mediated Gi1, GoA, and GoB activation (Busnelli et al., 2012) has still undefined pharmacological implications.
In this study, we show that the synthetic peptide TGOT has remarkable OTR-versus-V1aR/V1bR selectivity in terms of affinity binding and coupling for mice receptors. TGOT was originally demonstrated to be highly selective for rat OTR by means of in vivo bioassays (Lowbridge et al., 1977). However, this enhanced OTR/V1a selectivity is lost in humans, in whom the affinities of OT and TGOT to OTR and V1a receptors are comparable, thus indicating that the use of TGOT does not have any advantage over OT as far as OTR/V1a selectivity is concerned (Chini and Manning, 2007). As shown in Table 2, comparison of OT and TGOT affinities for human, rat, and mouse OTRs indicates that OT has the same affinity in the three species, whereas the affinity of TGOT increases by a factor of 100 going from human to rat to mouse. Concerning its coupling features, TGOT binding to the OTR led to the activation of Gq and all the members of the Gi and Go family exactly as the endogenous OT ligand (Busnelli et al., 2012). This is particularly relevant in neuronal cells, where it has been shown that OTR coupling to Gq and Gi/Go results in opposite effects on cell excitability via inhibition or activation of potassium channels (Gravati et al., 2010). Furthermore, TGOT promotes β-arrestin1 and β-arrestin2 recruitment as efficiently as OT, suggesting similar desensitization and internalization properties.
TABLE 2.
Affinity constants of OT and TGOT for the human, rat, and mouse OTR
|
Ki (mean ± S.D.) for: |
|||
|---|---|---|---|
| Human OTR | Rat OTR | Mouse OTR | |
| nM | |||
| OT | 0.79 ± 0.22a | 1.0 ± 0.1b | 0.83 ± 0.14c |
| TGOT | 6.62 ± 1.22d | 0.8 ± 0.2b | 0.04 ± 0.01c |
From Chini et al. (1995).
From Elands et al. (1988).
From the current study.
From Chini et al. (1996).
Concerning our pharmacological screening for mOTR-selective antagonists, three peptides were shown to be very selective: atosiban, OTA2, and OTA3. In our hands, the most promising antagonist is OTA3, which we found to be highly selective for mOTR. Unfortunately, among the V1aR antagonists tested, we didn’t identify any ligand with a Ki for the murine V1a receptor subtype at least 2 orders of magnitude lower than that for the other two receptor subtypes, a condition previously set as the minimal requirement for a selective ligand (Chini et al., 2008). The Manning compound, originally described as a potent V1aR-selective antagonist in rat, also was found to be not selective in humans (Manning et al., 2012) or in mice (this study) and can’t be used as a selective V1a antagonist in these species.
Revealing variations in ligand binding and signaling among OTR orthologs in in vitro models has an impact on various aspects of OT/AVP pharmacology, including the identification of pharmacological tools that can be used in single species of particular translational interest, such as genetically modified mouse models. Our study identified analogs that lack selectivity for human receptors but are highly selective for mouse OTRs and therefore very valuable for preclinical studies. However, as we used transfected cells, one major issue is to verify whether the selectivity profile observed in transfected cells is maintained in vivo. In this regard, when used in mice at a dose of 0.0008 ng/animal, OTA3 specifically blocked the mOTR-mediated rescue of sociability defects in heterozygous Oxtr+/− animals, suggesting the validity of this approach to identify mOTR antagonists (Sala et al., 2013). TGOT has been previously used in mice mainly in electrophysiology experiments, which found evidence of its selectivity for OTR- versus V1a-mediated responses (Huber et al., 2005; Gozzi et al., 2010). However, in slices, the effective dose was very similar to the OT doses used, and the half-maximally effective concentration was only slightly more potent than OT (Huber et al., 2005). A similar discrepancy between in vitro and in vivo potencies of TGOT was also observed in social behavioral rescue experiments recently performed in OTR-null mice (Sala et al., 2011, 2013). TGOT rescued the social deficit at a dose of 0.0005 ng/animal in Oxtr+/− mice, which is consistent with a selective action of TGOT through OTRs. However, at a dose of 0.05 ng/animal, TGOT also rescued the social deficit of Oxtr−/− mice, suggesting that despite its very low affinity for the V1a and V1b receptors in vitro, TGOT was still active on these receptors in vivo (Sala et al., 2013). Several factors may be responsible for the discrepancy in TGOT potency observed in vitro and in vivo. Diffusion and enzymatic degradation may greatly affect peptides’ stability in tissues. In the brain, the aminopeptidase oxytocinase hydrolyzes OT, AVP, enkephalins, and other neuropeptides. The enzyme is present in soluble and membrane-bound forms, and its distribution varies greatly in different brain regions (Fernando et al., 2005). In addition, binding affinity and velocity of catalysis for different substrates could also account for significant differences in local neuropeptide concentrations and final neurobiological effects.
Finally, studying the pharmacology of receptor orthologs may contribute to optimizing the design of selective analogs because subtle differences in ortholog activation can reveal crucial ligand-receptor interactions involved in binding and activation processes. As the conservation of the OTR sequence in the three species is very high (>90%), it should be feasible, in principle, to identify the variable receptor residue(s) responsible for differences in affinity binding among the three species. By analyzing an OTR ortholog sequence alignment (Fig. 7) and by molecular modeling of mOTR (Fig. 8), we explored the distribution and potential relevance of nonconserved residues to high agonist binding. Most of the substitutions among the subspecies are located at the N terminus and in ICL3. So far as is known, these receptor parts have not directly related to ligand affinity and selectivity in OTR species (Wesley et al., 2002). Three varying positions are located in close spatial proximity to the putative ligand binding region, whereby only the residue at TMH5 directly participates in the constitution of the ligand binding pocket. However, at none of these positions is present a residue that is different in all the three species, suggesting that the increased TGOT affinity observed in rodent OTR probably does not result from a single substitution but is more likely due to the combination of particular variation(s) close to the ligand binding site and/or at other distinct receptor parts. With respect to this topic, it will be of future interest to explore the effect of multiple amino-acid substitutions in OTR subspecies.
In conclusion, our results indicate that the selectivity profile of OTR/V1a/V1b receptors for the natural OT and AVP ligands is conserved in humans and rodents (rats and mice). However, subtle differences between receptor orthologs are responsible for an increase in the affinity of the synthetic agonist TGOT for mOTR. We also identified a number of OTAs characterized by very high selectivity for mOTR. TGOT and OTAs are therefore valuable molecular tools for investigating specific mOTR-mediated effects.
Abbreviations
- AVP
vasopressin
- BRET
bioluminescence resonance energy transfer
- ECL
extracellular loop
- GPCR
G-protein-coupled receptor
- HTRF
homogeneous time-resolved fluorescence
- ICL
intracellular loop
- IP1
myoinositol 1-phosphate
- LVA
linear vasopressin antagonist
- OT
oxytocin
- OTA
oxytocin receptor antagonist
- OTR
oxytocin receptor
- PBS
phosphate-buffered saline
- RLuc
Renilla luciferase
- SR49059
(2S)1-[(2R,3S)-1,5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxybenzene-sulfonyl)-3-hydroxy-2,3-dihydro-1H-indole-2-carbonyl]-pyrrolidine-2-carboxamide
- t1/2
half-time
- TGOT
[Thr4Gly7]OT
- TMH
transmembrane helix
- V1aR
vasopressin 1a receptor
- V1bR
vasopressin 1b receptor
- YFP
yellow fluorescent protein
Authorship Contributions
Participated in research design: Busnelli, Chini.
Conducted experiments: Busnelli, Bulgheroni, Kleinau.
Contributed reagents: Manning.
Performed data analysis: Busnelli, Bulgheroni, Kleinau, Chini.
Wrote or contributed to the writing of the manuscript: Busnelli, Manning, Kleinau, Chini.
Footnotes
This study was supported by Cariplo Foundation (Grant 2008.2314), Regione Lombardia (Progetto TerDisMental, ID 16983–Rif. SAL-50), and Telethon Foundation (Grant GGP12207).
References
- Barberis C, Morin D, Durroux T, Mouillac B, Guillon G, Seyer R, Hibert M, Tribollet E. (1999) Molecular pharmacology of AVP and OT receptors and therapeutic potential. Drug News Perspect 12:279–292 [Google Scholar]
- Birnbaumer M. (2000) Vasopressin receptors. Trends Endocrinol Metab 11:406–410 [DOI] [PubMed] [Google Scholar]
- Busnelli M, Saulière A, Manning M, Bouvier M, Galés C, Chini B. (2012) Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J Biol Chem 287:3617–3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini B, Manning M. (2007) Agonist selectivity in the oxytocin/vasopressin receptor family: new insights and challenges. Biochem Soc Trans 35:737–741 [DOI] [PubMed] [Google Scholar]
- Chini B, Manning M, Guillon G. (2008) Affinity and efficacy of selective agonists and antagonists for vasopressin and oxytocin receptors: an “easy guide” to receptor pharmacology. Prog Brain Res 170:513–517 [DOI] [PubMed] [Google Scholar]
- Chini B, Mouillac B, Ala Y, Balestre MN, Trumpp-Kallmeyer S, Hoflack J, Elands J, Hibert M, Manning M, Jard S, et al. (1995) Tyr115 is the key residue for determining agonist selectivity in the V1a vasopressin receptor. EMBO J 14:2176–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S. (2012) Homology modeling of class A G protein-coupled receptors. Methods Mol Biol 857:259–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X, Standfuss J. (2011) Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol 21:541–551 [DOI] [PubMed] [Google Scholar]
- Elands J, Barberis C, Jard S. (1988) [3H]-[Thr4,Gly7]OT: a highly selective ligand for central and peripheral OT receptors. Am J Physiol 254:E31–E38 [DOI] [PubMed] [Google Scholar]
- Engelmann M, Wotjak CT, Neumann I, Ludwig M, Landgraf R. (1996) Behavioral consequences of intracerebral vasopressin and oxytocin: focus on learning and memory. Neurosci Biobehav Rev 20:341–358 [DOI] [PubMed] [Google Scholar]
- Ferguson JN, Aldag JM, Insel TR, Young LJ. (2001) Oxytocin in the medial amygdala is essential for social recognition in the mouse. J Neurosci 21:8278–8285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. (2000) Social amnesia in mice lacking the oxytocin gene. Nat Genet 25:284–288 [DOI] [PubMed] [Google Scholar]
- Fernando RN, Larm J, Albiston AL, Chai SY. (2005) Distribution and cellular localization of insulin-regulated aminopeptidase in the rat central nervous system. J Comp Neurol 487:372–390 [DOI] [PubMed] [Google Scholar]
- Galés C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol 13:778–786 [DOI] [PubMed] [Google Scholar]
- Gozzi A, Jain A, Giovannelli A, Bertollini C, Crestan V, Schwarz AJ, Tsetsenis T, Ragozzino D, Gross CT, Bifone A. (2010) A neural switch for active and passive fear. Neuron 67:656–666 [DOI] [PubMed] [Google Scholar]
- Gravati M, Busnelli M, Bulgheroni E, Reversi A, Spaiardi P, Parenti M, Toselli M, Chini B. (2010) Dual modulation of inward rectifier potassium currents in olfactory neuronal cells by promiscuous G protein coupling of the oxytocin receptor. J Neurochem 114:1424–1435 [DOI] [PubMed] [Google Scholar]
- Hanson MA, Stevens RC. (2009) Discovery of new GPCR biology: one receptor structure at a time. Structure 17:8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida H, Yokoyama S, Munesue T, Kikuchi M, Minabe Y, Lopatina O. (2011) CD38 gene knockout juvenile mice: a model of oxytocin signal defects in autism. Biol Pharm Bull 34:1369–1372 [DOI] [PubMed] [Google Scholar]
- Huber D, Veinante P, Stoop R. (2005) Vasopressin and oxytocin excite distinct neuronal populations in the central amygdala. Science 308:245–248 [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Costanzi S. (2012) New insights for drug design from the X-ray crystallographic structures of G-protein-coupled receptors. Mol Pharmacol 82:361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Alphonse F, Perkovska S, Frantz MC, Durroux T, Méjean C, Morin D, Loison S, Bonnet D, Hibert M, Mouillac B, et al. (2009) Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J Am Soc Nephrol 20:2190–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, et al. (2007) CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446:41–45 [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2011) Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 336:296–302 [DOI] [PubMed] [Google Scholar]
- Kobilka B, Schertler GF. (2008) New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci 29:79–83 [DOI] [PubMed] [Google Scholar]
- Kratochwil NA, Gatti-McArthur S, Hoener MC, Lindemann L, Christ AD, Green LG, Guba W, Martin RE, Malherbe P, Porter RH, et al. (2011) G protein-coupled receptor transmembrane binding pockets and their applications in GPCR research and drug discovery: a survey. Curr Top Med Chem 11:1902–1924 [DOI] [PubMed] [Google Scholar]
- Lowbridge J, Manning M, Haldar J, Sawyer WH. (1977) Synthesis and some pharmacological properties of [4-threonine, 7-glycine]oxytocin, [1-(L-2-hydroxy-3-mercaptopropanoic acid), 4-threonine, 7-glycine]oxytocin (hydroxyl[Thr4, Gly7]oxytocin), and [7-glycine]oxytocin: peptides with high oxytocic-antidiuretic selectivity. J Med Chem 20:120–123 [DOI] [PubMed] [Google Scholar]
- MacKinnon AC, Tufail-Hanif U, Wheatley M, Rossi AG, Haslett C, Seckl M, Sethi T. (2009) Targeting V1A-vasopressin receptors with [Arg6, D-Trp7,9, NmePhe8]-substance P (6-11) identifies a strategy to develop novel anti-cancer therapies. Br J Pharmacol 156:36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning M, Misicka A, Olma A, Bankowski K, Stoev S, Chini B, Durroux T, Mouillac B, Corbani M, Guillon G. (2012) Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J Neuroendocrinol 24:609–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning M, Stoev S, Chini B, Durroux T, Mouillac B, Guillon G. (2008) Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin V1a, V1b, V2 and OT receptors: research tools and potential therapeutic agents. Prog Brain Res 170:473–512 [DOI] [PubMed] [Google Scholar]
- Michino M, Abola E, Brooks CL, 3rd, Dixon JS, Moult J, Stevens RC, GPCR Dock 2008 participants (2009) Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov 8:455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. (2013) Neuroscience. The promise and perils of oxytocin. Science 339:267–269 [DOI] [PubMed] [Google Scholar]
- Modi ME, Young LJ. (2012) The oxytocin system in drug discovery for autism: animal models and novel therapeutic strategies. Horm Behav 61:340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa S, Tanoue A, Koshimizu TA, Kitagawa Y, Tsujimoto G. (2004) Vasopressin stimulates insulin release from islet cells through V1b receptors: a combined pharmacological/knockout approach. Mol Pharmacol 65:623–629 [DOI] [PubMed] [Google Scholar]
- Pittman QJ, Spencer SJ. (2005) Neurohypophysial peptides: gatekeepers in the amygdala. Trends Endocrinol Metab 16:343–344 [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reversi A, Rimoldi V, Marrocco T, Cassoni P, Bussolati G, Parenti M, Chini B. (2005) The oxytocin receptor antagonist atosiban inhibits cell growth via a “biased agonist” mechanism. J Biol Chem 280:16311–16318 [DOI] [PubMed] [Google Scholar]
- Ring RH, Schechter LE, Leonard SK, Dwyer JM, Platt BJ, Graf R, Grauer S, Pulicicchio C, Resnick L, Rahman Z, et al. (2010) Receptor and behavioral pharmacology of WAY-267464, a non-peptide oxytocin receptor agonist. Neuropharmacology 58:69–77 [DOI] [PubMed] [Google Scholar]
- Sala M, Braida D, Donzelli A, Martucci R, Busnelli M, Bulgheroni E, Rubino T, Parolaro D, Nishimori K, Chini B. (2013) Mice heterozygous for the oxytocin receptor gene (Oxtr(+/-)) show impaired social behaviour but not increased aggression or cognitive inflexibility: evidence of a selective haploinsufficiency gene effect. J Neuroendocrinol 25:107–118 [DOI] [PubMed] [Google Scholar]
- Sala M, Braida D, Lentini D, Busnelli M, Bulgheroni E, Capurro V, Finardi A, Donzelli A, Pattini L, Rubino T, et al. (2011) Pharmacologic rescue of impaired cognitive flexibility, social deficits, increased aggression, and seizure susceptibility in oxytocin receptor null mice: a neurobehavioral model of autism. Biol Psychiatry 69:875–882 [DOI] [PubMed] [Google Scholar]
- Serradeil-Le Gal C, Raufaste D, Derick S, Blankenstein J, Allen J, Pouzet B, Pascal M, Wagnon J, Ventura MA. (2007) Biological characterization of rodent and human vasopressin V1b receptors using SSR-149415, a nonpeptide V1b receptor ligand. Am J Physiol Regul Integr Comp Physiol 293:R938–R949 [DOI] [PubMed] [Google Scholar]
- Takayanagi Y, Yoshida M, Bielsky IF, Ross HE, Kawamata M, Onaka T, Yanagisawa T, Kimura T, Matzuk MM, Young LJ, et al. (2005) Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc Natl Acad Sci USA 102:16096–16101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13 [DOI] [PubMed] [Google Scholar]
- Wallis M. (2012) Molecular evolution of the neurohypophysial hormone precursors in mammals: comparative genomics reveals novel mammalian oxytocin and vasopressin analogues. Gen Comp Endocrinol 179:313–318 [DOI] [PubMed] [Google Scholar]
- Wesley VJ, Hawtin SR, Howard HC, Wheatley M. (2002) Agonist-specific, high-affinity binding epitopes are contributed by an arginine in the N-terminus of the human oxytocin receptor. Biochemistry 41:5086–5092 [DOI] [PubMed] [Google Scholar]
- Wichard JD, Ter Laak A, Krause G, Heinrich N, Kühne R, Kleinau G. (2011) Chemogenomic analysis of G-protein coupled receptors and their ligands deciphers locks and keys governing diverse aspects of signalling. PLoS ONE 6:e16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wu BL. (2012) Ice breaking in GPCR structural biology. Acta Pharmacol Sin 33:324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg HH, Laporte SA. (2003) The oxytocin receptor. Trends Endocrinol Metab 14:222–227 [DOI] [PubMed] [Google Scholar]