

Abstract
N-methyl D-aspartate (NMDA) receptors (NMDARs) mediate fast excitatory synaptic transmission and play a critical role in synaptic plasticity associated with learning and memory. NMDAR hypoactivity has been implicated in the pathophysiology of schizophrenia, and clinical studies have revealed reduced negative symptoms of schizophrenia with a dose of pregnenolone that elevates serum levels of the neuroactive steroid pregnenolone sulfate (PregS). This report describes a novel process of delayed-onset potentiation whereby PregS approximately doubles the cell’s response to NMDA via a mechanism that is pharmacologically and kinetically distinct from rapid positive allosteric modulation by PregS. The number of functional cell-surface NMDARs in cortical neurons increases 60–100% within 10 minutes of exposure to PregS, as shown by surface biotinylation and affinity purification. Delayed-onset potentiation is reversible and selective for expressed receptors containing the NMDAR subunit subtype 2A (NR2A) or NR2B, but not the NR2C or NR2D, subunits. Moreover, substitution of NR2B J/K helices and M4 domain with the corresponding region of NR2D ablates rapid allosteric potentiation of the NMDA response by PregS but not delayed-onset potentiation. This demonstrates that the initial phase of rapid positive allosteric modulation is not a first step in NMDAR upregulation. Delayed-onset potentiation by PregS occurs via a noncanonical, pertussis toxin–sensitive, G protein–coupled, and Ca2+-dependent mechanism that is independent of NMDAR ion channel activation. Further investigation into the sequelae for PregS-stimulated trafficking of NMDARs to the neuronal cell surface may uncover a new target for the pharmacological treatment of disorders in which NMDAR hypofunction has been implicated.
Introduction
N-methyl D-aspartate (NMDA) receptors (NMDARs) play a critical role in synaptic plasticity underlying learning and memory, in part by dynamically regulating the trafficking of 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA) receptors via NMDAR channel–mediated Ca2+ transport into dendritic spines. NMDA receptor trafficking is also regulated in response to neuronal activity, via both phosphorylation-dependent and phosphorylation-independent pathways (Chen and Roche, 2007; Lau et al., 2009).
NMDAR antagonists reproduce both the positive and negative symptoms of schizophrenia in humans and worsen the symptoms of nonmedicated patients (Gaspar et al., 2009), suggesting that NMDAR hypofunction contributes to the symptoms of schizophrenia. Consistent with this hypothesis, enhancing NMDAR function by administering agonists acting at the NMDAR glycine site has been reported to be effective in reducing symptoms of schizophrenia (Labrie and Roder, 2010). The negative symptoms of schizophrenia, including cognitive deficits, blunted affect, poverty of speech, anhedonia, asociality, and lack of motivation, are highly predictive of poor clinical outcome (Milev et al., 2005), and are largely refractory to conventional antidopaminergic pharmacotherapy. Pharmacological options for enhancing NMDAR function are currently limited to acute agonism of receptor activity.
The results of clinical trials indicate that the positive and negative symptoms of schizophrenia are ameliorated in patients receiving adjunctive treatment with pregnenolone (Marx et al., 2011), concordant with elevated serum levels of the neuroactive steroid pregnenolone sulfate (PregS) (Marx et al., 2009). PregS enhances learning and memory in animal models (Gibbs et al., 2006; Marx et al., 2011) and enhances long-term potentiation (LTP) at hippocampal CA1 synapses (Sliwinski et al., 2004; Chen et al., 2007; Sabeti et al., 2007). PregS at micromolar concentrations acts as an allosteric modulator for a variety of neurotransmitter receptors, including glutamate, glycine, and GABAA receptors (Majewska and Schwartz, 1987; Wu et al., 1991). In particular, PregS enhances activation of NMDARs containing nuclear receptor (NR)2A (NR1/2A receptors) or NR2B (NR1/2B receptors) while inhibiting receptors containing NR2C (NR1/2C receptors) or NR2D (NR1/2D receptors) (Wu et al., 1991). Rapid enhancement of NMDAR function by PregS is evident on a time scale of seconds, consistent with direct allosteric modulation of cell-surface NMDARs (Wu et al., 1991; Park-Chung et al., 1997). Analysis of recombinant NMDAR chimeras expressed in Xenopus laevis oocytes (henceforth oocytes) has identified a steroid modulatory domain and possible PregS binding site on the NR2 subunit that includes the J/K helices in the S2 region of the glutamate recognition site and the contiguous fourth membrane domain (Jang et al., 2004). While exploring the effects of PregS in vivo, we found a higher potency process whereby low nanomolar concentrations of sulfated steroid released dopamine from striatal synaptosomes (Whittaker et al., 2008) ex vivo and the striatum in vivo contingent upon NMDAR activity (Sadri-Vakili et al., 2008). However, the cellular mechanism by which PregS may act at high potency is unknown.
This report shows that PregS pharmacologically stimulates NMDA efficacy by enhancing receptor trafficking to the cell surface. This occurs via a novel target and pathway that is dependent upon intracellular Ca2+ signaling but not NMDAR channel activation or entry of extracellular Ca2+. Instead, PregS stimulates release of intracellular Ca2+ via a pertussis toxin (PTx)–sensitive and phospholipase C (PLC)–dependent pathway. Therefore, in addition to ionotropic signaling, NMDARs are capable of regulating their own surface expression through a noncanonical mode of signaling (Rozas et al., 2003) that utilizes an intracellular transduction pathway more typical of G protein–coupled receptors (GPCRs).
Materials and Methods
Rat NMDAR subunit subtype NR1-1a (hereafter referred to as NR1) cDNAs were provided by Dr. S. Nakanishi, and the cDNAs of NR2A, NR2B, NR2C, and NR2D subunits were gifts from Dr. P. H. Seeburg. Stock solutions of steroids were prepared in dimethylsulfoxide (DMSO). The final concentration of DMSO in all buffer and drug solutions was 0.5%. Chemicals were from Sigma-Aldrich (St. Louis, MO). Restriction enzymes were from New England Biolabs (Beverly, MA).
Preparation of Complementary RNA.
Plasmids were linearized with appropriate restriction enzymes prior to in vitro transcription using mMESSAGE mMACHINE High Yield Capped RNA Transcription kits (Ambion, Inc., Austin, TX). Plasmids containing NMDAR subunits were linearized with NotI (NR1), KpnI (NR2B), or XhoI (NR2A). Linearized DNAs were subjected to phenol/CHCl3 extraction and ethanol precipitation. The T7 in vitro transcription kit was used for NR1 and NR2A complementary RNAs (cRNAs), and the SP6 kit was used for NR2B cRNAs.
Receptor Expression in Oocytes.
Expression of NMDA and GABAA receptors in oocytes was carried out as previously described (Berezhnoy et al., 2008; Kostakis et al., 2011). Briefly, oocytes from X. laevis frogs (Nasco, Fort Atkinson, WI) were microinjected with cRNAs transcribed in vitro from plasmids containing cDNAs of desired NMDAR or GABAA receptor subunits, and maintained in Barth’s solution [in mM: 84 NaCl, 2.4 NaHCO3, 0.82 MgSO4, 1 KCl, 0.33 Ca(NO3)2, 0.41 CaCl2, 7.5 Tris/HCl, 2.5 pyruvate, 100 U/ml penicillin/streptomycin, pH 7.4] at 18°C for 2–4 days before recording.
Oocyte Electrophysiology.
Two-electrode voltage clamp recordings from oocytes were conducted (23–25°C; holding potential −70 mV) and membrane currents were filtered (1 kHz) and digitized and sampled (100 Hz) as described previously (Malayev et al., 2002). Ca2+- and Mg2+-free Ba-Ringer perfusion buffer contained (in mM): 96 NaCl, 2 KCl, 1.8 BaCl2, 5 HEPES, 0.5% DMSO, pH 7.5. NMDA was applied in combination with a saturating concentration of glycine (50 µM). Oocytes were preincubated with externally applied inhibitors in Barth’s solution followed by inclusion of inhibitor in the recording solution. PregS was applied as 100 µM, except where indicated, and all concentrations of PregS appeared soluble in 0.5% DMSO.
Data Analysis.
To pool results from multiple oocytes, baseline responses to NMDA were established, and peak NMDA responses in the presence of PregS were expressed to reveal response augmentation. Rapid enhancement of the NMDA response by PregS was defined as %P(rapid) = [INMDA+PregS(simultaneous)/INMDA – 1] × 100, the percentage increase in peak current (%P) induced by NMDA + PregS, as compared with the peak current induced by application of NMDA in the absence of PregS. For the purpose of analysis, delayed enhancement of the NMDA response is defined as %P(delayed) = [INMDA+PregS(10 minute)/INMDA+PregS(simultaneous) – 1] × 100, the percentage increase in the response to NMDA + PregS when applied after a 10-minute preincubation with PregS, as compared with the response elicited when NMDA and PregS are applied simultaneously. For example, if rapid potentiation increases the response by 100% and delayed enhancement of the NMDA response also increases the response by an additional 100%, then the NMDA response after a 10-minute exposure to PregS is 400% of the response in the absence of PregS. Significance testing was performed by 2-tailed t test. EC50 values were estimated by nonlinear regression using the logistic equation.
Cell Culture.
Primary rat neocortical cultures were prepared from E18 embryos maintained 7 days in vitro, as described previously (McLean et al., 2000).
Confocal Ca2+ Imaging of Xenopus Oocytes and Neocortical Cells in Culture.
Oocytes were injected with 50 nl of 1 mM fluo-3 (penta ammonium salt, F1240; Molecular Probes, Eugene, OR) (Jaconi et al., 1997). Confocal images were acquired in real time using a Zeiss Axiovert 150M laser scanning confocal microscope with a Plan-neofluor 10×/0.3 objective lens (Carl Zeiss AG, Oberkochen, Germany) at 8 seconds per frame. Oocytes were positioned in a 14-mm glass microwell containing 2 ml of Ca2+-free Ba-Ringer, and the focus was adjusted to image the cross-section of greatest diameter, approximately through the center of the oocyte. Oocytes exhibited a low level of fluorescence in the absence of fluo-3 injection. Gain was adjusted such that fluo-3–injected oocytes exhibited minimal visible fluorescence, and this gain setting was used for all imaging. The oocyte was initially imaged in saline to establish a stable baseline, and then 2 ml of PregS in Ba-Ringer was superfused to yield a final concentration of 100 µM PregS. Fluorescence was measured with an argon laser (488-nm wavelength) and an emission filter (band path 505–530 nm). The fluorescence signal intensity for each oocyte was calculated using the measurement histogram function of the Carl Zeiss LSM Image Examiner software (Carl Zeiss AG). Values for each oocyte were divided by the first value from the imaging of that oocyte to give a change in fluorescence from the baseline.
Neocortical cells were plated on polylysine-treated glass-bottom dishes and maintained in vitro for 7 days prior to imaging. The cells were incubated for 20 minutes with fluo-4 acetoxymethyl ester, the cell permeable ester of the fluorescent calcium indicator (F-14201; Molecular Probes). The confocal images were acquired in real time using a Zeiss Axiovert 150M laser-scanning confocal microscope with a C-Apochromat 40×/1.2 40× water immersion objective at 1 frame/s. Culture dishes initially contained 2 ml of Ca2+-free HEPES-buffered saline. The gain was kept constant. After baseline imaging, 2 ml of PregS was perfused into the culture dish to yield a final concentration 50 μM. Fluo-4 fluorescence was measured with an argon laser, and an emission filter and changes in Ca2+ levels were determined as described earlier.
Labeling and Purification of Surface Proteins.
Primary rat neocortical cells were treated for 10 minutes at room temperature with 100 μM PregS or vehicle (0.1% DMSO) in Ca+2-free HEPES-buffered saline. Cell-surface proteins were biotinylated with membrane-impermeant biotinamidocaproate N-hydroxysuccinamide ester, and extracts were prepared as described previously (Martin et al., 1999) with the following modifications: Cortical cultures were biotinylated for 10 minutes at 25°C. Protein extracts were prepared in ice-cold 1% (v/v) Nonidet P-40 (Sigma-Aldrich), 0.1% (w/v) SDS, 10% (v/v) glycerol, 50 mM Tris-HCl, and 150 mM sodium chloride with protease inhibitors (Roche, Basel, Switzerland). Extract protein concentration was determined, and equal amounts of protein from vehicle and PregS-treated cells were retained (total) for Western blotting. Equal amounts of protein from the remaining lysate of each treatment group were affinity-purified with NeutrAvidin Agarose Resin (Thermo Fisher Scientific, Waltham, MA) at 4°C overnight, then pelleted and resuspended in extraction buffer three times to remove unbound protein. The final pellet was heated at 80°C in 1:20 (v/v) β-mercaptoethanol–supplemented Tris-Glycine SDS Sample buffer (Invitrogen, Carlsbad, CA) to dissociate the neutravidin-biotin conjugate, and equal volumes of the resulting samples (biotinylated) were used in Western blots.
Antibodies.
The following antibodies were used: mouse anti–glutamate receptor NR1 [1:1000 (clone 54.1); BD Pharmingen, Franklin Lakes, NJ], mouse anti–sodium potassium ATPase [1:1000 (clone 464.6); Abcam, Cambridge, MA], and mouse anti–valosin-containing peptide [1:5000 (clone 5); Abcam]. Antibodies were detected with the appropriate horseradish peroxidase (HRP)–conjugated secondary antibody, Protein A-HRP [1:4000; Zymed (Invitrogen)] or goat anti-mouse IgG F(ab)2 fragment-HRP (1:4000; Abcam).
Western Blots.
Protein extracts were separated by SDS polyacrylamide gel electrophoresis on 6% tris-glycine gels and transferred to nitrocellulose membranes (Invitrogen), and polypeptides were detected by immunoblotting followed by enhanced chemiluminescence (GE Healthcare, Waukesha, WI) according to standard methods (Martin et al., 1999). X-ray film (CL-Xposure; Fisher Scientific) was scanned using a Molecular Dynamics Personal Densitometer SI Model 375 (Molecular Dynamics, Sunnyvale, CA) and quantified using Image Quant TL software [Amersham Biosciences (GE Healthcare)]. Increasing amounts of total protein were run on each gel, probed with anti-NR1 antibody, and quantified to verify that densitometric analysis was performed within the linear range of the detection system. Surface NR1 band intensity was quantified only if band intensity was within the linear range of total NR1 band intensity. Surface protein is defined as the ratio of biotinylated to total protein band intensity, and surface and total protein is expressed as percentage vehicle-treated control.
Electrophysiological Recordings of Primary Neurons.
Dissociated 7-day chick embryo spinal cord neurons (2–4 weeks in culture) were prepared as described (Farb et al., 1979). Whole-cell currents were recorded by as described (Park-Chung et al., 1997). Patch electrodes (5–10 MΩ) were filled with intracellular solution containing (in mM) 153 CsCl, 10 EGTA, and 10 HEPES (pH to 7.2 with CsOH). Bath solution contained (in mM): 150 NaCl, 4 KCl, 1 CaCl2, and 10 HEPES (pH to 7.2 with NaOH). For some experiments, a Ca2+-free solution was used in which 1 mM BaCl2 was substituted for CaCl2 (Ba buffer). Drug solutions were applied to single neurons (23–25°C; holding potential −70 mV) by pressure ejection (15 psi) from seven-barrel borosilicate glass pipettes positioned approximately 50 µm from the neuronal soma.
Results
Enhancement of the NMDA-Induced Current by PregS Exhibits Rapid and Delayed Phases.
As previously reported (Wu et al., 1991), simultaneous application of NMDA and PregS results in rapid potentiation of the NMDA response of chick spinal cord neurons in culture, reflecting allosteric modulation of NMDAR activation. The peak current response to a 10-second application of 30 µM NMDA + 100 µM PregS was increased by 167% ± 29% (n = 3) over the response to 30 µM NMDA alone. However, with continued exposure to PregS, a further, delayed phase of potentiation became apparent (Fig. 1A), with the NMDA response after 180 seconds increased by 359% ± 58% over the initial response to NMDA alone (i.e., 4.6 times the initial NMDA response). Both rapid and delayed enhancement of the NMDA response by PregS are reversible, with the NMDA-induced current returning to control 4.5 minutes after washout of PregS. Replacement of Ca2+ with Ba2+ in the extracellular solution had no significant effect (P > 0.05, unpaired t test) on the delayed potentiation of the NMDA response by PregS; the response to a 10-second application of NMDA + PregS was increased by 87% ± 36% compared with NMDA alone, which increased to 261% ± 51% potentiation (n = 4) after 180 seconds of exposure to PregS (Fig. 1B).
Fig. 1.

PregS induces delayed-onset potentiation of the NMDA response in neurons. PregS (100 µM) and NMDA are applied by pressure ejection to primary embryonic spinal cord neurons in culture while recording by whole-cell voltage clamp. (A) Potentiation of the NMDA response by PregS increases upon continued exposure to PregS and typically levels off after about 85 seconds. Typical results are shown. Two minutes elapsed between the initial control application of NMDA and the beginning of PregS application; 4.5 minutes elapsed between the termination of PregS application and the final control NMDA response. (B) Delayed enhancement of the NMDA-induced current by PregS persists when Ba2+ is substituted for Ca2+ in the perfusion buffer. (C) Delayed increase in the NMDA-induced current by PregS persists when PregS is included in the intracellular electrode buffer. A minimum of 1 minute was allowed after establishment of the high-resistance seal on the chick spinal cord neurons to permit equilibration of PregS.
To determine whether the delay in enhancement is imposed by the time required for PregS to reach an intracellular target, 100 µM PregS was added to the intracellular buffer in the whole-cell recording electrode to saturate intracellular sites for at least 60 seconds after obtaining the whole-cell configuration and before extracellular application of the same concentration of PregS. The time course of PregS potentiation was, however, unaltered (P > 0.05, unpaired t test) by the inclusion of PregS in the intracellular solution (Fig. 1C). Potentiation by PS was 128% ± 40% after 10 seconds and 481% ± 140% (n = 4) after 180 seconds, arguing that delayed potentiation, similar to rapid potentiation, is mediated by a site associated with the extracellular membrane surface.
The NR2 Subunit Controls the Delayed Phase of Potentiation.
NMDARs composed of various subunit combinations were expressed in oocytes to determine whether the NR2 subunit might affect delayed potentiation. Similar to observations in neurons, both rapid and delayed phases of potentiation are observed. PregS rapidly and reversibly potentiates the peak current elicited by NMDA on cells expressing NR1/2A receptors. However, the NMDA response continues to increase with a time constant of ∼3 minutes, resulting in a total potentiation of 200–370% (Fig. 2, A and B). The delayed-onset NMDA response returns to control 2–3 minutes after removal of PregS, showing that delayed potentiation is reversible (Fig. 2B). Whereas the response to application of NMDA alone typically exhibited a rapid rise and a “flat-topped” response profile that was stable over a 10-second application interval, responses in the presence of PregS typically exhibited a profile with a slight upward slope, likely reflecting the progress of delayed potentiation (Fig. 2A). Repetitive application of NMDA, as shown in Figs. 1 and 2, was not necessary, as application of PregS for 10 minutes prior to NMDA + PregS was sufficient to elicit delayed potentiation. For subsequent experiments, the magnitudes of rapid and delayed potentiation were assessed by comparing the response to NMDA alone, the response to NMDA + PregS, and the response to NMDA + PregS applied after 10 minutes of exposure to PregS.
Fig. 2.

Delayed-onset potentiation by PregS is recapitulated by recombinant NMDARs expressed in oocytes. PregS and NMDA are applied by bath perfusion to oocytes expressing NR1/2A subunits while recording in the two-electrode whole-cell voltage clamp mode. (A) Potentiation of the NMDA response by PregS increases upon continued exposure to PregS and typically levels off after 7–10 minutes. Black bars indicate successive applications of NMDA (300 µM). Breaks between traces are 1.3–1.8 minutes (see B for times). PregS (100 µM) was added to the perfusion buffer at t = 0 and included for 7 minutes (overlying black bar). (B) Peak NMDA-induced currents determined as in (A) are normalized to the average response before application of PregS beginning at t = 0. Smooth curve reflects an exponential fit (τ = 184 seconds). Error bars indicate S.E.M. (n = 3). Arrows indicate rapid and delayed components of potentiation. Unless otherwise indicated, NMDA (300 µM) and PregS (100 µM) in 0.5% DMSO were used with Xenopus oocytes as the standard test concentrations. Oocytes were perfused with Ba-Ringer and voltage-clamped at –70 mV.
Delayed potentiation of the NMDA response by PregS is observed in oocytes expressing NR1/2A or NR1/2B receptors but not in oocytes expressing NR1/2C or NR1/2D receptors (Fig. 3A). This is in contrast to rapid modulation, wherein PregS initiates rapid positive modulation of NR1/2A and NR1/2B receptors but negative modulation of NR1/2C and NR1/2D receptors (Malayev et al., 2002). In the case of NR1/2A receptors, the concentration-response curve for delayed potentiation by PregS exhibits a 25-fold greater potency (EC50 = 850 nM) than for rapid potentiation (EC50 = 21 µM) (Fig. 3B). Thus, PregS exhibits substantial potentiation and selectivity at a much lower concentration: at 100 nM PregS (Fig. 3B), there is substantial delayed potentiation yet little or no rapid potentiation. By contrast, in the case of NR1/2B receptors, 100 nM PregS exhibits no detectable delayed or rapid potentiation (Fig. 3C).
Fig. 3.

Delayed potentiation by PregS is independent of fast potentiation and selective for NR1/2A (and NR1/2B). (A) Delayed potentiation is observed with NR1/(2A or 2B) but not NR1/(2C or 2D) or the NR1/NR2D–B chimera (χ4) that is inactive with respect to fast potentiation. NR1 cRNA was coexpressed in oocytes with cRNA for the NR2A, NR2B, NR2C, NR2D, or χ4 subunit. Peak currents elicited by application of NMDA + PregS, either simultaneously or after 10-minute perfusion with PregS, are expressed relative to the NMDA-induced current in the absence of PregS. *Indicates a significant (P < 0.001, paired 2-tail t test) increase over simultaneous addition of PregS and NMDA. Error bars indicate S.E.M. (n = 7–12). (B) Concentration-effect curves for rapid (EC50 = 21 µM) and delayed (EC50 = 850 nM) increase in NR1/2A receptors. To measure rapid potentiation, oocytes were exposed simultaneously to 300 µM NMDA plus the indicated concentration of PregS. To measure delayed potentiation, oocytes were perfused for 10 minutes with the indicated concentration of PregS, then exposed to NMDA + PregS. Delayed potentiation is defined as the additional percentage potentiation of the peak NMDA response after 10 minutes relative to the response when PregS and NMDA are applied simultaneously. Arrows indicate EC50. Error bars indicate S.E.M. (n = 5–7) (C) Concentration-effect curves for rapid (EC50 = 19 µM) and delayed (EC50 = 33 µM) potentiation with NR1/2B receptors indicate lower potency but higher efficacy for delayed potentiation by PregS. Arrows indicate EC50. Error bars indicate S.E.M. (n = 5–7 oocytes).
Delayed Potentiation Is Independent of Rapid Potentiation.
The observation that the PregS EC50 for delayed potentiation of NR1/2A receptors is 25-fold lower than for rapid potentiation suggests that rapid and delayed potentiation are independent processes. As shown in Fig. 4, PregS at a concentration of 100 nM produces no rapid potentiation of the 10 µM NMDA response of NR1/2A receptors, yet nevertheless induces 45% ± 3% delayed potentiation.
Fig. 4.

Delayed potentiation but not rapid potentiation is induced by 100 nM PregS in oocytes expressing NR1/2A receptors. Using a reduced concentration of 100 nM PregS, substantial delayed potentiation is observed without detectable rapid potentiation. (A) Typical responses of oocytes expressing NR1/2A receptors to 10 µM NMDA alone, 10 µM NMDA + 100 nM PregS (applied simultaneously), and 10 µM NMDA + 100 nM PregS after 10-minute preincubation with 100 nM PregS. (B) Averaged values of normalized peak current responses for rapid and delayed increase in NR1/2A receptors (n = 8–10). When added simultaneously with NMDA, PregS produced negligible potentiation of the NMDA response (1% ± 2%), whereas after 10-minute preincubation with 100 nM PregS, the response to NMDA was enhanced by 45% ± 3%. Error bars represent S.E.M (n = 8–10). *Indicates a significant difference between rapid and delayed potentiation (P < 0.0005).
As previously reported, rapid PregS potentiation is eliminated in NR1-1a/χ4 receptors, where χ4 is a chimeric NR2 subunit in which steroid modulatory domain 1 encompassing the J/K helices and the M4 domain (S759–I836) of NR2B is replaced with the equivalent region of NR2D (Jang et al., 2004). As shown in Fig. 3A, delayed potentiation by PregS persists with this construct, arguing that delayed potentiation is not contingent on rapid positive allosteric modulation and acts via a novel NMDAR site that is independent of steroid modulatory domain 1.
GABAA Receptors Do Not Exhibit Delayed Potentiation.
To determine whether delayed potentiation is limited to NMDARs, α1β2γ2s GABAA receptors and NR1/2A receptors were coexpressed in oocytes. Whereas the response to 300 µM NMDA was enhanced when NMDA was applied in combination with 100 µM PregS, the response to GABA was substantially inhibited but still readily measurable. Moreover, whereas a 10-minute pre-exposure of oocytes to PregS produced further enhancement of the NMDA response, the amplitude of the GABA response was similar to that observed when GABA and PregS were applied simultaneously, indicating the absence of a delayed effect of PregS on GABAA receptors (Supplemental Fig. 1).
G Protein Activation Is Involved in Delayed Potentiation.
The extended time scale of delayed potentiation suggests the involvement of intracellular signal transduction pathways. To test for GPCR involvement, potentiation by PregS was examined ∼4 hours after injection of oocytes with 500 pg of PTx. Delayed potentiation was significantly diminished in PTx-injected oocytes. To test for the involvement of PLC, we examined the effects of 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U-73122), an inhibitor of G protein–mediated PLC activation (Smith et al., 1990; Thompson et al., 1991; Smallridge et al., 1992). Oocytes treated with U-73122 (20 µM, 1 hour) exhibit reduced responses to NMDA (∼10% of control, unpublished data), and delayed potentiation is absent. Oocytes treated with the inactive analog 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-2,5-pyrrolidinedione (U-73343) are unaffected (Table 1). Notably, none of the pharmacological treatments affected rapid potentiation by PregS (Table 1).
TABLE 1.
Second messenger pathways
Cells were treated with the indicated inhibitor prior to measuring the increase in peak response to NMDA (300 µM) produced by PregS (100 µM). Treatment conditions were as follows: U-73122/U-73343, 1 hour, 20 µM in Barth’s solution plus 0.4% DMSO; PTx, 50 nl injection of 500 pg of PTx in 5 mM Tris, 0.05 mM EDTA, 0.015% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (CHAPS), pH 8.
| Treatment | Target | Total Potentiation | Rapid Potentiation | Delayed Potentiation |
|---|---|---|---|---|
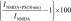 |
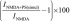 |
 |
||
| % | % | % | ||
| Vehicle | 203 ± 15 | 68 ± 4 | 76 ± 7 (15)a | |
| PTx | Gi/Go | 140 ± 13 | 72 ± 4 | 40 ± 6 (13)* |
| U-73343 | Inactive control | 182 ± 17 | 62 ± 6 | 75 ± 7 (6) |
| U-73122 | PLC | 83 ± 6 | 60 ± 5 | 14 ± 5 (8)** |
The number of oocytes is given in parentheses.
P < 0.001; **P < 0.005 [statistically significant versus vehicle (2-tailed t test)]
Protein Kinase C Mediates Delayed but Not Rapid Potentiation.
To investigate the role of protein kinases in delayed potentiation by PregS, we examined the effect of PregS in oocytes treated with protein kinase inhibitors and activators. Pretreatment of cells expressing NR1/2A receptors with the protein kinase C (PKC) activator phorbol 12-myristate 13-acetate (PMA) substantially increases the NMDA response in the absence of PregS (404% ± 19%, n = 8), but does not significantly affect rapid potentiation by PregS, whereas delayed potentiation by PregS is abolished (Table 2). Because PMA itself enhances the NMDA response (Supplemental Fig. 2), this likely reflects saturation of the PKC-dependent pathway by PMA.
TABLE 2.
Intracellular signal transduction pathways
Cells were treated with the indicated pharmacological inhibitor prior to measuring the rapid and delayed (10 minutes) increase in peak response to NMDA (300 µM) produced by PregS (100 µM). Treatment conditions were as follows: PMA, 10 minutes, 2 µM in Barth’s solution plus 0.5% DMSO; bisindolylmaleimide I, 2–3 hours, 2 µM in Barth’s solution plus 0.5% DMSO; H-89, 1–2 hours, 4 µM in Barth’s solution plus 0.5% DMSO; staurosporine, 2 hours, 2 µM in Barth’s solution plus 0.5% DMSO; DRB, 2 hours, 60 µM in Barth’s solution plus 0.5% DMSO; genistein, 2 hours, 50 µM in Barth’s solution plus 0.5% DMSO; U-0126, 1 hour, 20 µM in Barth’s solution plus 1% DMSO; tyrphostin A47, 20 minutes, 100 µM in Barth’s solution plus 0.5% DMSO.
| Treatment | Target | Total Potentiation | Rapid Potentiation | Delayed Potentiation |
|---|---|---|---|---|
| % | % | % | % | |
| Vehicle | 189 ± 8 | 65 ± 3 | 75 ± 2 (7)a | |
| PMA | PKC | 76 ± 5 | 65 ± 3 | 6 ± 1 (8)* |
| Staurosporine | Protein kinase | 111 ± 7 | 67 ± 2 | 25 ± 3 (8) |
| DRB | Casein kinase | 173 ± 3 | 60 ± 5 | 71 ± 6 (4) |
| Genistein | Nonreceptor protein kinase | 184 ± 6 | 64 ± 4 | 73 ± 3 (5) |
| Vehicle | 199 ± 8 | 66 ± 5 | 81 ± 5 (8) | |
| Bisindolylmaleimide I | PKC | 118 ± 12 | 65 ± 3 | 31 ± 7 (8)** |
| H-89 | PKA | 204 ± 9 | 63 ± 3 | 87 ± 4 (6) |
| Vehicle | 226 ± 18 | 70 ± 4 | 91 ± 6 (8) | |
| U-0126 | MEK | 232 ± 22 | 77 ± 8 | 91 ± 6 (6) |
| Vehicle | 194 ± 19 | 64 ± 8 | 79 ± 8 (6) | |
| Tyrphostin A47 | Insulin receptor Tyr kinase | 210 ± 24 | 72 ± 8 | 80 ± 9 (6) |
DRB, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole; H-89, N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide; MEK, mitogen-activated protein kinase.
The number of oocytes is given in parentheses.
P < 0.01; **P < 0.005 [statistically significant versus vehicle (2-tailed t test)].
Pretreatment of cells with staurosporine, a broad-spectrum inhibitor of serine/threonine protein kinases, does not affect the magnitude of the control NMDA response, but significantly reduces delayed potentiation (Table 2). Similar results are observed with the PKC inhibitor bisindolylmaleimide I. In contrast, the nonreceptor protein kinase inhibitor genistein, the casein kinase II inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (Litchfield, 2003), and the mitogen-activated protein kinase inhibitor 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) are without effect on delayed potentiation. Delayed potentiation by PregS is unaffected by treatment with the insulin receptor tyrosine kinase inhibitor 3,4-dihydroxy-α-cyanothio-cinnamamide (tyrphostin A47) (10 minutes, 100 µM), which blocks insulin-induced potentiation of the NMDA response (Skeberdis et al., 2001b) (Table 2). None of the tested inhibitors altered rapid potentiation, demonstrating that distinct mechanisms mediate rapid and delayed potentiation.
σ Receptors Do Not Mediate Delayed Potentiation.
To test for involvement of σ1 receptors, oocytes were pretreated with the σ receptor ligands 1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride (BD 1063) (30 µM) or haloperidol (10 µM). Delayed potentiation was unaffected (control: 80% ± 6%; BD-1063, 74% ± 9%; haloperidol: 72% ± 5%; n = 6–13), indicating that σ1 receptors do not play a role in potentiation of the NMDA response by PregS.
Delayed Potentiation by PregS Requires Ca2+ Release from Intracellular Stores.
Pharmacological evidence for the involvement of PLC and PKC in delayed potentiation suggests a Ca2+-mediated pathway. To evaluate the role of Ca2+, oocytes were injected with EGTA 30 minutes prior to recording. As shown in Table 3, delayed potentiation is significantly reduced by intracellular chelation of Ca2+. Because recordings were routinely carried out in Ca2+-free Ba-Ringer, the most likely source of Ca2+ is release from intracellular stores. We therefore examined the effect of the endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin, which depletes Ca2+ stores and elevates [Ca2+]i. After incubation with thapsigargin (1 mM for 1 hour), oocytes exhibit ∼700% increase in the NMDA current compared with untreated oocytes (unpublished data), in agreement with previous reports (Nishizaki et al., 1999; Skeberdis et al., 2001a). Rapid potentiation is unaltered after thapsigargin treatment, but delayed potentiation is eliminated. Delayed potentiation is abolished in oocytes injected with 50 nl of 1 mM heparin (Table 3), an inhibitor of the oocyte inositol 1,4,5-trisphosphate (IP3) receptor, but persists in control oocytes injected with the inactive analog de-N-sulfated heparin (Noh et al., 1998).
TABLE 3.
Calcium signaling
Cells were treated with the indicated inhibitor prior to measuring the rapid and delayed (10 minutes) increase in peak response to NMDA (300 µM) produced by PregS (100 µM). Treatment conditions were as follows: EGTA, 50 nl injection of 5 mM EGTA in water; heparin/de-N-sulfated heparin, 1 mM, 50 nl injection; thapsigargin, 1 hour, 1 mM in Barth’s solution plus 0.1% DMSO.
| Treatment | Target | Total Potentiation | Rapid Potentiation | Delayed Potentiation |
|---|---|---|---|---|
| % | % | % | % | |
| De-N-sulfated heparin | Inactive control | 227 ± 26 | 77 ± 7 | 91 ± 9 (8)a |
| Heparin | IP3 receptor | 110 ± 8 | 86 ± 7 | 12 ± 5 (9)* |
| Vehicle | 208 ± 19 | 61 ± 7 | 91 ± 7 (10) | |
| EGTA | [Ca2+]i | 111 ± 9 | 69 ± 5 | 25 ± 5 (11)* |
| Vehicle | 197 ± 15 | 72 ± 5 | 74 ± 6 (7) | |
| Thapsigargin | SERCA | 101 ± 6 | 73 ± 5 | 16 ± 4 (8)** |
The number of oocytes is given in parentheses.
P < 0.005; **P < 0.001 [statistically significant versus vehicle (2-tailed t test)].
PregS Increases Intracellular [Ca2+].
To test the hypothesis that PS stimulates release of Ca2+ from intracellular stores, [Ca2+]i was visualized in oocytes by confocal microscopy using fluo-3 (Fig. 5). Oocytes expressing NR1/2A receptors exhibit a transient increase in Ca2+ fluorescence beginning within seconds of PregS addition, but not vehicle addition, peaking after about 50 seconds and subsiding to baseline after 3–4 minutes. PregS had no effect on Ca2+ fluorescence in oocytes that were uninjected or injected with vehicle, demonstrating that NMDA receptors are essential for PregS-induced Ca2+ release. Oocytes expressing NR1/2D receptors failed to exhibit an increase in Ca2+ fluorescence, demonstrating subunit specificity.
Fig. 5.
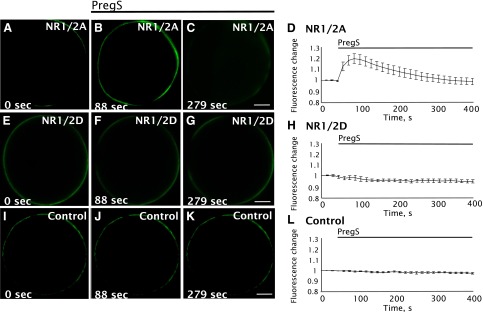
PregS induces intracellular Ca2+ increases in oocytes expressing NR1/2A but not NR1/2D receptors. Cells were injected with 50 nl of 1 mM fluo-3 Ca2+ indicator and changes in fluorescence during an exposure to PregS were monitored by confocal microscopy. Cells expressing NR1/2A (A–D) exhibit a visible increase in fluorescence near the membrane after PregS application. However, oocytes expressing NR1/2D (E–H) and control oocytes (injected with water vehicle without cRNA) (I–L) do not respond to PregS. Images show NR1/2A (A–C), NR1/2D (E–G), and control oocytes (I–K) before PregS (A, E, and I) and after PregS application (B, C, F, G, J, and K). The average response to PregS for NR1/2A (D), NR1/2D (H), and control oocytes (L) is represented to the right of the Xenopus oocyte images (n = 8). Scale bar, 200 μm.
The increase in Ca2+ fluorescence is primarily localized close to the membrane, consistent with the location of IP3-releasable Ca2+ stores in oocytes (Yao et al., 1995). The PregS-induced increase in Ca2+ fluorescence was observed in Ca2+-free Ba-Ringer, indicating that the increase in [Ca2+]i fluorescence is due to Ca2+ release from intracellular stores, and not entry of Ca2+ via NMDAR channels. A control experiment in which an oocyte expressing NR1/2A receptors was exposed to PregS in Ca2+-free/Ba2+-free Ringer containing 1 mM EGTA yielded a similar increase in fluorescence.
To determine whether PregS is similarly capable of increasing intracellular Ca2+ in neurons, primary rat neocortical neurons in culture were incubated with the membrane-permeant Ca2+ indicator fluo-4 acetoxymethyl ester and exposed to various concentrations of PregS. As shown in Fig. 6, PregS elicits a large initial increase in neuronal Ca2+ fluorescence that diminishes with time but persists over baseline for at least 3 minutes.
Fig. 6.

PregS induces Ca2+ release in neocortical neurons. Neocortical cells isolated from E18 rat maintained in vitro for 1 week were incubated with fluo-4 Ca2+ indicator. Change in fluorescence (∆F, arbitrary units) during an exposure to PregS (50 µM in 0.05% DMSO) was monitored by confocal microscopy. Images show neocortical cells in phase (A), phase with fluo-4 (B), and with fluo-4 (C). Neocortical cells exhibit a visible increase in fluorescence in the cell body and processes after PregS treatment (D, E, H, and I). Cells exhibit no significant change in fluorescence after vehicle treatment (K and L). Images of cells before (D and H) and after (E and I) PregS treatment and before (K) and after (L) vehicle treatment are shown. The time course of the responses to PregS of selected cell bodies, designated by arrowheads in (D), and of the entire field are shown in (F) and (G), respectively. The time course of the response to PregS of a neocortical cell process, designated by the arrowhead in (H), is shown in (J). The time course of the response to vehicle application of selected cell bodies (designated by arrowheadss in K) and of the entire field are shown in (M) and (N), respectively. Scale bar, 10 µm.
Exocytosis Is Required for Delayed Potentiation.
Delayed potentiation by PregS was blocked by injection of oocytes with botulinum toxin type A (BoNT A), a zinc endoprotease produced by bacteria of the genus Clostridium that cleaves SNAP-25 and prevents SNARE-regulated exocytosis (Pellizzari et al., 1999; Lan et al., 2001a). Rapid potentiation was insensitive to BoNT A (Fig. 7), indicating that delayed, but not rapid, potentiation requires exocytosis. Treatment with BoNT A alone did not reduce the amplitude of the NMDA response, arguing that most NMDARs do not exhibit rapid recycling in the absence of PregS. Similarly, delayed potentiation was significantly inhibited by brefeldin A (BFA) (control: 70% ± 3%; BFA: 42% ± 5%; P < 0.01; n = 13), which disassembles the Golgi complex and inhibits vesicular transport (Misumi et al., 1986; Oda et al., 1987; Klausner et al., 1992).
Fig. 7.

Delayed potentiation of the NMDA response by PregS requires exocytosis of NMDARs. (A) Prior exposure to PregS for 10 minutes enhances whole-cell currents elicited by coapplication of NMDA and PregS to oocytes expressing NR1/2A receptors. Vh = −70 mV. (B) Microinjection of BoNT A (50 ng, 5 hours before recording) reduces PregS-induced delayed potentiation without affecting rapid potentiation. BoNT A was reduced with 20 mM dithiothreitol (DTT) at room temperature 1 hour before use. Controls were injected with 10 mM DTT in 1 mg/ml bovine serum albumin (BSA) (n = 10). (C) BoNT A reduces delayed potentiation (*P < 0.00001; n = 9) with no effect on rapid potentiation. Error bars represent S.E.M. based upon the number of oocytes tested.
PregS Increases the Functional Cellular Response to NMDA via Increasing Surface Receptors.
To determine whether delayed potentiation is due to an increase in the number of functional NMDARs on the cell membrane, we examined tail currents elicited by a depolarizing voltage jump following complete NMDAR blockade by the open-channel blocker 9-aminoacridine (9-AA). Block by 9-AA traps activated NMDARs in a nonconducting state from which the coagonists NMDA and glycine are unable to dissociate, synchronizing all functional NMDAR channels in a blocked open conformation (Fig. 8A) (Benveniste and Mayer, 1995). Block by 9-AA is voltage-dependent, such that a depolarizing voltage step to +70 mV induces rapid (<3 milliseonds) dissociation of 9-AA, such that all of the NMDAR channels that have accumulated in the blocked state reopen nearly simultaneously after depolarization (Benveniste and Mayer, 1995; Chen et al., 1999). The resulting tail current is independent of the efficacy of NMDA and proportional to the number of activatable receptors (Horak et al., 2006). The amplitude of the tail current after a voltage jump from −70 to +70 mV is more than doubled following 10-minute exposure to PregS (Fig. 8B), indicating that the number of functional surface NMDARs is increased. This is similar to the magnitude of delayed potentiation, arguing that delayed potentiation is due to an increase in the number of functional surface receptors.
Fig. 8.

PregS increases the number of functional surface NMDARs in oocytes expressing NR1/2A receptors. (A) NMDA (300 µM) and 9-AA (100 µM) coapplied to oocytes expressing NR1/2A receptors results in a transient inward current as NMDA-activated channels are blocked by 9-AA (a voltage-dependent open-channel blocker). As the holding potential is switched from –70 to +70 mV, an outward tail current reflecting 9-AA unblock of NMDAR channels ensues (black traces). Cells were then exposed to vehicle (Ba-Ringer) (B) or PregS (C) for 10 minutes, and the 9-AA block and unblock sequences were repeated (blue traces). Peak tail currents after baseline subtraction are expressed relative to the control current (black trace) from the same cell (p2/p1). (D) The peak current ratio p2/p1 for PregS-treated oocytes (blue bar; n = 8) is significantly higher than for vehicle-treated oocytes (white bar; n = 6). *Relative current P < 0.00001, unpaired 2-tail t test.
PregS Increases Surface NMDARs in Neurons.
To determine whether PregS increases surface NMDARs in neurons, rat neocortical neurons were exposed to PregS for 10 minutes and surface proteins labeled with biotin. Following affinity purification on an avidin column, levels of total and biotinylated surface NR1 were assessed by Western blot. As quantified in Fig. 9, PregS increases surface NR1 by 62% ± 25% (P = 0.033, Student’s t test; n = 6). Levels of total NR1 were not increased with PregS treatment (total NR1 for PregS-treated as a percentage of control: 89% ± 10%; P = 0.291, Student’s t test; n = 6). Na/K ATPase (Kaplan, 2002; Zhang et al., 2009) and valosin-containing protein (Zhang et al., 1994; Kobayashi et al., 2002) were used as surface and intracellular markers, respectively. Biotinylated Na/K ATPase was observed in all experiments (n = 6), whereas biotinylated valosin-containing protein was below the limit of detection.
Fig. 9.
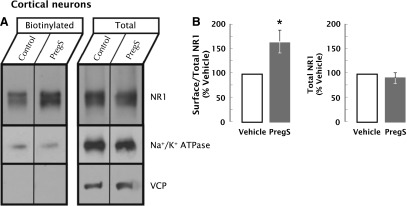
PregS increases surface NR1 subunits on neocortical neurons. (A) Representative immunoblot of surface NR1 levels in neocortical neurons exposed to PregS for 10 minutes as compared with vehicle. Surface biotinylated proteins were affinity purified and analyzed with a fraction of total cellular proteins by immunoblotting with antisera to NR1, surface Na+/K+ ATPase (positive control), or cytoplasmic valosin-containing protein (VCP) (negative control). (B) Quantitation of the effect of PregS (50 μM, 0.1% DMSO, 10 minutes) on total and surface-biotinylated NR1. (Left panel) Immunoreactivity in biotinylated aliquots as a fraction of total NR1 expressed as a percentage of vehicle. PregS induced a 62% ± 25% increase (*P = 0.033, n = 6 experiments, Student’s t test) in the biotinylated fraction of NR1. (Right panel) There was no change in total NR1 levels with PregS treatment (P = 0.291, n = 6 experiments, Student’s t test).
Discussion
The ability of nerve cells to regulate receptor number, composition, and distribution is central to nervous system function. Neuronal activity regulates NMDA receptor trafficking via both phosphorylation-dependent and phosphorylation-independent pathways (Chen and Roche, 2007; Lau et al., 2009), and insulin potentiates the NMDA response via the insulin receptor–activated tyrosine kinase (Skeberdis et al., 2001b).
The hypothesis of a linkage between ionotropic glutamate receptors and metabotropic Gi protein–coupled pathways via a “noncanonical” signal transduction mechanism that is not triggered by ion current suggests a pathway for cross-talk between the large classes of ionotropic and metabotropic systems. Results consistent with this can be found in several reports (Rodríguez-Moreno and Sihra, 2007). For example, the treatment of rat cortical neurons in culture with AMPA in the absence of extracellular permeant cations decreases PTx-mediated ADP glycosylation of Gαi1 and induces rapid association of Gαi1 with glutamate receptor subunit subtype 1 (GluR1) AMPA receptors (Wang et al., 1997). Similarly, exposure of mouse dorsal root ganglion neurons to kainate elevates [Ca2+]i via a PTx-sensitive, phospholipase C–dependent mechanism that is not dependent upon ion permeation, and this effect is absent in neurons from GlurR5-deficient mice (Rozas et al., 2003). A direct interaction with a G protein seems unlikely, as NMDARs do not exhibit homology to GPCR G protein–interacting domains and are not known to interact directly with G proteins. Direct protein-protein coupling and functional cross-talk between NR1/2A NMDARs and D1 dopamine receptors (Pei et al., 2004; Martina and Bergeron, 2008; Nai et al., 2010 ) is suggestive of a receptor subtype selective mechanism for cross-talk. This raises the possibility of cell signaling–mediated cross-talk.
In this report, the results show that PregS stimulates a sustained increase in the NMDA response that develops over several minutes, rather than milliseconds to seconds, and results from the incorporation of additional functional receptors into the surface membrane. The delayed-onset PregS-induced augmentation of the NMDA response is subunit-specific and, unlike the rapid phase of potentiation, requires G protein–coupled activation of PKC and PLC and increased [Ca2+]i. When combined with the effect of rapid positive allosteric modulation, total enhancement of the NMDA response averages 200% but can reach up to 400% in a given neuron.
Delayed potentiation of the electrophysiological response is mediated by the release of Ca2+ from intracellular stores. EGTA, heparin, and thapsigargin inhibit delayed potentiation, suggesting a role for Ca2+. Consistent with the results of inhibitors, Ca2+ imaging using fluo-3 shows that PregS releases Ca2+ from intracellular stores in oocytes expressing NR1/2A receptors. In contrast to some other agents that release intracellular Ca2+, such as acetylcholine (Dascal et al., 1985) and lysophosphatidic acid (Jaconi et al., 1997), PregS does not elicit detectable activation of Ca2+-sensitive chloride current in oocytes, suggesting that PregS-induced Ca2+ release is localized or limited in magnitude. Oocytes expressing NR1/2D receptors did not exhibit delayed modulation of the NMDA response or PregS-induced Ca2+ release, suggesting that the interaction of PregS with NR1/2D receptors is unable to activate the signaling pathway that releases intracellular Ca2+ and leads to increased NMDAR surface expression.
BoNT A blocks delayed potentiation, arguing that PregS stimulates delivery of NMDARs to the cell surface by SNARE-mediated exocytosis. The vesicular trafficking inhibitor BFA (Klausner et al., 1992), which inhibits NMDA-induced LTP and rapid membrane insertion of AMPA receptor subunits (Broutman and Baudry, 2001), similarly inhibits delayed potentiation, arguing that NMDARs are inserted into the membrane from an intracellular pool via a functional secretory pathway. The ability of PregS to increase delivery of NMDARs to the cell surface, as confirmed by immunoprecipitation of surface-labeled NMDARs, demonstrates a significant increase in surface NR1 subunits following PregS treatment of cortical neurons in culture.
Delayed potentiation is reversed ∼3–5 minutes after washout of PregS, suggesting that the additional surface NMDARs are rapidly reinternalized. The PregS-induced increase in surface NMDARs could be due to either stimulation of NMDAR exocytosis or inhibition of NMDAR endocytosis (Nong et al., 2004). However, BoNT A did not reduce the NMDA response in the absence of PregS (unpublished data), arguing that the latter is not the case.
Injection of PTx inhibits delayed potentiation, implicating a PTx-sensitive Gi/Go protein, and blockade of delayed potentiation by U-73122 implicates G protein–mediated PLC activation. Although PLC activation is primarily associated with Gq-coupled GPCRs, there is evidence that Gi can also participate in PLC-dependent mobilization of intracellular Ca2+, likely via the βγ subunit (Mizuta et al., 2011). How PregS activates G protein–coupled signaling pathways to increase surface NMDARs remains unclear. PregS interacts with σ1 receptors that couple to Gi in mouse brain membranes (Ueda et al., 2001), but delayed potentiation is unaffected by the σ1 antagonists BD 1063 and haloperidol, indicating that delayed potentiation in oocytes is not mediated by σ1 receptors.
Application of PregS for 10 minutes in the absence of NMDA or glutamate increases the response to a subsequent application of NMDA. As PregS alone does not induce a detectable current, activation of the NMDAR channel is not required for delayed potentiation. For the same reason, the increase in [Ca2+]i observed when PregS alone is applied to oocytes cannot be due to entry of Ca2+ via the NMDAR channel or voltage-gated Ca2+ channels. Moreover, delayed potentiation by PregS persists when extracellular Ca2+ is replaced with Ba2+, which does not stimulate PKC-mediated enhancement of the NMDA response (Zheng et al., 1997). These findings indicate that PregS induces increased [Ca2+]i and delayed potentiation of the NMDA response via a pathway that is not dependent upon ion permeation through the NMDAR channel.
Taken together, the results support the model diagrammed in Fig. 10, in which PregS, acting through the NMDAR, stimulates Gi/o-dependent activation of PLC, resulting in production of diacylglycerol and IP3, thereby triggering Ca2+ release from intracellular stores and stimulating Ca2+-dependent insertion of NMDARs into the surface membrane. This is similar to the pathway described for potentiation of NMDA responses and NMDAR surface expression by the metabotropic GluR agonist (1S,3R)-1-amino-cyclopentane-1,3-dicarboxylic acid (ACPD) in oocytes coexpressing NR1-4b/NR2A receptors and mGluR1α glutamate receptors (Skeberdis et al., 2001a), which has been reported to mediate postsynaptic long-term potentiation at hippocampal mossy fiber synapses (Kwon and Castillo, 2008). As with delayed potentiation by PregS, enhancement of NMDAR surface expression by ACPD is antagonized by staurosporine, BoNT A, U-73122, or intracellular Ca2+ chelation. However, potentiation by ACPD requires coexpression of GluR1α (Lan et al., 2001b; Skeberdis et al., 2001a), whereas delayed potentiation and release of intracellular Ca2+ by PregS requires only expression of NR1/2A receptors.
Fig. 10.

Model of a signal transduction pathway for delayed-onset potentiation of the NMDA response by PregS. Diagram illustrates the proposed intracellular pathways that participate in the PregS-stimulated trafficking of functional NMDA receptors to the cell surface via a noncanonical G protein-, PLC-, Ca2+-, and PKC-dependent mechanism. DAG, diacylglycerol; IP3R, IP3 receptor; PIP2, phosphatidylinositol 4,5-bisphosphate.
The location of the recognition site responsible for delayed potentiation by PregS remains to be identified. The recognition site for delayed potentiation is likely distinct from the site responsible for rapid potentiation, based on 1) substantially higher affinity of PregS at inducing delayed compared with rapid potentiation of NR1/2A receptors, 2) delayed potentiation of NR1/2A receptors by 100 nM PregS in the absence of rapid potentiation, and 3) delayed potentiation of NR1-1a/χ4 receptors without rapid potentiation. Another possibility is that the PregS recognition site that mediates delayed potentiation and [Ca2+]i increase is physically located on another protein, with the NMDAR playing a subunit-specific yet permissive role to enable this interaction.
Movement of receptors from the subsynaptic receptor pool to the synaptic population and the constitutive trafficking of receptors is essential to the viability and functionality of neurons (Lau and Zukin, 2007). Membrane fusion events contribute to NMDA receptor–dependent LTP, which in adult rat brain slices is associated with rapid sarcoma family kinases (Src)- and PKC-dependent membrane insertion of NMDARs, pointing to a potential role for NMDAR trafficking in synaptic plasticity (Grosshans et al., 2002).
Neuroactive steroids exert modulatory effects on neurotransmission in a variety of systems. PregS is endogenously present in the human brain, but its concentration in the extracellular space is unknown (Liere et al., 2004; Gibbs et al., 2006). Although most neurosteroids are neutral hydrophobic molecules, PregS is a negatively charged sulfate ester, potentially capable of being stored and selectively released. A PregS-like factor may act as a retrograde messenger to mediate long-term enhancement of AMPA receptor function in neonatal rat hippocampal slices via presynaptic NMDARs (Mameli and Valenzuela, 2006). PregS acts as a cognitive enhancer in vivo (Gibbs et al., 2006; Schumacher et al., 2008) and acts both presynaptically and postsynaptically to enhance synaptic transmission in multiple neurotransmitter systems (Zheng, 2009), but the mechanism of these diverse effects is not well understood.
Regulation of intracellular Ca2+ release or sequestration is potentially a unifying mechanism to account for the diverse effects of this neuroactive steroid on synaptic transmission, many of which can be understood in terms of alterations in Ca2+-dependent processes, such as the vesicular release of transmitters or delivery of receptors to the cell surface. The results suggest that, in addition to the well-established ionotropic mechanism whereby NMDARs directly transport Ca2+ ions, NMDARs are also capable of regulating [Ca2+]i by engaging a noncanonical metabotropic signaling cascade that in turn increases NMDAR surface expression. PregS-induced NMDAR upregulation constitutes a novel mechanism whereby a sulfated neurosteroid or related molecule could act synergistically with direct allosteric modulation of receptor function to profoundly influence synaptic transmission. Such a feed-forward mechanism could provide a positive regulatory mechanism for the augmentation of synaptic activity independent of NMDAR channel activity, with downstream consequences for NMDAR-dependent synaptic plasticity and memory.
Regulated trafficking of NMDARs between membrane and intracellular pools is critical for maintenance and plasticity of synaptic connections, and dysregulation of NMDAR trafficking has been implicated in neuropsychiatric disorders (Lau and Zukin, 2007). The present results suggest that NMDAR surface expression is subject to a novel mode of regulation mediated by neuroactive steroids via the NMDA receptor itself, which may play a role in disorders of NMDAR trafficking or provide a basis for development of therapeutic interventions.
Supplementary Material
Acknowledgments
This work is based on unpublished work by Dr. Mijeong Park-Chung, who first observed delayed potentiation by PregS in neurons; Dr. Nader Yaghoubi, who first replicated delayed potentiation in the Xenopus oocyte system; and Dr. Andrew Malayev, who obtained initial evidence indicating a role for PKC.
Abbreviations
- 9-AA
9-aminoacridine
- ACPD
(1S,3R)-1-amino-cyclopentane-1,3-dicarboxylic acid
- AMPA
2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid
- BD 1063
1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrochloride
- BFA
brefeldin A
- BoNT A
botulinum toxin type A
- cRNA
complementary RNA
- DMSO
dimethylsulfoxide
- GluR
glutamate receptor subunit
- GPCR
G protein–coupled receptor
- HRP
horseradish peroxidase
- IP3
inositol 1,4,5-trisphosphate
- LTP
long-term potentiation
- NMDA
N-methyl D-aspartate
- NMDAR
NMDA receptor
- NR
NMDAR subunit subtype
- PKC
protein kinase C
- PLC
phospholipase C
- PMA
phorbol 12-myristate 13-acetate
- PregS
pregnenolone sulfate
- PTx
pertussis toxin
- U0126
1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene
- U-73122
1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione
- U-73343
1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-2,5-pyrrolidinedione
Authorship Contributions
Participated in research design: Kostakis, Smith, Jang, Martin, Russek, Gibbs, Farb.
Conducted experiments: Kostakis, Smith, Jang, Martin, Richards.
Performed data analysis: Kostakis, Smith, Jang, Richards, Russek, Martin, Gibbs, Farb.
Wrote or contributed to the writing of the manuscript: Kostakis, Smith, Russek, Gibbs, Farb.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of Mental Health [Grant R01MH049469]; and the National Institutes of Health National Institute of General Medical Sciences [Grant T32GM008541] (to D.H.F.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Benveniste M, Mayer ML. (1995) Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J Physiol 483:367–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezhnoy D, Gravielle MC, Downing S, Kostakis E, Basile AS, Skolnick P, Gibbs TT, Farb DH. (2008) Pharmacological Properties of DOV 315,090, an ocinaplon metabolite. BMC Pharmacol 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutman G, Baudry M. (2001) Involvement of the secretory pathway for AMPA receptors in NMDA-induced potentiation in hippocampus. J Neurosci 21:27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Roche KW. (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53:362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Miyamoto Y, Furuya K, Mori N, Sokabe M. (2007) PREGS induces LTP in the hippocampal dentate gyrus of adult rats via the tyrosine phosphorylation of NR2B coupled to ERK/CREB [corrected] signaling. J Neurophysiol 98:1538–1548 [DOI] [PubMed] [Google Scholar]
- Chen N, Luo T, Raymond LA. (1999) Subtype-dependence of NMDA receptor channel open probability. J Neurosci 19:6844–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N, Gillo B, Lass Y. (1985) Role of calcium mobilization in mediation of acetylcholine-evoked chloride currents in Xenopus laevis oocytes. J Physiol 366:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farb DH, Berg DK, Fischbach GD. (1979) Uptake and release of [3H]γ-aminobutyric acid by embryonic spinal cord neurons in dissociated cell culture. J Cell Biol 80:651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar PA, Bustamante ML, Silva H, Aboitiz F. (2009) Molecular mechanisms underlying glutamatergic dysfunction in schizophrenia: therapeutic implications. J Neurochem 111:891–900 [DOI] [PubMed] [Google Scholar]
- Gibbs TT, Russek SJ, Farb DH. (2006) Sulfated steroids as endogenous neuromodulators. Pharmacol Biochem Behav 84:555–567 [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. (2002) LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci 5:27–33 [DOI] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Chodounska H, Vyklický L., Jr (2006) Subtype-dependence of N-methyl-D-aspartate receptor modulation by pregnenolone sulfate. Neuroscience 137:93–102 [DOI] [PubMed] [Google Scholar]
- Jaconi M, Pyle J, Bortolon R, Ou J, Clapham D. (1997) Calcium release and influx colocalize to the endoplasmic reticulum. Curr Biol 7:599–602 [DOI] [PubMed] [Google Scholar]
- Jang MK, Mierke DF, Russek SJ, Farb DH. (2004) A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc Natl Acad Sci USA 101:8198–8203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JH. (2002) Biochemistry of Na,K-ATPase. Annu Rev Biochem 71:511–535 [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol 116:1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Tanaka K, Inoue K, Kakizuka A. (2002) Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J Biol Chem 277:47358–47365 [DOI] [PubMed] [Google Scholar]
- Kostakis E, Jang MK, Russek SJ, Gibbs TT, Farb DH. (2011) A steroid modulatory domain in NR2A collaborates with NR1 exon-5 to control NMDAR modulation by pregnenolone sulfate and protons. J Neurochem 119:486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. (2008) Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron 57:108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie V, Roder JC. (2010) The involvement of the NMDA receptor D-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci Biobehav Rev 34:351–372 [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. (2001a) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4:382–390 [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Zheng X, Bennett MV, Zukin RS. (2001b) Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J Neurosci 21:6058–6068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Takeuchi K, Rodenas-Ruano A, Takayasu Y, Murphy J, Bennett MV, Zukin RS. (2009) Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem Soc Trans 37:1369–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8:413–426 [DOI] [PubMed] [Google Scholar]
- Liere P, Pianos A, Eychenne B, Cambourg A, Liu S, Griffiths W, Schumacher M, Sjövall J, Baulieu EE. (2004) Novel lipoidal derivatives of pregnenolone and dehydroepiandrosterone and absence of their sulfated counterparts in rodent brain. J Lipid Res 45:2287–2302 [DOI] [PubMed] [Google Scholar]
- Litchfield DW. (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Schwartz RD. (1987) Pregnenolone-sulfate: an endogenous antagonist of the gamma-aminobutyric acid receptor complex in brain? Brain Res 404:355–360 [DOI] [PubMed] [Google Scholar]
- Malayev A, Gibbs TT, Farb DH. (2002) Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDA receptors by sulphated steroids. Br J Pharmacol 135:901–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Valenzuela CF. (2006) Alcohol increases efficacy of immature synapses in a neurosteroid-dependent manner. Eur J Neurosci 23:835–839 [DOI] [PubMed] [Google Scholar]
- Martina M, Bergeron R. (2008) D1 and D4 dopaminergic receptor interplay mediates coincident G protein-independent and dependent regulation of glutamate NMDA receptors in the lateral amygdala. J Neurochem 106:2421–2435 [DOI] [PubMed] [Google Scholar]
- Martin SC, Russek SJ, Farb DH. (1999) Molecular identification of the human GABABR2: cell surface expression and coupling to adenylyl cyclase in the absence of GABABR1. Mol Cell Neurosci 13:180–191 [DOI] [PubMed] [Google Scholar]
- Marx CE, Bradford DW, Hamer RM, Naylor JC, Allen TB, Lieberman JA, Strauss JL, Kilts JD. (2011) Pregnenolone as a novel therapeutic candidate in schizophrenia: emerging preclinical and clinical evidence. Neuroscience 191:78–90 [DOI] [PubMed] [Google Scholar]
- Marx CE, Keefe RS, Buchanan RW, Hamer RM, Kilts JD, Bradford DW, Strauss JL, Naylor JC, Payne VM, Lieberman JA, et al. (2009) Proof-of-concept trial with the neurosteroid pregnenolone targeting cognitive and negative symptoms in schizophrenia. Neuropsychopharmacology 34:1885–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Shpektor D, Bandyopadhyay S, Russek SJ, Farb DH. (2000) A minimal promoter for the GABA(A) receptor alpha6-subunit gene controls tissue specificity. J Neurochem 74:1858–1869 [DOI] [PubMed] [Google Scholar]
- Milev P, Ho BC, Arndt S, Andreasen NC. (2005) Predictive values of neurocognition and negative symptoms on functional outcome in schizophrenia: a longitudinal first-episode study with 7-year follow-up. Am J Psychiatry 162:495–506 [DOI] [PubMed] [Google Scholar]
- Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. (1986) Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem 261:11398–11403 [PubMed] [Google Scholar]
- Mizuta K, Mizuta F, Xu D, Masaki E, Panettieri RA, Jr, Emala CW. (2011) Gi-coupled γ-aminobutyric acid-B receptors cross-regulate phospholipase C and calcium in airway smooth muscle. Am J Respir Cell Mol Biol 45:1232–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nai Q, Li S, Wang SH, Liu J, Lee FJ, Frankland PW, Liu F. (2010) Uncoupling the D1-N-methyl-D-aspartate (NMDA) receptor complex promotes NMDA-dependent long-term potentiation and working memory. Biol Psychiatry 67:246–254 [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Matsuoka T, Nomura T, Kondoh T, Tamaki N, Okada Y. (1999) Store Ca2+ depletion enhances NMDA responses in cultured human astrocytes. Biochem Biophys Res Commun 259:661–664 [DOI] [PubMed] [Google Scholar]
- Noh SJ, Kim MJ, Shim S, Han JK. (1998) Different signaling pathway between sphingosine-1-phosphate and lysophosphatidic acid in Xenopus oocytes: functional coupling of the sphingosine-1-phosphate receptor to PLC-xbeta in Xenopus oocytes. J Cell Physiol 176:412–423 [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Salter MW. (2004) NMDA receptors are movin’ in. Curr Opin Neurobiol 14:353–361 [DOI] [PubMed] [Google Scholar]
- Oda K, Hirose S, Takami N, Misumi Y, Takatsuki A, Ikehara Y. (1987) Brefeldin A arrests the intracellular transport of a precursor of complement C3 before its conversion site in rat hepatocytes. FEBS Lett 214:135–138 [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Wu FS, Purdy RH, Malayev AA, Gibbs TT, Farb DH. (1997) Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol Pharmacol 52:1113–1123 [DOI] [PubMed] [Google Scholar]
- Pei L, Lee FJ, Moszczynska A, Vukusic B, Liu F. (2004) Regulation of dopamine D1 receptor function by physical interaction with the NMDA receptors. J Neurosci 24:1149–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzari R, Rossetto O, Schiavo G, Montecucco C. (1999) Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos Trans R Soc Lond B Biol Sci 354:259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Sihra TS. (2007) Metabotropic actions of kainate receptors in the CNS. J Neurochem 103:2121–2135 [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV, Lerma J. (2003) Noncanonical signaling by ionotropic kainate receptors. Neuron 39:543–553 [DOI] [PubMed] [Google Scholar]
- Sabeti J, Nelson TE, Purdy RH, Gruol DL. (2007) Steroid pregnenolone sulfate enhances NMDA-receptor-independent long-term potentiation at hippocampal CA1 synapses: role for L-type calcium channels and sigma-receptors. Hippocampus 17:349–369 [DOI] [PubMed] [Google Scholar]
- Sadri-Vakili G, Janis GC, Pierce RC, Gibbs TT, Farb DH. (2008) Nanomolar concentrations of pregnenolone sulfate enhance striatal dopamine overflow in vivo. J Pharmacol Exp Ther 327:840–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M, Liere P, Akwa Y, Rajkowski K, Griffiths W, Bodin K, Sjövall J, Baulieu EE. (2008) Pregnenolone sulfate in the brain: a controversial neurosteroid. Neurochem Int 52:522–540 [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Opitz T, Zheng X, Bennett MV, Zukin RS. (2001a) mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology 40:856–865 [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Zheng X, Zukin RS, Bennett MV. (2001b) Insulin promotes rapid delivery of N-methyl-D- aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci USA 98:3561–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwinski A, Monnet FP, Schumacher M, Morin-Surun MP. (2004) Pregnenolone sulfate enhances long-term potentiation in CA1 in rat hippocampus slices through the modulation of N-methyl-D-aspartate receptors. J Neurosci Res 78:691–701 [DOI] [PubMed] [Google Scholar]
- Smallridge RC, Kiang JG, Gist ID, Fein HG, Galloway RJ. (1992) U-73122, an aminosteroid phospholipase C antagonist, noncompetitively inhibits thyrotropin-releasing hormone effects in GH3 rat pituitary cells. Endocrinology 131:1883–1888 [DOI] [PubMed] [Google Scholar]
- Smith RJ, Sam LM, Justen JM, Bundy GL, Bala GA, Bleasdale JE. (1990) Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J Pharmacol Exp Ther 253:688–697 [PubMed] [Google Scholar]
- Thompson AK, Mostafapour SP, Denlinger LC, Bleasdale JE, Fisher SK. (1991) The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. A role for Gp in receptor compartmentation. J Biol Chem 266:23856–23862 [PubMed] [Google Scholar]
- Ueda H, Yoshida A, Tokuyama S, Mizuno K, Maruo J, Matsuno K, Mita S. (2001) Neurosteroids stimulate G protein-coupled sigma receptors in mouse brain synaptic membrane. Neurosci Res 41:33–40 [DOI] [PubMed] [Google Scholar]
- Wang Y, Small DL, Stanimirovic DB, Morley P, Durkin JP. (1997) AMPA receptor-mediated regulation of a Gi-protein in cortical neurons. Nature 389:502–504 [DOI] [PubMed] [Google Scholar]
- Whittaker MT, Gibbs TT, Farb DH. (2008) Pregnenolone sulfate induces NMDA receptor dependent release of dopamine from synaptic terminals in the striatum. J Neurochem 107:510–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. (1991) Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol Pharmacol 40:333–336 [PubMed] [Google Scholar]
- Yao Y, Choi J, Parker I. (1995) Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J Physiol 482:533–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Hou Q, Wang M, Lin A, Jarzylo L, Navis A, Raissi A, Liu F, Man HY. (2009) Na,K-ATPase activity regulates AMPA receptor turnover through proteasome-mediated proteolysis. J Neurosci 29:4498–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ashendel CL, Becker GW, Morré DJ. (1994) Isolation and characterization of the principal ATPase associated with transitional endoplasmic reticulum of rat liver. J Cell Biol 127:1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P. (2009) Neuroactive steroid regulation of neurotransmitter release in the CNS: action, mechanism and possible significance. Prog Neurobiol 89:134–152 [DOI] [PubMed] [Google Scholar]
- Zheng X, Zhang L, Wang AP, Bennett MV, Zukin RS. (1997) Ca2+ influx amplifies protein kinase C potentiation of recombinant NMDA receptors. J Neurosci 17:8676–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.