

Abstract
Retinoid X receptor α [RXRα; nuclear receptor (NR)2B1] is a crucial regulator in the expression of a broad array of hepatic genes under both normal and pathologic conditions. During inflammation, RXRα undergoes rapid post-translational modifications, including c-Jun N-terminal kinase (JNK)-mediated phosphorylation, which correlates with a reduction in RXRα function. A small ubiquitin-like modifier (SUMO) acceptor site was recently described in human RXRα, yet the contributors, regulators, and consequences of SUMO-RXRα are not well understood. Inflammation and other stressors alter nuclear receptor function in liver and induce SUMOylation of several NRs as part of proinflammatory gene regulation, but linkages between these two pathways in liver, or for RXRα directly, remain unexplored. We sought to determine if inflammation induces SUMOylation of RXRα in human liver-derived (HuH-7) cells. Lipopolysaccharide, interleukin-1β, and tumor necrosis factor α (TNFα) rapidly and substantially stimulated SUMOylation of RXRα. Two RXRα ligands, 9-cis retinoic acid (9cRA) and LG268, induced SUMOylation of RXRα, whereas both inflammation- and ligand-induced SUMOylation of RXRα require the K108 residue. Pretreatment with 1,9-pyrazoloanthrone (SP600125), a potent JNK inhibitor, abrogates TNFα- and 9cRA-stimulated RXRα SUMOylation. Pretreatment with SUMOylation inhibitors markedly augmented basal expression of several RXRα-regulated hepatobiliary genes. These results indicate that inflammatory signaling pathways rapidly induce SUMOylation of RXRα, adding to the repertoire of RXRα molecular species in the hepatocyte that respond to inflammation. SUMOylation, a newly described post-translational modification of RXRα, appears to contribute to the inflammation-induced reduction of RXRα-regulated gene expression in the liver that affects core hepatic functions, including hepatobiliary transport.
Introduction
Retinoid X receptor α (RXRα), a member of the nuclear receptor (NR) superfamily (Germain et al., 2006), is essential for a range of physiologic functions in multiple tissues via crucial roles in development (Mark et al., 2009), metabolism (Shulman and Mangelsdorf, 2005), energy homeostasis (Imai, 2003; Grun and Blumberg, 2006), and inflammation (Ghose et al., 2004, 2007; Kuenzli et al., 2004; Zimmerman et al., 2006; Nunez et al., 2010). In liver, RXRα, the principal RXR isoform, plays a central role in liver physiology, particularly with respect to driving the expression of genes involved in intermediary metabolism, detoxification, as well as hepatobiliary transport and bile formation (Wagner et al., 2010). RXRα (and other RXRs) are unique among the NRs by being able to both homodimerize and, more relevantly, heterodimerize with over a dozen NRs (Lefebvre et al., 2010). Many aspects of liver function depend upon the ability of RXRα to partner with other Type II NRs, including the farnesoid X receptor (FXR), liver X receptors (LXRs), peroxisome proliferator associated receptors (PPARs), the pregnane X receptor (PXR), retinoic acid receptors, and the vitamin D receptor (Lefebvre et al., 2010). It is through the obligate dimerization with these NR family members that RXRα serves as a critical, integrating gene regulator, particularly with respect to the control of hepatic transport and bile acid synthesis (Kosters and Karpen, 2010). Thus, modulation of RXRα protein has the likelihood of both broad and specific consequences to RXRα:NR partner heterodimer functioning, a feature that has led to some current pharmacological therapeutics and that is a ripe area for further development.
Suppression of RXRα function is a major component of the negative hepatic acute phase response, manifested by marked downregulation of RXRα target gene expression in response to inflammation (Ghose et al., 2004; Kim et al., 2007). This downregulation of RXRα target genes—such as bile salt export pump (BSEP; ABCB11) and other transporters—often results in cholestasis, which can lead to jaundice and progression of hepatocellular damage from retention of bile acids and other small molecules destined for export (Kosters and Karpen, 2008). Our laboratory and others have shown that the downregulation of RXRα-target genes is at least in part attributable to post-translational modifications (PTMs) of RXRα, including c-Jun N-terminal kinase (JNK)-mediated phosphorylation (Zimmerman et al., 2006). Higher molecular weight forms of RXRα rapidly appear in both mouse liver and cell culture models in response to either lipopolysaccharide (LPS) or interleukin-1β (IL-1β) signaling (Kosters et al., 2009). However, the identity and function of these high molecular weight RXRα species are essentially unknown, and the possibility that inflammatory mediators could induce PTM with ubiquitin or small ubiquitin-like modifier (SUMO) in addition to phosphorylation has not been explored. Choi et al. (2006) identified one SUMOylation site in human RXRα, at K108, but the mediators and modifiers of SUMO-RXRα, especially in response to inflammatory signals, remain undetermined.
SUMOylation is the process whereby SUMO, a small ∼100 amino acid peptide, is covalently linked to a target protein; usually, this occurs at the consensus target sequence ψKxE, where ψ is any hydrophobic amino acid residue (Johnson, 2004). There are three known SUMO proteins in mammals: SUMO1, SUMO2, and SUMO3 (Treuter and Venteclef, 2011). Similar to ubiquitinylation, SUMO proteins are attached to their target proteins by using E1, E2, and E3 ligases; albeit with individual ligases that are distinct from those used in ubiquitinylation. Although a protein may be SUMOylated under basal conditions, often less than 1% of a particular protein pool is SUMOylated at any given time, and this SUMOylation can be in response to various cellular stressors or require the presence of ligand (Johnson, 2004). Inflammation and engagement of SUMOylation pathways has been linked with several NRs expressed in the liver, including PPARs (Pascual et al., 2005; Ghisletti et al., 2007; Leuenberger et al., 2009), LXRs (Ghisletti et al., 2007; Venteclef et al., 2010), PXR (Staudinger et al., 2011), and LRH1 (Venteclef et al., 2010), but not RXRα to date. In each of these NRs, SUMOylation results in a (trans)repression of gene expression (Pascual et al., 2005; Ghisletti et al., 2007; Leuenberger et al., 2009; Hu et al., 2010; Venteclef et al., 2010). In addition to inflammation and SUMOylation, JNK signaling and NR SUMOylation are linked to one NR’s function, the glucocorticoid receptor (GR), which leads to repression of GR-dependent gene expression (Davies et al., 2008).
Whether there are intersecting roles for inflammation-induced signaling and modulation of RXRα function by SUMOylation is unknown in any cell type or tissue. In the studies presented here, we first sought to determine if inflammatory signals induce SUMOylation of RXRα in a human liver cell line given the broad roles for RXRα in liver biology. Moreover, although recent reports indicate that SUMOylation of several NR family members are components of the response to inflammation (Treuter and Venteclef, 2011), it was not known if RXRα is a SUMOylation target of inflammation-based signaling. And finally, we sought to determine not only if cytokines or RXRα ligands can induce SUMOylation of RXRα, but if inhibition of SUMOylation could attenuate its suppressive effects on RXRα-regulated gene expression. All together, the aim of these studies was to determine if inflammatory mediators induce SUMOylation of RXRα and whether these pathways could possibly modulate RXRα-mediated hepatocellular gene targets.
Materials and Methods
Cell Culture.
HuH-7 cells, a human hepatocellular carcinoma-derived cell line, were a gift from the MK Estes laboratory (Department of Molecular Virology and Microbiology, Baylor College of Medicine) and maintained in minimum essential medium without l-glutamine containing Earle’s salts (VWR, Radnor, PA) and supplemented with 10% certified fetal bovine serum (Equitech Bio, Kerrville, TX), penicillin-streptomycin (Invitrogen, Carlsbad, CA), and l-glutamine (Cellgrow, Manassas, VA). For cytokine or ligand treatment, cells were maintained in serum-free conditions for the duration of treatment.
Cytokine, Ligand, and Inhibitor Treatments.
For cytokine treatments, cells were treated with either 1 mg/ml LPS (Sigma-Aldrich, St. Louis, MO), 10 ng/ml IL-1β (R&D Systems, Minneapolis, MN), 10 ng/ml tumor necrosis factor α (TNFα) (R&D Systems), or the equivalent volume of vehicle (water) for 0-4 hours. For ligand treatments, cells were treated with either 1 μM 9-cis-retinoic acid (9cRA; Sigma-Aldrich), 1 μM LG268 (kind gift from Ligand Pharmaceuticals, La Jolla, CA) or vehicle (dimethyl sulfoxide; DMSO) for 30 minutes. For SUMOylation inhibitor experiments, cells were pretreated with 100 μM anacardic acid (AA; Sigma-Aldrich) or 25 μM ginkgolic acid (GA; Sigma-Aldrich) or vehicle (DMSO) for 4 hours and then treated with 10 ng/ml TNFα for 30 minutes. For gene expression experiments, cells were pretreated with vehicle (DMSO), 10 μM GA, or 50 μM AA for 4 hours and then exposed to 50 μM chenodeoxycholic acid (CDCA) and/or 10 ng/ml TNFα. For JNK inhibitor experiments, cells were pretreated with 30 μM SP600125 (1,9-pyrazoloanthrone; Calbiochem, Gibbstown, NJ) for 30 minutes and then 10 ng/ml TNFα or 1 μM 9cRA for 30 minutes before harvest.
Cell Fractionation, His-Tag Isolation, and Immunoblotting.
Total cell protein was isolated by washing cells with ice-cold phospate-buffered saline and then lysing cells with the addition of 300 μl/10 cm plate of lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 5 mM EDTA, pH 8.0; 0.5% NP-40; 25 mM N-ethylmaleimide; Calbiochem Protease Inhibitor Cocktail Set I; Calbiochem Phosphatase Inhibitor Cocktail Sets I and II). Protein concentrations were determined with bicinchoninic acid assay. His-Tag isolation was performed with Dynabeads (Invitrogen) using 500 μg total protein and according to manufacturer’s protocol. After elution, 100 μl Laemmli Buffer was added to the samples. Cell lysates (10–25 μg/lane) or 20 μl of His-tag isolated samples were separated by 10% SDS-PAGE gels and electrophoretically transferred to nitrocellulose membrane. The membranes were blocked in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 at 25°C for a minimum of 1 hour. Incubation in a 1:1000 dilution of monoclonal anti-FLAG M2 antibody (Sigma-Aldrich), 1:1000 dilution of c-myc monoclonal antibody (Clontech, Mountain View, CA), 1:50,000 dilution of β-actin (Sigma-Aldrich) in blocking buffer or a 1:1000 dilution of anti-phospho-cjun (ser63) (Cell Signaling Technology, Danvers, MA) in 5% bovine serum albumin/Tris-buffered saline with 0.1% Tween 20 were carried out at 4°C for a minimum of 12 hours. After washing, membranes were incubated in 1:20,000 anti-mouse (Sigma-Aldrich) or 1:2000 anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA) horseradish peroxidase-linked antibody in the blocking buffer for a minimum of 1 hour at 25°C. Membranes were then washed and analyzed using Pierce Enhanced Chemiluminescence or Thermoscientific SuperSignal and exposed to Blue Autoradiography Film (ISC Bioexpress, Kaysville, UT) for detection. Quantitation of bands was determined using Carestream molecular imaging 5.0 (Carestream Health, Inc., Rochester, NY), and SUMO-RXRα bands were analyzed relative to FLAG-RXRα bands. Because of the differences in intensity between the two bands, multiple exposures were done and quantification was analyzed in the linear range.
Luciferase Assays.
After transient transfections, cells were maintained in complete media for a minimum of 8 hours. The media were then changed to serum-free media containing vehicle (DMSO) or ligand (1 μM 9cRA) for 20-24 hours. Cells were lysed and prepared as per manufacturer’s protocol (Promega Dual-Luciferase Reporter Assay System; Madison, WI). All data were corrected for renilla expression and untreated, reporter plasmid-only transfected samples were normalized to 1.
Plasmid Constructs.
An N-terminal dually FLAG- and His-tagged human RXRα construct was a kind gift from the J. M. Kurie laboratory (Department of Thoracic/Head and Neck Medical Oncology, M. D. Anderson Cancer Center) (Mann et al., 2005). The K108R mutant was derived from this RXRα plasmid using site-directed mutagenesis with the Quick Change Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA). UBC9, N-terminal c-myc–tagged SUMO1 and SUMO2 plasmids were kind gifts from David Owerbach (Molecular Diabetes and Metabolism Section and the Harry B. and Aileen B. Gordon Diabetes Research Center, Department of Pediatrics, Baylor College of Medicine) (Bohren et al., 2004).
Transient Transfections.
After plating and reaching 50–60% confluency, HuH-7 cells were transfected using 1.5 μl of FuGENE (Roche Applied Science, Indianapolis, IN) per microgram of plasmid DNA in serum-free media. Ten micrograms of total plasmid DNA was used for 10-cm plates. After a minimum of 16 hours, cells were washed with warm phosphate-buffered saline and the media changed to serum-containing media. After a minimum of 16 hour in complete media, cells were treated in serum-free media and harvested as described above.
Quantitative Real-Time Polymerase Chain Reaction.
Total RNA was isolated from HuH-7 cells tissue using Qiagen’s RNeasy kit and Qiashredder according to the manufacturer’s protocol. Complementary DNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). Quantitative real-time polymerase chain reaction (PCR) was performed with a StepOne Plus real-time PCR System (Applied Biosystems) using SYBR Green PCR master mix (Applied Biosystems). Primers were obtained from Sigma Genosys and sequences are listed in Supplemental Table 1. Quantitative expression values were extrapolated from standard curves and were normalized to 18s. Data are expressed as relative to vehicle treatments, and all treatments were compared with the control group, which was set to 1.
Statistical Analysis.
Data were expressed as the means ± S.E. from at least three independent experiments. Differences between experimental groups were evaluated for statistical significance using Student’s t test where P < 0.05 were considered to be statistically significant.
Results
SUMOylation of RXRα Is Induced by Inflammatory Mediators and RXRα Ligands.
We first sought to determine if inflammatory mediators could influence the SUMOylation of RXRα. HuH-7 cells, a human hepatocellular carcinoma-derived cell line, were cotransfected with a plasmid that expresses a dual FLAG-His-tagged RXRα (F-RXRα; ∼molecular weight 70 kDa); UBC9 (the only known E2 SUMO-conjugating enzyme); and either a plasmid that expresses myc-tagged SUMO1 or SUMO2. Controls were transfected with F-RXRα, UBC9, and the empty SUMO vector plasmids. Transfected cultures were subjected to cytokine or LPS treatments for 0.5–4 hours (Fig. 1A). Although 4 hours of exposure to either LPS or IL-1β did not lead to increased SUMO(2)ylation of RXRα, both treatments induced SUMO(1)ylation of RXRα (Fig. 1, A and B; ∼molecular weight 110 kDa). TNFα-treatment of transfected cell cultures significantly induced SUMOylation of RXRα with either SUMO1 or SUMO2 at 30 minutes (Fig. 1, A and B). Because of the necessary overexposure of the F-RXRα band to obtain a detectable SUMOylated F-RXRα band, each band was quantified at different exposures and normalized to the untreated samples for comparison. Furthermore, the higher molecular weight F-RXRα “smear” is believed to be a result of PTMs, including ubiquitinylation or phosphorylation. Because the degree of this modified RXRα varied, and was often increased in response to inflammatory mediators or decreased in response to inhibitors, quantification of the Western band omitted these higher molecular weight smears of RXRα. Furthermore, total levels of F-RXRα increased in response to TNFα and IL-1β, but not LPS, suggesting the possibility that regulation may be different among these inflammatory inducers. (We controlled for these minor differences in expression by always quantifying the FLAG-RXRα band relative to the total RXRα band.) Both baseline and TNFα-induced higher molecular weight bands were abolished when F-RXRα- and SUMO1-transfected HuH-7 cells were pretreated with either general SUMOylation inhibitor, AA, or GA (Fig. 1C), thus demonstrating that these higher molecular weight species are SUMO-RXRα. Furthermore, His-tag purification of F-RXRα followed by immunodetection with anti-c-myc (to identify c-myc–tagged SUMO1), led to the appearance of a prominent 110-kDa band in response to TNFα (Fig. 1D). Together, these provide strong evidence that cytokines, particularly TNFα, induce SUMOylation of RXRα in liver cells.
Fig. 1.
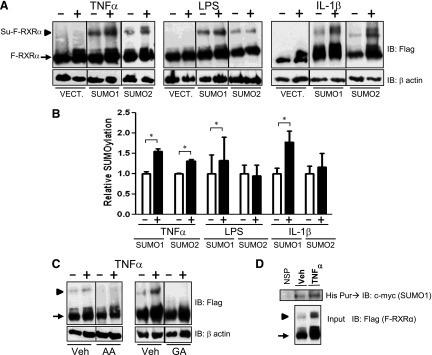
Inflammatory mediators increase SUMOylation of RXRα. (A) HuH-7 cells were transfected with F-RXRα, UBC9 and c-myc (empty vector; Vect.), SUMO1, or SUMO2. Cells were treated for 4 hours with vehicle or LPS, 1 mg/ml, or IL-1β, 10 ng/ml, for 30 minutes with TNFα, 10 ng/ml, and harvested and immunoblotted with anti-FLAG. (B) Densitometric analysis of blots treated as per A and including a minimum of three separate experiments, where the SUMO-F-RXRα band intensity is plotted relative to the F-RXRα band. Vehicle-treated samples are normalized to 1.0, data are presented as mean + S.E. and *P < 0.05, relative to the vehicle-treated control. Empty vector samples are not graphed because there is no detectable SUMO–F-RXRα band. (C) HuH-7 cells were transfected with F-RXRα and SUMO1 and pretreated with vehicle (Veh) or SUMO inhibitors 100 μM AA or 25 μM GA for 4 hours prior to treatment with vehicle or TNFα as in A. (D) HuH-7 cells transfected and treated with His-tagged RXRα and myc-tagged SUMO1 as in A then His-purified and immunoblotted with anti–c-myc antibody. Blot shows three different samples for both vehicle- and TNFα-treated cells. Note: all images are representative blots from a minimum of three experiments. Arrow denotes F-RXRα, and arrowhead denotes Su–F-RXRα. NSP, nonspecific.
Some NR ligands induce SUMOylation of target NR family members (e.g., LXRα and PPARγ), but this is not known for RXRα (Ghisletti et al., 2007). HuH-7 cells were transfected with F-RXRα and SUMO1 and treated with either vehicle (DMSO) or the naturally occurring RXRα ligand, 9cRA, for 30 minutes before harvest. 9cRA treatment significantly increased SUMO(1)ylation of RXRα compared with vehicle (Fig. 2, A and B). Because 9cRA has multiple effects on the cell in addition to functioning as an RXRα ligand, we treated F-RXRα/SUMO1 transfected HuH-7 cells with the specific and highly potent rexinoid LG268, which also led to induction of RXRα SUMO(1)ylation (Fig. 2, A and B). Taken together, either 9cRA or LG268 can induce SUMOylation of RXRα.
Fig. 2.

Ligand treatment increases SUMO(1)ylation of RXRα. (A) HuH-7 cells were transfected with F-RXRα and SUMO1 and treated with vehicle (veh; DMSO), 1 μM 9cRA, or 1 μM LG268 (LG) for 30 minutes; harvested; and immunoblotted as described in Fig. 1. (B) Graph of the averaged densitometry showing an increase in SUMOylation with either ligand. Data are presented as mean + S.E. and *P < 0.05, relative to the vehicle-treated control. Arrow denotes F-RXRα, and arrowhead denotes Su-F-RXRα.
The K108 Residue of RXRα Is the Primary SUMOylation Site in TNFα- and 9cRA-Induced SUMOylation.
It was previously reported that human RXRα contains multiple SUMO consensus sequences and that one site, K108, is the main, and likely sole, SUMOylation site (Choi et al., 2006). However, this was determined under basal conditions in a nonliver (human embryonic kidney–derived) cell line and it was not known if this one site was SUMOylated in liver cells or if there was a response to inflammatory mediators. We therefore sought to determine whether K108 is the primary SUMOylation site in RXRα under inflammatory conditions or in the presence of ligand. HuH-7 cells were transfected with either F-RXRα or a SUMOylation-deficient mutant, F-RXRαK108R, as well as SUMO1 and treated with either vehicle or TNFα. As shown in Fig. 3A, TNFα induces SUMO(1)ylation of RXRα, but not in F-RXRαK108R-transfected cells. Next, we investigated whether K108 was necessary for 9cRA-induced SUMO(1)ylation of RXRα. Similarly, when HuH-7 cells were transfected with either F-RXRα or F-RXRαK108R, as well as SUMO1, and then treated with either vehicle or 9cRA, induction of SUMO(1)ylation of RXRα was evident in cells transfected with wild-type F-RXRα plasmid, but not F-RXRαK108R (Fig. 3B). These experiments indicate that K108 is the principle, and likely sole, SUMOylation site in human RXRα and is necessary for cytokine- and ligand-induced SUMOylation of RXRα.
Fig. 3.

TNFα- and 9cRA-induced SUMOylation is dependent upon K108 residue. HuH-7 cells were transfected with SUMO1 and either F-RXRα or F-RXRαK108R and treated with vehicle or 10 ng/ml TNFα (A; as in Fig. 1) or 9cRA (B; as in Fig. 2). Note absence of a SUMO band in K108R-transfected lane with either treatment. Representative blots from a minimum of three experiments are shown. WT, wild-type.
JNK Is Critically Involved in TNFα-Induced and 9cRA-Induced SUMO(1)ylation of RXRα.
JNK signaling appears necessary for stressor and inflammation-induced phosphorylation of RXRα (Zimmerman et al., 2003, 2004). In at least a few signaling pathways, JNK-mediated phosphorylation can positively regulate the SUMOylation of a target protein (Davies et al., 2008; Leitao et al., 2010). Given the role for JNK-dependent suppression of RXRα function, we hypothesized that JNK modulates TNFα-induced SUMO(1)ylation of RXRα. HuH-7 cells that were transfected with SUMO1 and F-RXRα–expressing plasmids, pretreated with the potent pan-JNK inhibitor SP600125, and exposed to TNFα for 30 minutes before harvest. TNFα induced SUMO(1)ylation of RXRα in the presence of vehicle, whereas SP600125 pretreatment abrogated this effect (Fig. 4A). JNK does not appear to be required for basal SUMO(1)ylation of RXRα, because baseline SUMOylation was present, albeit at lower levels than vehicle-treated cells; however, no TNFα-induction occurred when JNK activity was inhibited with SP600125 (Fig. 4A). Control experiments with the downstream target of JNK (phospho-c-jun) verified the capability of SP600125 to effectively inhibit JNK signaling in HuH-7 cells (Fig. 4, A and B).
Fig. 4.

TNFα- and 9cRA-induced SUMO(1)ylation of RXRα involves activation of JNK. (A) HuH-7 cells were transfected with F-RXRα and SUMO1, pretreated with either vehicle (Veh) or 30 μM SP600125 for 30 minutes prior to vehicle or 10 ng/ml TNFα for 30 minutes; harvested; and immunoblotted with either antibodies specific for FLAG, p-cjun (phospho-c-jun), or β-actin. (B) Huh-7 cells were transfected as in A and treated with vehicle or 1 μM 9cRA for 30 minutes prior to harvest and immunoblotting. (C) HuH-7 cells were transfected with F-RXRα and SUMO1 pretreated with either vehicle or 20 μM PD98059 for 30 minutes prior to vehicle or 10 ng/ml TNFα for 30 minutes, harvested, and immunoblotted with either FLAG, p-ERK, or β-actin. Note: all images are representative blots from a minimum of three experiments. Arrow denotes F-RXRα, and arrowhead denotes Su-F-RXRα. (D) Densitometric analysis of blots in A–C in which the SUMO–F-RXRα band intensity is plotted relative to the F-RXRα band. Vehicle-treated samples (i.e., absence of TNFα or 9cRA) are normalized to 1.0, data are presented as mean + S.E. and *P < 0.05, relative to the vehicle-treated control.
To determine whether JNK could also be involved with ligand-induced SUMO(1)ylation of RXRα, HuH-7 cells transfected with SUMO1 and F-RXRα plasmids were pretreated for 30 minutes with SP600125 and then exposed to 9cRA for 30 minutes. Similarly to TNFα-induced SUMOylation, 9cRA induced SUMO(1)ylation of RXRα in the presence of vehicle, whereas SP600125 pretreatment abrogated this effect (Fig. 4B).
To determine if TNFα-induced SUMOylation is specific to JNK, the potential role of extracellular signal-regulated kinases (ERK) signaling in TNFα-induced SUMOylation of RXRα was also explored. Cells were transfected with SUMO1 and F-RXRα, pretreated with the ERK inhibitor 2-[2-amino-3-methoxyphenyl]-4H-1-benzopyran-4-one (PD98059), and exposed to TNFα for 30 minutes before harvest. ERK inhibition had no effect on TNFα-stimulated RXRα SUMOylation (Fig. 4C), indicating that JNK is likely the main MAP kinase involved with inflammation-induced SUMOylation of RXRα.
Inhibition of SUMOylation Increases Expression of RXRα Target Genes.
To determine if SUMOylation impacts gene expression of RXRα liver target genes, we investigated the effects of SUMO inhibitors on RXRα/FXR target genes expressed in HuH-7 cells. The bile salt export pump (BSEP; ABCB11), which exports bile acids from the hepatocyte into the canaliculi, is strongly upregulated in response to FXR ligands such as the primary bile acid CDCA (Ananthanarayanan et al., 2001; Plass et al., 2002). Inhibition of SUMOylation with GA had the most profound effect on basal transcription, with a 90-fold increase in transcription (P < 0.05). Similarly, significant upregulation of BSEP RNA was seen with another SUMOylation inhibitor, AA (P < 0.01) (Fig. 5A). In addition, GA treatment increased RNA levels of other RXRα-dependent genes [SHP (short heterodimer partner) and Cyp2B6] but not the canalicular transporter MDR1 or housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH), neither of which are known as significantly RXRα-regulated genes. SHP is upregulated by RXRα/FXR and Cyp2B6 by RXRα/CAR (Goodwin et al., 2000; Tompkins and Wallace, 2007; Piton et al., 2010). These data demonstrate a significant increase or decrease of gene expression with GA-treated cells of genes that are known to be significantly up- or downregulated, respectively, by RXRα. However, genes that are not known RXRα targets, or that are only minimally regulated by RXRα, such as MDR1 (Fig. 5A), are not significantly changed, although globally inhibiting SUMOylation may have a slight effect on gene expression. Taken together, inhibition of SUMOylation upregulated RXRα-dependent gene expression, but did not induce a global increase in gene expression in HuH-7 cells.
Fig. 5.

Inhibition of SUMOylation enhances RXRα target gene expression. Each graph shows a representative experiment, performed in triplicate, of a minimum of three experiments. (A) HuH-7 cells were pretreated with vehicle, 10 μM GA, or 50 μM AA for 4 hours prior to CDCA (if applicable) for an additional 20 hours. cDNA was made from RNA extractions, and real-time polymerase chain reaction was performed with primers for each listed gene. All expression, except glyceraldehyde 3-phosphate dehydrogenase (GAPDH), is relative to 18s and normalized to vehicle-treated cells. (B) HuH-7 cells were transfected with empty vector, RXRα, or RXRαK108R (SUMOylation deficient) plasmids and treated with vehicle or 9cRA 24 hours prior to harvest and measuring of luminescence. Data are normalized to empty vector, vehicle-treated cells. (C) HuH-7 cells were pretreated with vehicle and 10 μM GA for 4 hours prior to 9cRA (if applicable) for an additional 20 hours. cDNA was made from RNA extractions, and real-time polymerase chain reaction was performed with primers for each listed gene. All expression, except GAPDH, is relative to 18s and normalized to vehicle-treated cells. Data are presented as mean + S.E. and *P < 0.05; **P < 0.01; and ***P < 0.001.
To investigate the role of SUMOylation of RXRα in ligand-dependent gene expression, we transfected HuH-7 cells with both wild-type and SUMOylation-deficient plasmids and evaluated expression on a RXRα-specific promotor. As expected, expression was significantly (P < 0.05) higher in cells with the SUMOylation-deficient RXRα (Fig. 5B). HuH-7 cells were also treated with both GA and the RXRα ligand 9cRA for gene expression studies. As before, GA alone markedly induced BSEP RNA expression, but substantially enhanced 9cRA-induced BSEP RNA (from ∼3- to ∼30-fold, P < 0.05; Fig. 5C), thus indicating that SUMOylation of RXRα is playing a major role in the regulation of ligand-induced BSEP RNA expression. Altogether, SUMOylation seems to be playing a major role maintaining a low level of expression in the nonliganded state.
Discussion
The liver orchestrates the systemic inflammatory response and the acute phase response—namely by producing acute phase proteins and by downregulating expression of many genes, including hepatobiliary transporters regulated by RXRα heterodimers (Beigneux et al., 2000; Ruminy et al., 2001; Zimmerman et al., 2006; Ghose et al., 2007). These inflammation-induced changes lead to toxin accumulation, further damaging liver integrity and function (Kosters and Karpen, 2010). RXRα function in the inflamed liver is reduced through MAP kinase activation, particularly JNK, which phosphorylates RXRα and inhibits its transcriptional activity (Zimmerman et al., 2004, 2006; Mann et al., 2005; Macoritto et al., 2008; Kosters et al., 2009). Here we show that another PTM, SUMOylation, modulates RXRα activity in response to inflammation. Moreover, we show a linkage between inflammation-induced JNK activation and RXRα SUMOylation. These are the first studies identifying inflammation-mediated SUMOylation of hepatic RXRα, providing novel explanations for the suppression of RXRα heterodimer function in response to inflammation, thus broadening the stage for potential pharmacological targets.
MAP kinases have been implicated in PTM of proteins, including SUMOylation. Here, JNK activation is linked to RXRα SUMOylation, thereby adding RXRα to the ranks of other NRs that undergo JNK-modulated SUMOylation, GR (Davies et al., 2008) and PR (Leitao et al., 2010). ERK, a known RXRα kinase (Solomon et al., 1999; Macoritto et al., 2008), has also been shown to regulate SUMOylation (Utsubo-Kuniyoshi et al., 2007; Arito et al., 2008; Guo and Sharrocks, 2009). Although both JNK and ERK phosphorylate RXRα, only JNK induces RXRα SUMOylation (Fig. 4). This places RXRα alongside other NRs that undergo ligand-induced SUMOylation, including LXRα and PPARγ (Ghisletti et al., 2007), which may also allow for RXRα-mediated transrepression. These data suggest additional layers of crosstalk between RXRα heterodimers and signaling pathways and is an important area for future experimentation.
JNK signaling mediates TNFα-induced SUMOylation of RXRα (Fig. 4); however, this could involve direct or indirect targeting. Because it is known that JNK phosphorylates RXRα (Adam-Stitah et al., 1999; Zimmerman et al., 2003, 2004), JNK-induced phosphorylation could influence RXRα SUMOylation. This needs to be explored in appropriate future experiments. Furthermore, it has been shown that phosphorylation sites within close proximity to SUMOylation sites can positively affect SUMOylation (Hietakangas et al., 2006); although the amino acid sequence of RXRα contains neighboring serine residues, to date, no reports have been published regarding phosphorylation at these serines.
Interestingly, in addition to the established RXRα SUMOylation site (K108), two other potential SUMO consensus sequences (K201, K245) could have been engaged during activation of SUMOylation pathways. However, these studies strongly indicate that only the K108 residue is targeted and necessary for both TNFα- and ligand-induced SUMOylation of RXRα (Fig. 3). Thus, with respect to TNFα- and ligand-induced SUMOylation of RXRα, the two additional potential consensus SUMOylation sites do not participate in the response to these signals.
In other NRs, ligands can induce SUMOylation (Pascual et al., 2005; Ghisletti et al., 2007; Leuenberger et al., 2009; Hu et al., 2010). This also seems to be true for RXRα (Fig. 2). Uniquely, ligand-induced SUMOylation of RXRα is modulated by JNK (Fig. 4). Although JNK augments GR SUMOylation, GR SUMOylation seems to be ligand independent (Davies et al., 2008). Similar to PXR SUMOylation (Hu et al., 2010) is that both inflammatory stimuli and ligand can induce RXRα SUMOylation. Thus, the final common pathway of RXRα SUMOylation appears to converge at K108 via JNK signaling, either with ligand or cytokine activation. Interestingly, previous work has shown no impact of 9cRA on RXRα phosphorylation status in COS-1 cells (Adam-Stitah et al., 1999). This raises the possibility JNK could be inducing SUMOylation of RXRα indirectly; that is, without phosphorylating RXRα. Further experiments are needed to determine whether ligand and TNFα-stimulated pathways act synergistically or distinctly and to evaluate whether each stimulus has a similar or different systemic effect.
These data also show that SUMOylation likely modulates the expression of hepatobiliary transporter genes in diverse regulatory contexts. Bile acid-activated RXRα/FXR target genes (BSEP, SHP) augmented gene expression in the presence of GA and CDCA (Fig. 5), indicating SUMOylation normally represses transcription, particularly in basal (absence of ligand) states. This is not surprising because SUMOylation of most NRs represses gene expression (Treuter and Venteclef, 2011). Considering that any decrease in BSEP significantly contributes cholestasis development and possibly liver cancer (Mulder et al., 2009; Wang et al., 2011), this identifies SUMOylation as one of the potentially modifiable regulators of hepatobiliary function. Other inflammatory models (Cherrington et al., 2004; Le Vee et al., 2008; Wauters et al., 2010) have reported a significant reduction in BSEP expression during inflammation; however, technical experimental considerations precluded the combinatorial, prolonged testing of TNFα, GA, and CDCA, in our model system. Other models need to be used to address this question. Regardless, considering the marked augmentation of gene expression with GA treatment, targeting SUMOylation could be of potential therapeutic benefit in inflammatory cholestatic conditions.
Contributing to a suggestive role for RXRα SUMOylation in regulation of hepatic gene expression are the observations that 1) similar effects are obtained with CDCA or 9cRA; 2) the inhibition of SUMOylation, and the resultant increase in gene expression, is not limited to RXRα/FXR target genes; and 3) luminescence is increased when using a SUMOylation-deficient RXRα. The comparatively less dramatic 9cRA-induced BSEP RNA induction than with the potent FXR ligand CDCA is expected given the subordinate nature of RXRα ligand (Shulman et al., 2004). Importantly, 9cRA combined with GA significantly augmented BSEP RNA transcription (Fig. 5). This suggests that RXRα SUMOylation is physiologically relevant. CYP2B6, a drug metabolizer, is upregulated through RXRα and partners CAR and PXR (Wang et al., 2004; Tompkins and Wallace, 2007; Piton et al., 2010). Inhibiting SUMOylation with GA augmented CYP2B6 gene expression, which parallels the effect seen on RXRα/FXR target genes (Fig. 5). Unlike BSEP and SHP, 9cRA only slightly, but significantly, increased in CYP2B6 RNA expression. This is also expected because with conditional partners such as CAR, RXRα ligands will not increase transcription, whereas with permissive partners such as FXR and PXR, RXRα ligands do increase transcription (Shulman et al., 2004). RXRα is the common transcriptional regulator among these genes, thus supporting the hypothesis that RXRα SUMOylation is significantly impacting gene expression, although the promoter context may modulate the effect. Also suggesting that RXRα SUMOylation inhibits transcription is that in luciferase assays, cells transfected with SUMOylation-deficient RXRα exhibited significantly higher luminescence (Fig. 5B). Although multiple factors, including SUMOylation of coregulators, may regulate RXRα-target genes, the evidence suggests that RXRα SUMOylation is significantly involved in gene expression.
A relevant caveat to these studies is the reliance, as for others in the field, upon transient overexpression to better reveal SUMOylation. This protein overexpression may cause artifact because of a larger protein pool and possible effects secondary to endoplasmic reticulum stress or the transfection itself. Because SUMOylated protein is often less than 1% of a protein pool and this modification is transient (Johnson, 2004), overexpression is relied upon to demonstrate effect and plausibility. When more sensitive reagents are developed, we expect to delineate the genes, interacting proteins, and chromatin sites where SUMO-RXRα acts. We primarily were interested not in the array of SUMO-RXRα’s effects natively, but as a component of the inflammatory response. Future experiments will place SUMO-RXRα in context of the broad hepatocellular inflammatory response such as increases in specific miRNAs and other signaling pathways (Davidson-Moncada et al., 2010). Although our experiments necessarily focused on SUMOylation, we cannot exclude that miRNAs or another inflammatory process could be contributing. One caveat to the gene expression experiments is that we used general SUMOylation inhibitors, GA and AA, and cannot completely exclude that contributions to the observed effects may include SUMOylation of proteins other than RXRα, such as coregulators. However, given that RXR is a key component in the regulation of the analyzed genes, SUMOylation of RXRα likely plays at least a partial role, if not a complete role, in the modulation of gene expression. Moreover, GA did not have a global effect on gene transcription because MDR1 and glyceraldehyde 3-phosphate dehydrogenase showed no gene expression changes with GA treatment (Fig. 5). Finally, the present studies were done in a cancer cell line that, by definition, has pathophysiology. Although we cannot definitively exclude a cancer-related phenotype, we did use HuH7 cells, which are among the most differentiated liver cell lines and which have been shown to have an intact TNFα signaling pathway (Saez-Rodriguez et al., 2011), which minimizes this possibility.
In summary, these data indicate inflammatory signaling induces JNK modulated RXRα SUMOylation. Additionally, RXRα ligands induce SUMOylation of RXRα, providing a novel layer of regulation of RXRα-dependent gene expression. Furthermore, inhibition of SUMOylation markedly upregulates RXRα-dependent gene expression by at least three different RXRα heterodimer partners—FXR, CAR, and PXR. Together, these are the first studies identifying inflammation-induced SUMOylation of RXRα. Given the multiply partnered roles for RXRα and the SUMOylation-induced suppression of RXRα-regulated hepatic gene expression, SUMOylation could be a feasible new target to ameliorate the clinical changes in liver in response to inflammation.
Supplementary Material
Acknowledgments
The authors thank Astrid Kosters, David Owerbach, Sundararajah Thevananther, and Zhining Den for valuable advice and discussions, and Michelle Tovar for technical assistance.
Abbreviations
- AA
anacardic acid
- BSEP
bile salt export pump
- CDCA
chenodeoxycholic acid, 9cRA, 9-cis retinoic acid
- DMSO
dimethylsulfoxide
- ERK
extracellular signal-regulated kinases
- FXR
farnesoid X receptor
- F-RXRα
FLAG-His-tagged RXRα
- GA
ginkgolic acid
- GR
glucocorticoid receptor
- IL-1β
interleukin-1β
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- LXRs
liver X receptors
- NR
nuclear receptor
- PCR
polymerase chain reaction
- PD98059
2-[2-amino-3-methoxyphenyl]-4H-1-benzopyran-4-one
- PPARs
peroxisome proliferator associated receptors
- PTM
post-translation modification
- PXR
pregnane X receptor
- RXRα
retinoid X receptor α
- SHP
short heterodimer partner
- SP600125
1,9-pyrazoloanthrone
- SUMO
small ubiquitin-like modifier
- TNFα
tumor necrosis factor α
Authorship Contributions
Participated in research design: Schneider Aguirre, Karpen.
Conducted experiments: Schneider Aguirre.
Contributed new reagents or analytic tools: Schneider Aguirre.
Performed data analysis: Schneider Aguirre, Karpen.
Wrote or contributed to the writing of the manuscript: Schneider Aguirre, Karpen.
Footnotes
This work was supported by grants from National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK56239]; the Texas Gulf Coast Digestive Disease Center [Grant DK56338]; and the Medical Scientist Training Program Grant [T32GM007330-34].
Portions of this work were previously presented at the following meeting: Aguirre RS and Karpen SJ (2010) Inflammation-associated SUMOylation of hepatic RXRα: a new modification of a central liver gene regulator. American Association for the Study of Liver Diseases; 2010 Oct 29–Nov 2; Boston, MA (abstract 588).
Aguirre RS (2011) SUMOylation of Liver RXRα: A New Modification of a Central Liver Gene Regulator. Doctoral dissertation, Baylor College of Medicine, Houston, TX.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Adam-Stitah S, Penna L, Chambon P, Rochette-Egly C. (1999) Hyperphosphorylation of the retinoid X receptor alpha by activated c-Jun NH2-terminal kinases. J Biol Chem 274:18932–18941 [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. (2001) Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem 276:28857–28865 [DOI] [PubMed] [Google Scholar]
- Arito M, Horiba T, Hachimura S, Inoue J, Sato R. (2008) Growth factor-induced phosphorylation of sterol regulatory element-binding proteins inhibits sumoylation, thereby stimulating the expression of their target genes, low density lipoprotein uptake, and lipid synthesis. J Biol Chem 283:15224–15231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. (2000) The acute phase response is associated with retinoid X receptor repression in rodent liver. J Biol Chem 275:16390–16399 [DOI] [PubMed] [Google Scholar]
- Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D. (2004) A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J Biol Chem 279:27233–27238 [DOI] [PubMed] [Google Scholar]
- Cherrington NJ, Slitt AL, Li N, Klaassen CD. (2004) Lipopolysaccharide-mediated regulation of hepatic transporter mRNA levels in rats. Drug Metab Dispos 32:734–741 [DOI] [PubMed] [Google Scholar]
- Choi SJ, Chung SS, Rho EJ, Lee HW, Lee MH, Choi HS, Seol JH, Baek SH, Bang OS, Chung CH. (2006) Negative modulation of RXRalpha transcriptional activity by small ubiquitin-related modifier (SUMO) modification and its reversal by SUMO-specific protease SUSP1. J Biol Chem 281:30669–30677 [DOI] [PubMed] [Google Scholar]
- Davidson-Moncada J, Papavasiliou FN, Tam W. (2010) MicroRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci 1183:183–19420146715 [Google Scholar]
- Davies L, Karthikeyan N, Lynch JT, Sial EA, Gkourtsa A, Demonacos C, Krstic-Demonacos M. (2008) Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol 22:1331–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain P, Staels B, Dacquet C, Spedding M, Laudet V. (2006) Overview of nomenclature of nuclear receptors. Pharmacol Rev 58:685–704 [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. (2007) Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell 25:57–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose R, Mulder J, von Furstenberg RJ, Thevananther S, Kuipers F, Karpen SJ. (2007) Rosiglitazone attenuates suppression of RXRalpha-dependent gene expression in inflamed liver. J Hepatol 46:115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose R, Zimmerman TL, Thevananther S, Karpen SJ. (2004) Endotoxin leads to rapid subcellular re-localization of hepatic RXRalpha: A novel mechanism for reduced hepatic gene expression in inflammation. Nucl Recept 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526 [DOI] [PubMed] [Google Scholar]
- Grün F, Blumberg B. (2006) Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 147(6, Suppl)S50–S55 [DOI] [PubMed] [Google Scholar]
- Guo B, Sharrocks AD. (2009) Extracellular signal-regulated kinase mitogen-activated protein kinase signaling initiates a dynamic interplay between sumoylation and ubiquitination to regulate the activity of the transcriptional activator PEA3. Mol Cell Biol 29:3204–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L. (2006) PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci USA 103:45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Xu C, Staudinger JL. (2010) Pregnane X receptor is SUMOylated to repress the inflammatory response. J Pharmacol Exp Ther 335:342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T. (2003) Functional genetic dissection of nuclear receptor signalling in obesity, diabetes and liver regeneration using spatiotemporally controlled somatic mutagenesis in the mouse. Keio J Med 52:198–203 [DOI] [PubMed] [Google Scholar]
- Johnson ES. (2004) Protein modification by SUMO. Annu Rev Biochem 73:355–382 [DOI] [PubMed] [Google Scholar]
- Kim MS, Sweeney TR, Shigenaga JK, Chui LG, Moser A, Grunfeld C, Feingold KR. (2007) Tumor necrosis factor and interleukin 1 decrease RXRalpha, PPARalpha, PPARgamma, LXRalpha, and the coactivators SRC-1, PGC-1alpha, and PGC-1beta in liver cells. Metabolism 56:267–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosters A, Karpen SJ. (2008) Bile acid transporters in health and disease. Xenobiotica 38:1043–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosters A, Karpen SJ. (2010) The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis 30:186–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosters A, White DD, Sun H, Thevananther S, Karpen SJ. (2009) Redundant roles for cJun-N-terminal kinase 1 and 2 in interleukin-1beta-mediated reduction and modification of murine hepatic nuclear retinoid X receptor alpha. J Hepatol 51:898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenzli S, Tran C, Saurat JH. (2004) Retinoid receptors in inflammatory responses: a potential target for pharmacology. Curr Drug Targets Inflamm Allergy 3:355–360 [DOI] [PubMed] [Google Scholar]
- Le Vee M, Gripon P, Stieger B, Fardel O. (2008) Down-regulation of organic anion transporter expression in human hepatocytes exposed to the proinflammatory cytokine interleukin 1beta. Drug Metab Dispos 36:217–222 [DOI] [PubMed] [Google Scholar]
- Lefebvre P, Benomar Y, Staels B. (2010) Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab 21:676–683 [DOI] [PubMed] [Google Scholar]
- Leitao B, Jones MC, Fusi L, Higham J, Lee Y, Takano M, Goto T, Christian M, Lam EW, Brosens JJ. (2010) Silencing of the JNK pathway maintains progesterone receptor activity in decidualizing human endometrial stromal cells exposed to oxidative stress signals. FASEB J 24:1541–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuenberger N, Pradervand S, Wahli W. (2009) Sumoylated PPARalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J Clin Invest 119:3138–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macoritto M, Nguyen-Yamamoto L, Huang DC, Samuel S, Yang XF, Wang TT, White JH, Kremer R. (2008) Phosphorylation of the human retinoid X receptor alpha at serine 260 impairs coactivator(s) recruitment and induces hormone resistance to multiple ligands. J Biol Chem 283:4943–4956 [DOI] [PubMed] [Google Scholar]
- Mann KK, Padovani AM, Guo Q, Colosimo AL, Lee HY, Kurie JM, Miller WH., Jr (2005) Arsenic trioxide inhibits nuclear receptor function via SEK1/JNK-mediated RXRalpha phosphorylation. J Clin Invest 115:2924–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark M, Ghyselinck NB, Chambon P. (2009) Function of retinoic acid receptors during embryonic development. Nucl Recept Signal 7:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J, Karpen SJ, Tietge UJ, Kuipers F. (2009) Nuclear receptors: mediators and modifiers of inflammation-induced cholestasis. Front Biosci 14:2599–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez V, Alameda D, Rico D, Mota R, Gonzalo P, Cedenilla M, Fischer T, Boscá L, Glass CK, Arroyo AG, et al. (2010) Retinoid X receptor alpha controls innate inflammatory responses through the up-regulation of chemokine expression. Proc Natl Acad Sci USA 107:10626–10631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437:759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton A, Rauch C, Langouet S, Guillouzo A, Morel F. (2010) Involvement of pregnane X receptor in the regulation of CYP2B6 gene expression by oltipraz in human hepatocytes. Toxicol In Vitro 24:452–459 [DOI] [PubMed] [Google Scholar]
- Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL, Müller M. (2002) Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 35:589–596 [DOI] [PubMed] [Google Scholar]
- Ruminy P, Gangneux C, Claeyssens S, Scotte M, Daveau M, Salier JP. (2001) Gene transcription in hepatocytes during the acute phase of a systemic inflammation: from transcription factors to target genes. Inflamm Res 50:383–390 [DOI] [PubMed] [Google Scholar]
- Saez-Rodriguez J, Alexopoulos LG, Zhang M, Morris MK, Lauffenburger DA, Sorger PK. (2011) Comparing signaling networks between normal and transformed hepatocytes using discrete logical models. Cancer Res 71:5400–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R. (2004) Structural determinants of allosteric ligand activation in RXR heterodimers. Cell 116:417–429 [DOI] [PubMed] [Google Scholar]
- Shulman AI, Mangelsdorf DJ. (2005) Retinoid x receptor heterodimers in the metabolic syndrome. N Engl J Med 353:604–615 [DOI] [PubMed] [Google Scholar]
- Solomon C, White JH, Kremer R. (1999) Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor alpha. J Clin Invest 103:1729–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger JL, Xu C, Biswas A, Mani S. (2011) Post-translational modification of pregnane x receptor. Pharmacol Res 64:4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins LM, Wallace AD. (2007) Mechanisms of cytochrome P450 induction. J Biochem Mol Toxicol 21:176–181 [DOI] [PubMed] [Google Scholar]
- Treuter E, Venteclef N. (2011) Transcriptional control of metabolic and inflammatory pathways by nuclear receptor SUMOylation. Biochim Biophys Acta 1812:909–918 [DOI] [PubMed] [Google Scholar]
- Utsubo-Kuniyoshi R, Terui Y, Mishima Y, Rokudai A, Mishima Y, Sugimura N, Kojima K, Sonoda Y, Kasahara T, Hatake K. (2007) MEK-ERK is involved in SUMO-1 foci formation on apoptosis. Cancer Sci 98:569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, Nilsson LM, Parini P, Jänne OA, Gustafsson JA, et al. (2010) GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev 24:381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Zollner G, Trauner M. (2010) Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin Liver Dis 30:160–177 [DOI] [PubMed] [Google Scholar]
- Wang H, Faucette S, Moore R, Sueyoshi T, Negishi M, LeCluyse E. (2004) Human constitutive androstane receptor mediates induction of CYP2B6 gene expression by phenytoin. J Biol Chem 279:29295–29301 [DOI] [PubMed] [Google Scholar]
- Wang R, Sheps JA, Ling V. (2011) ABC transporters, bile acids, and inflammatory stress in liver cancer. Curr Pharm Biotechnol 12:636–646 [DOI] [PubMed] [Google Scholar]
- Wauters J, Mesotten D, Van Zwam K, van Pelt J, Thiessen S, Dieudonné AS, Vander Borght S, Van den Berghe G, Wilmer A. (2010) The impact of resuscitated fecal peritonitis on the expression of the hepatic bile salt transporters in a porcine model. Shock 34:508–516 [DOI] [PubMed] [Google Scholar]
- Zimmerman TL, Ghose R, Thevananther S, Karpen SJ. (2003) Il-1 beta mediated modification and nucleo-cytoplasmic translocation of RXR alpha involves JNK. Hepatology 38:391A–391A [Google Scholar]
- Zimmerman TL, Ghose R, Thevananther S, Karpen SJ. (2004) IL-1 beta induced RXR alpha nuclear export requires JNK activated cell signaling and involves serine. Hepatology 40:213A–213A [Google Scholar]
- Zimmerman TL, Thevananther S, Ghose R, Burns AR, Karpen SJ. (2006) Nuclear export of retinoid X receptor alpha in response to interleukin-1beta-mediated cell signaling: roles for JNK and SER260. J Biol Chem 281:15434–15440 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.