

Abstract
Objective:
To evaluate plasma 8-hydroxy-deoxy-guanosine (8OHdG) levels as a potential biomarker of premanifest and early Huntington disease (HD).
Methods:
Personnel from 2 independent laboratories quantified 8OHdG in blinded longitudinal plasma samples taken 24 months apart from 160 TRACK-HD participants, as well as samples containing control plasma with added (“spiked”) 8OHdG. One laboratory used a liquid chromatography–electrochemical array (LCECA) assay, and the other used liquid chromatography–mass spectrometry (LCMS).
Results:
The LCMS assay was more accurate than the LCECA assay for measurements of “spiked” 8OHdG levels in plasma. Neither assay demonstrated cross-sectional differences in plasma 8OHdG among controls, premanifest HD, and early symptomatic HD. Similarly, neither assay showed longitudinal changes in any disease group over 24 months.
Conclusions:
Plasma concentration of 8OHdG is not a biomarker of disease state or progression in HD. We recommend that future putative biomarker studies use blinded sample analysis, standard curves, independent analytical methods, and strict quality control of sample collection and storage.
Huntington disease (HD), an autosomal dominant neurodegenerative disease, is caused by an abnormally expanded trinucleotide (CAG) repeat in the huntingtin (HTT) gene.1 The disorder is typically diagnosed at onset of motor symptoms, but there is a “premanifest” period associated with clinical signs, including brain atrophy, psychiatric symptoms, and cognitive decline.2–4
PREDICT-HD5 and TRACK-HD,6 both longitudinal observational studies, aim to identify early HD biomarkers. These and other studies have shown differences in neuroimaging, cognitive, and motor measures among control, premanifest, and clinically diagnosed cohorts.7,8 Although premanifest and clinically diagnosed groups show robust longitudinal changes in neuroimaging measures, the latter shows more easily detectable declines in other measures compared with controls and premanifest HD (preHD).9–14
Currently, there are no validated candidate blood, CSF, or urine biomarkers that track the progression of HD.15 8-Hydroxy-deoxy-guanosine (8OHdG) has been proposed as a biomarker of HD progression.16 8OHdG is a product of the oxidation of guanine by reactive oxygen species, and its levels in blood and urine may correlate with the degree of oxidative DNA damage.17,18 Although increased levels of 8OHdG have been reported in patients with HD and mouse models indicating such oxidative DNA damage, the role of such damage in HD pathogenesis remains obscure.19–23
Using our recently developed liquid chromatography–mass spectrometry (LCMS) assay, we were unable to replicate the modest increase previously demonstrated with the liquid chromatography–electrochemical array (LCECA) assay in plasma 8OHdG in PREDICT-HD participants compared with controls.22,24–26 To re-evaluate 8OHdG as an HD biomarker, we conducted a head-to-head, blinded, comparative study of the 2 assays using a large set of rigorously prepared plasma samples.
METHODS
Participants.
The TRACK-HD cohort comprised 366 participants at the baseline visit and 332 participants at the 24-month visit at 4 sites (Leiden, Netherlands; London, UK; Paris, France; and Vancouver, Canada). Each site recruited equal numbers of participants in 3 groups: preHD, early HD, and controls. At the time of recruitment, preHD participants had a disease-burden score ≥250 (disease burden is [number of CAG repeats − 35.5] × age) and a total motor score (TMS) ≤5 in the Unified Huntington's Disease Rating Scale (UHDRS). The preHD cohort was split at the median predicted years to diagnosis at the first visit (10.8 years).27 PreHD-A and preHD-B participants were those nearer and further from predicted diagnosis, respectively. Diagnosed HD patients included HD stage 1 (HD1) and HD stage 2 (HD2) based on the baseline UHDRS total functional capacity score. Participants in the control group were balanced for age and sex to the combined preHD and HD groups.
Standard protocol approvals and patient consents.
All participants provided written informed consent, and local ethics committees approved the study protocol.
Clinical assessment.
All participants underwent the full TRACK-HD clinical assessment battery at each visit, as described previously.6,9,10 This included collection of demographic and clinical data as well as UHDRS-99; 3-tesla brain MRI; and cognitive, quantitative motor, oculomotor, and neuropsychiatric assessments.
Plasma sample collection and storage.
At each visit, TRACK-HD study personnel collected blood samples from the cubital vein of unfasted participants at approximately 10 am into 5–7 × 6-mL EDTA-coated tubes. Tubes were immediately inverted 10 times, stored briefly on ice, and centrifuged at 2,000g at 4°C for 15 minutes. If visual inspection revealed discoloration of sample in an individual tube, indicating hemolysis, it was discarded. If all plasma tubes were discolored or turbid, all tubes were recentrifuged. Plasma was transferred from each EDTA tube into 15-mL conical base tubes, leaving 5 mm of liquid above pellets. These tubes were centrifuged at 3,000g at 4°C for 15 minutes, and then plasma was carefully transferred into a 50-mL tube, leaving 5 mm above pellets. This tube was sealed, agitated gently, placed on ice, and then 500-μL aliquots were pipetted into 1.2-mL cryotubes. Cryotubes were stored at −80°C for up to 1 month locally and then shipped on dry ice to a central biorepository (BioRep, Milan, Italy) where they were stored at −80°C.
Quality control of the plasma collection procedure included measurement of hemoglobin levels as an indicator of hemolysis, using multiwavelength spectrophotometric readings, from one plasma cryotube from each of the first 5 participants at each site during each year of the study, at random times throughout the study, and following changes in site staff. If hemoglobin levels exceeded 100 μg/mL, the correct sampling procedure was reviewed with study personnel and the quality-control procedures continued until levels were <100 μg/mL.
Plasma samples.
Three hundred twenty TRACK-HD plasma samples in their original 500-μL aliquots, along with 15 spiked samples, were randomized, blinded (numbered 1 to 335), and shipped on dry ice to the 2 laboratories for analysis. We included 32 participants from each of the 5 disease groups. With one exception, samples from each disease group included equal numbers of males and females and equal distribution across the 4 study sites. For each participant, 2 samples (baseline and 24-month visit) were sent to the 2 laboratories. Thus, there were, on average, 4 samples per sex per site per disease group per visit.
Spiked samples were prepared in identical cryotubes with control human plasma (Innovative Research reference no. IPLA-2-NO6-50, lot no. IR-10-1527) spiked with 8OHdG (Calbiochem reference no. 390582, lot no. D00104661) to final concentrations in triplicate of 0, 5, 10, 20, or 50 pg/mL.
LCMS assay.
We purchased 15N5-8OH2′dG (1 mg, 98%, catalog no. NLM-6715-0) from Cambridge Isotope Laboratories and unlabeled 8OHdG was purchased from Cayman Chemicals. All solvents were high-performance liquid chromatography grade.
As an internal standard, we spiked 400 μL of each sample with 62.5 pg of 15N5-8OH2′dG. Endogenous 8OHdG was calculated using peak area ratio as the spiked concentration of 15N5-8OH2′dG × (peak area endogenous 8OHdG/peak area 15N5-8OH2′dG). Use of the internal standard enabled normalization for variability in recovery and matrix effects. To each sample, we added 500 μL of water and 150 μL of 1 M ammonium acetate (pH 5.25). Samples were randomized, vortexed for 10 minutes, and centrifuged for 5 minutes at 10,000 rpm at room temperature. Supernatants were then loaded on a C18 cartridge for cleanup (SEP-PAC cartridge by Waters Corp., Milford, MA). Cartridges were washed with 3 mL of water, then 8OHdG was eluted with 1 mL of methanol. Eluates were dried in a SpeedVac and stored at −80°C.
Frozen samples were resolubilized in 72 μL of 10 mM ammonium acetate (pH 4.6), and 18 μL was injected into the LCMS system composed of a nanoAcquity LC pump from Waters Corporation and 4000 QTRAP AB Sciex (Foster City, CA) tandem mass spectrometer, operating in positive-ion mode. The MultiQuant software tool in the Analyst software bundle (AB Sciex, Framingham, MA) was used for data analysis. Sample run time was 20 minutes on a Zorbax SB C18 column, 35 × 0.5 mm, particle size 5 μm with guard column by Agilent (Santa Clara, CA) with a 15 μL/min flow rate and an 8-minute gradient from 0% to 50% B solvent, using A solvent composed of 10 mM ammonium acetate (pH 4.6) and B solvent composed of acetonitrile + 0.1% formic acid.
LCECA assay.
Plasma samples were analyzed for 8OHdG using the LCECA method as described previously.24–26
Statistical analysis.
We used simple linear regression analysis to assess accuracy in detection of spiked 8OHdG for each assay. Cross-sectional analyses were measured between relevant disease groups at the baseline visit. Within-subject change scores were used for comparison of longitudinal changes in disease groups to controls, using linear models and controlling for age and sex. Confidence intervals and p values for each comparison were not corrected to account for multiple comparisons. As a result, 1 of 20 of these comparisons was expected to be significant (p < 0.05) even if no true relationship was present.
RESULTS
Validation of LCMS assay for 8OHdG.
To validate the LCMS assay, we generated a standard curve by spiking 15N5-8OH2′dG at concentrations ranging from 1 to 1,000 pg/mL in pooled human plasma (Bioreclamation, LLC, Westbury, NY). The resultant standard calibration curve (figure 1A) is linear from 1 to 1,000 pg/mL (r2 = 0.999). The estimated lower limit of detection is approximately 1 pg/mL and the estimated lower limit of quantification is approximately 5 pg/mL. The coefficient of variation (CV) is 4% at concentrations above 10 pg/mL. Recovery of spiked standard was 79%.
Figure 1. LCMS calibration curve and spiked sample recovery for LCMS and LCECA assays.

Standard calibration curve for LCMS assay (A). Concentration of spiked 8OHdG measured in control plasma using LCMS assay (B) and LCECA assay (C). 8OHdG = 8-hydroxy-deoxy-guanosine; LCECA = liquid chromatography–electrochemical array; LCMS = liquid chromatography–mass spectrometry.
We first used the LCMS assay to measure 8OHdG in previously described plasma samples from the PREDICT-HD study.22 Our analysis, using a nonparametric Kruskal-Wallis test, and the previous report found no differences in 8OHdG levels among controls, preHD far from diagnosis, preHD near to diagnosis, and diagnosed HD participants (figure e-1 on the Neurology® Web site at www.neurology.org).
Blinded comparison of LCMS and LCECA assays.
Because our findings did not confirm those from the PREDICT-HD study using the LCECA assay,22 we compared the 2 assays head-to-head, blindly, using 320 TRACK-HD plasma samples and control samples spiked with 8OHdG as described above. The LCMS assay was remarkably accurate in quantifying spiked concentrations of 8OHdG (adjusted r2 > 0.999; figure 1B), whereas the fit of the LCECA assay for the spiked samples was not as good (adjusted r2 = 0.622; figure 1C). Precision of the spiked samples expressed as %CV was ≤5.5% and accuracy expressed as %bias was ≤10.8%.
Table 1 shows demographic characteristics of study participants. Boxplots for 8OHdG levels at baseline and 24-month follow-up visits are shown for the LCMS assay (figure 2A) and the LCECA assay (figure 2B). We found no differences between disease groups at the baseline visit (table 2, top 2 rows). Longitudinal analysis of scores for controls and the other disease groups revealed no robust changes in 8OHdG between baseline and 24-month follow-up visits (figure 2, A and B; table 3, top 2 rows).
Table 1.
Demographic characteristics of the cohort
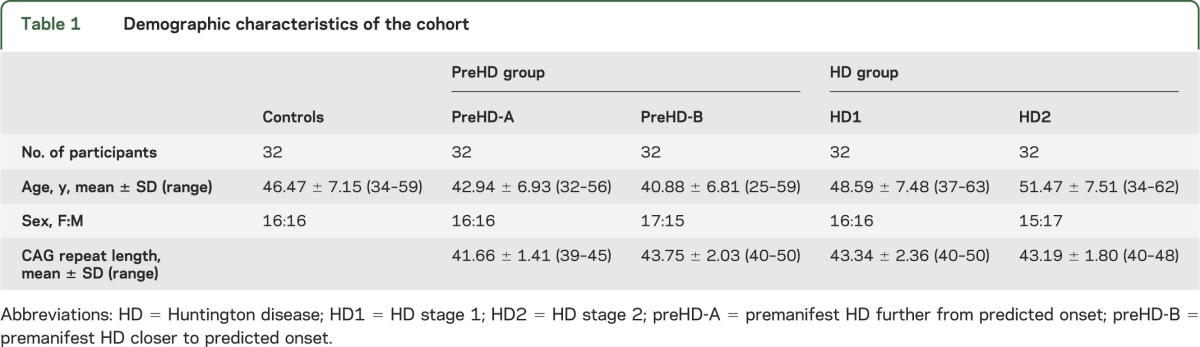
Figure 2. 8OHdG, putamen volume, and total motor score in TRACK-HD participants at baseline and 24 months.

Boxplots for each disease group showing baseline (B) and 24-month (24) 8OHdG levels measured with LCMS (A) and LCECA (B) assays, putamen volume as a percentage of ICV (C), and total motor score (D). One outlier (121 pg/mL, HD1 24 months) was excluded for graphical purposes only for the LCECA assay. 8OHdG = 8-hydroxy-deoxy-guanosine; HD = Huntington disease; HD1 = HD stage 1; HD2 = HD stage 2; ICV = total intracranial volume; LCECA = liquid chromatography–electrochemical array; LCMS = liquid chromatography–mass spectrometry; pre-A = premanifest HD further from predicted onset; pre-B = premanifest HD closer to predicted onset.
Table 2.
Adjusted between-group differences for baseline dataa

Table 3.
Differences in change over 24 months compared with controlsa

Analysis of other TRACK-HD measures.
To determine whether this study was appropriately conducted and sufficiently powered to show cross-sectional and longitudinal effects, we performed similar analyses of other TRACK-HD measures. Figure 2 shows boxplots for one imaging measure, putamen volume (figure 2C), and one clinical measure, TMS (figure 2D), from the same subset of participants at baseline and at 24-month visits. We found strong (p < 0.0001) between-group differences in both measures at the baseline visit (table 2, bottom rows). In addition, between-visit score changes differed between each group of gene-positive participants and controls for both measures, with the exception of TMS for the preHD-A group (table 3, bottom).
DISCUSSION
We set out to resolve conflicting published data regarding the utility of 8OHdG as a biomarker for HD progression by performing blinded cross-platform analyses of 8OHdG. Because samples from PREDICT-HD previously provided support for 8OHdG as an HD biomarker using the LCECA assay,22 we first developed a new LCMS assay and used it to evaluate samples from the same study. We found no differences in plasma 8OHdG among controls, near to onset premanifest, far from onset premanifest, and diagnosed early-stage HD participants. Similarly, using both the LCECA and LCMS assays, we found no differences in plasma 8OHdG among preHD gene carriers, early-stage HD participants, and age-matched controls from the TRACK-HD study, nor did 8OHdG levels change during a 2-year period. In contrast, the same participants showed large differences in clinical and imaging measures, both among disease groups and within individual participants across time.
Demonstrating excellent assay characteristics, the LCMS assay was more accurate than the LCECA assay in measuring the levels of 8OHdG in spiked samples (figure 1), showed good signal-to-noise ratio, was linear from 1 to 1,000 pg/mL, and above 10 pg/mL had a CV of 4%. LCMS instrumentation and techniques often replace previous analytic methods as the technology improves and proliferates (including those with ultraviolet or electrochemical readouts, such as LCECA) because they have better sensitivity, precision, and specificity.
Increased levels of leukocyte 8OHdG have previously been reported in HD participants compared with controls, but no stage-related changes were identified.21 Also, serum 8OHdG has been reported to be higher in patients with HD than in controls.16 Although using the LCECA assay longitudinal increases in plasma 8OHdG levels that varied with disease burden have been reported, this did not hold true when using the LCMS assay for the same analysis.22 More recently, it was reported that baseline plasma 8OHdG levels in a small number of participants in the 2-CARE study were not different between individuals with HD and controls.28
These previous reports came to different conclusions about the effects of HD status on 8OHdG levels at baseline and through disease progression.16,21,22 These analyses were not blinded, used the same LCECA assay, and did not include calibration curves or standardized methods of collection, preparation, and storage. Furthermore, plasma samples from the previous study were stored in larger volumes, requiring at least one freeze-thaw cycle before analysis.22 The present study used plasma samples from TRACK-HD, which employed a stringent, consistent sample collection and preparation protocol with quality-control measures and did not include a freeze-thaw cycle.6 We also included plasma spiked with various concentrations of 8OHdG to ensure the assays were measuring the same substrate and to better compare the accuracy of the 2 assays. Previous studies have not described sample-collection procedures in detail, making comparison difficult, but we consider proper sample handling critical to biomarker analysis efforts.
Although previous reports describing changes in 8OHdG in HD participants supported the view that HD-related neurodegeneration results from oxidative stress due to impaired mitochondrial function or excitotoxicity,19,21,29 our data do not support this hypothesis. Furthermore, we found no evidence that plasma 8OHdG is a useful measure of disease status, disease progression, or the efficacy of potential therapeutics.
Creatine treatment decreased and placebo treatment increased serum 8OHdG levels in patients with HD, and coenzyme Q10 treatment decreased plasma 8OHdG levels in both HD patients and controls.16,28 Although these observations raise the possibility that 8OHdG could be a pharmacodynamic biomarker for antioxidant drug action, in the absence of evidence that creatine or coenzyme Q10 has a clinical benefit for HD patients that can be monitored by 8OHdG levels, this should not be used as an efficacy biomarker in clinical trials.
Although our study strongly indicates that plasma 8OHdG is not a biomarker for HD progression, it may nonetheless reflect disease state when assessed in other fluids or tissues, or in other disorders. Two reports indicated that 8OHdG was elevated in HD in leukocytes and serum, whereas the current study and the recent report by Long et al. used plasma.16,21,22 Interestingly, in other studies, 8OHdG was elevated in leukocytes in Alzheimer disease and in urine in Friedreich ataxia30,31 and was higher in CSF from patients with Parkinson disease without dementia than in controls, but there were no differences between levels in controls and those with Parkinson disease without dementia, Alzheimer disease, or dementia with Lewy bodies.32 Furthermore, 8OHdG was higher in CSF, urine, and plasma from patients with amyotrophic lateral sclerosis compared with controls, whereas only the plasma levels and not the urine or CSF levels were elevated in a group of participants with other neurologic disorders.33
Identification and validation of biomarkers for HD clinical trials is extremely important and remains a challenging goal; this was a principal goal of the prospective, longitudinal, observational TRACK-HD study. Because the use of inappropriate biomarkers in clinical trials includes the risk of continuing trials that are likely to fail (potential false-positive signal) and stopping trials that may have led to a positive outcome (potential false-negative), potential biomarkers must be evaluated using the most careful and rigorous scientific principles and analysis tools. Based on our results, we recommend that future studies of putative biomarkers employ the following: 1) blinded sample analyses; 2) verification of measurements by independent analytic methods, particularly if the primary assay is not well-validated; 3) standard curves to estimate the accuracy and precision of measurements in the presence of the biological fluid(s); and 4) collection and storage of biological fluids under strict quality control. Using these strict criteria, our data challenge previous claims that 8OHdG is a useful clinical biomarker for HD progression.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the staff and study participants at each TRACK-HD site and all TRACK-HD coinvestigators. In particular, the authors thank Hans Johnson for measurement of putamen volume. The authors thank BioRep s.r.l. for maintenance of the biorepository and preparation of spiked samples; Wayne Matson for generating the LCECA assay results pursuant to a research agreement between CHDI Foundation and the Bedford VA Research Corporation, Inc., Bedford, MA; Jamshid Arjomand for input on study design; Simon Noble for reviewing the manuscript; and Sherry Lifer, Sue Juckett, and Mithra Mahmoudi for study support. The authors thank Michael Schirm and Kiang (Kevin) He for support in the design and performance of the LCMS assay.
GLOSSARY
- 8OHdG
8-hydroxy-deoxy-guanosine
- CV
coefficient of variation
- EDTA
ethylenediaminetetraacetic acid
- HD
Huntington disease
- LCECA
liquid chromatography–electrochemical array
- LCMS
liquid chromatography–mass spectrometry
- preHD
premanifest Huntington disease
- TMS
total motor score
- UHDRS
Unified Huntington’s Disease Rating Scale
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
B. Borowsky: drafting/revising manuscript, study concept and design, analysis and interpretation of data, study supervision and coordination, principal investigator. J. Warner: statistical analysis, drafting/revising manuscript. B.R. Leavitt: acquisition of data, drafting/revising manuscript. S.J. Tabrizi, R.A.C. Roos, and A. Durr: acquisition of data, reviewing/revising manuscript. C. Becker: analysis and interpretation of data, acquisition of data. C. Sampaio: drafting/revising manuscript. A.J. Tobin: drafting/revising manuscript, study concept and design, analysis and interpretation of data. H. Schulman: drafting/revising manuscript, study concept and design, analysis and interpretation of data, acquisition of data.
STUDY FUNDING
The CHDI Foundation, Inc., a not-for-profit biomedical research organization exclusively dedicated to developing therapies that slow the progression of Huntington disease, supported this study.
DISCLOSURE
B. Borowsky and J. Warner are employed by CHDI Management. B.R. Leavitt is employed by the University of British Columbia, and has served as a scientific advisor to Siena Biotech, Novartis, Isis Pharmaceuticals, Bristol-Myers Squibb, and Teva Pharmaceutical Industries. S.J. Tabrizi reports no disclosures. R.A.C. Roos is employed by LUMC and has received research grants from CHDI and Gossweiler Foundation. A. Durr received honoraria from Pfizer Inc. for participating in the Global TTR FAP Genetics Advisory Board. C. Becker reports no disclosures. C. Sampaio and A.J. Tobin are employed by CHDI Management. H. Schulman consults for Caprion Proteomics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72:971–983 [DOI] [PubMed] [Google Scholar]
- 2.Novak MJ, Tabrizi SJ. Huntington's disease: clinical presentation and treatment. Int Rev Neurobiol 2011;98:297–323 [DOI] [PubMed] [Google Scholar]
- 3.Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis 2010;5:40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington's disease. Neurology 2000;54:452–458 [DOI] [PubMed] [Google Scholar]
- 5.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD Study. J Neurol Neurosurg Psychiatry 2008;79:874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD Study: cross-sectional analysis of baseline data. Lancet Neurol 2009;8:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stout JC, Paulsen JS, Queller S, et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology 2011;25:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reilmann R, Bohlen S, Kirsten F, Ringelstein EB, Lange HW. Assessment of involuntary choreatic movements in Huntington's disease—toward objective and quantitative measures. Mov Disord 2011;26:2267–2273 [DOI] [PubMed] [Google Scholar]
- 9.Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011;10:31–42 [DOI] [PubMed] [Google Scholar]
- 10.Tabrizi SJ, Reilmann R, Roos RAC, et al. The TRACK-HD battery—potential endpoints for clinical trials in early stage Huntington’s disease and insights into future trials for premanifest HD. Lancet Neurol 2012;11:42–53 [DOI] [PubMed] [Google Scholar]
- 11.Aylward EH, Nopoulos PC, Ross CA, et al. Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry 2011;82:405–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowe KC, Paulsen JS, Langbehn DR, et al. Self-paced timing detects and tracks change in prodromal Huntington disease. Neuropsychology 2010;24:435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solomon AC, Stout JC, Weaver M, et al. Ten-year rate of longitudinal change in neurocognitive and motor function in prediagnosis Huntington disease. Mov Disord 2008;23:1830–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witjes-Ane MN, Mertens B, van Vugt JP, Bachoud-Levi AC, van Ommen GJ, Roos RA. Longitudinal evaluation of “presymptomatic” carriers of Huntington’s disease. J Neuropsychiatry Clin Neurosci 2007;19:310–317 [DOI] [PubMed] [Google Scholar]
- 15.Weir DW, Sturrock A, Leavitt BR. Development of biomarkers for Huntington's disease. Lancet Neurol 2011;6:573–590 [DOI] [PubMed] [Google Scholar]
- 16.Hersch SM, Gevorkian S, Marder K, et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology 2006;66:250–252 [DOI] [PubMed] [Google Scholar]
- 17.Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem 1997;272:19633–19636 [DOI] [PubMed] [Google Scholar]
- 18.Shigenaga MK, Gimeno CJ, Ames BN. Urinary 8-hydroxy-2′-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc Natl Acad Sci USA 1989;86:9697–9701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Browne SE, Bowling AC, MacGarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 1997;41:646–653 [DOI] [PubMed] [Google Scholar]
- 20.Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett 1999;272:53–56 [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Wu Y, Cheng M, et al. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem Biophys Res Commun 2007;359:335–340 [DOI] [PubMed] [Google Scholar]
- 22.Long JD, Matson WR, Juhl AR, et al. 8OHdG as a marker for Huntington disease progression. Neurobiol Dis 2012;46:625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem 2001;79:1246–1249 [DOI] [PubMed] [Google Scholar]
- 24.Bogdanov MB, Beal MF, McCabe DR, Griffin RM, Matson WR. A carbon column-based liquid chromatography electrochemical approach to routine 8-hydroxy-2′-deoxyguanosine measurements in urine and other biologic matrices: a one-year evaluation of methods. Free Radic Biol Med 1999;27:647–666 [DOI] [PubMed] [Google Scholar]
- 25.Matson WR, Langlais P, Volicer L, Gamache PH, Bird E, Mark KA. n-Electrode three-dimensional liquid chromatography with electrochemical detection for determination of neurotransmitters. Clin Chem 1984;30:1477–1488 [PubMed] [Google Scholar]
- 26.Matson WR, Gamache PG, Beal MF, Bird ED. EC array sensor concepts and data. Life Sci 1987;41:905–908 [DOI] [PubMed] [Google Scholar]
- 27.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 2004;65:267–277 [DOI] [PubMed] [Google Scholar]
- 28.Biglan KM, Dorsey ER, Evans RVV, et al. Plasma 8-hydroxy-2′-deoxyguanosine levels in Huntington disease and healthy controls treated with coenzyme Q10. J Huntingtons Dis 2012;1:73–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabrizi SJ, Workman J, Hart PE, et al. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol 2000;47:80–86 [DOI] [PubMed] [Google Scholar]
- 30.Mecocci P, Polidori MC, Ingegni T, et al. Oxidative damage to DNA in lymphocytes from AD patients. Neurology 1998;51:1014–1017 [DOI] [PubMed] [Google Scholar]
- 31.Schulz JB, Dehmer T, Schols L, et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000;55:1719–1721 [DOI] [PubMed] [Google Scholar]
- 32.Gmitterova K, Heinemann U, Gawinecka J, et al. 8-OHdG in cerebrospinal fluid as a marker of oxidative stress in various neurodegenerative diseases. Neurodegener Dis 2009;6:263–269 [DOI] [PubMed] [Google Scholar]
- 33.Bogdanov M, Brown RH, Matson W, et al. Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med 2000;29:652–658 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.