

Abstract
Objectives:
We sought to identify demographic and clinical features that were associated with expression of symptoms in the presence of Alzheimer disease (AD) neuropathologic changes.
Methods:
We studied 82 asymptomatic (Clinical Dementia Rating global score = 0) and 824 symptomatic subjects (Clinical Dementia Rating score >0) with low to high AD neuropathologic changes at autopsy who were assessed at 1 of 34 National Institute on Aging–funded Alzheimer’s Disease Centers. All subjects underwent a clinical examination within 1 year of death. Logistic regression was used to evaluate factors associated with the odds of being asymptomatic vs symptomatic.
Results:
Asymptomatic subjects tended to have low neurofibrillary tangle scores but a wide range of neuritic plaque frequencies. There were, however, a few asymptomatic subjects with very high tangle and neuritic plaque burden, as well as symptomatic subjects with few changes. In the multivariable model, asymptomatic subjects were older (odds ratio [OR] = 1.04; 95% confidence interval [CI] = 1.01–1.07), had lower clinical Hachinski Ischemic Score (OR = 0.82; 95% CI = 0.69–0.97), were less likely to have an APOE ε4 allele (OR = 0.36; 95% CI = 0.16–0.83), and had lower neurofibrillary tangle score (OR = 0.28; 95% CI = 0.17–0.45) compared with symptomatic subjects.
Conclusions:
Dissociating clinical symptoms from pathologic findings better allows for investigation of preclinical AD. Our results suggest that although the severity of the pathology, particularly neurofibrillary tangles, has a large role in determining the extent of symptoms, other factors, including age, APOE status, and comorbidities such as cerebrovascular disease also explain differences in clinical presentation.
The National Institute on Aging (NIA) recently urged prioritization of research to better define the preclinical stage of Alzheimer disease (AD) and to determine factors that predict emergence of clinical impairment.1 The reasons why persons with similar degrees of β-amyloid (Aβ) deposition and neurofibrillary tangles have different clinical expressions remain incompletely understood.1–4
The importance of this issue has been highlighted by recent changes in the neuropathologic definition of AD. Prior classification schemes such as CERAD (Consortium to Establish a Registry for Alzheimer's Disease)5 and NIA–Reagan Institute6 emphasized the presence of clinical symptoms along with AD neuropathologic features in order to assign a likelihood that the neuropathologic changes contributed to the clinical phenotype, thus discounting the presence of pathology in preclinical stages. The NIA–Alzheimer’s Association (AA) guidelines for the neuropathologic assessment of AD were released in 2012.7,8 These guidelines dissociate the clinical syndrome of dementia of the Alzheimer type (DAT) from the underlying AD neuropathologic change, which is an important step toward the acknowledgment that AD neuropathologic change precedes the onset of symptoms by several years.1,7
The new neuropathologic definitions give us an opportunity to better define and characterize persons with AD who have not yet developed DAT symptoms. We sought to use this opportunity to identify factors that were associated with clinical manifestations of mild cognitive impairment (MCI) due to AD and overt AD dementia in a large database of persons with autopsy findings consistent with AD.
METHODS
Study sample.
Data for this analysis came from the National Alzheimer's Coordinating Center (NACC) Uniform Data Set (UDS) and Neuropathology Data Set, which had been collected at 34 current and past Alzheimer's Disease Centers between September 2005 and September 2012. The UDS contains clinical and demographic information on subjects with normal cognition, MCI, AD dementia, and other etiologies. Detailed descriptions of UDS data have been published previously.9 UDS subjects may also consent to autopsy. For those who have an autopsy, neuropathologic features are recorded and submitted to the NACC.
The NIA-AA criteria for neuropathologic AD introduce an “ABC score,” which served as the basis for defining neuropathologic AD in this cohort.7 Braak stage (B score) for neurofibrillary tangles10 and CERAD neuritic plaque frequency5 (C score) were recorded in the Neuropathology Data Set. However, a Thal phase11 for Aβ plaques (A score) is not currently included. Thus, to include the most frequent plaque type, we included “diffuse plaque,” which is most likely an early form of Aβ plaque formation and is defined as plaques with no apparent dystrophic neurites, as detected by silver impregnation methods, ubiquitin, or tau immunohistochemistry. All types of Aβ plaques, including diffuse plaques, are also readily identified using Aβ immunohistochemistry.
Using the variable descriptions in the database, subjects with “sparse,” “moderate,” or “frequent” diffuse plaques were considered to have a Thal Aβ plaque phase of 1 or higher and met inclusion criteria for this study. Likewise, subjects with sparse, moderate, or frequent neuritic plaques had a neuritic plaque C score of 1 or higher and also met study inclusion criteria. Limiting the sample to subjects with either diffuse or neuritic plaques approximates to the inclusion of all subjects meeting NIA-AA criteria for low to high AD neuropathologic change. Thus, the study sample included only those subjects with amyloid plaques, excluding those without, regardless of clinical diagnosis.
Participants with incidental or amygdala-only Lewy bodies were included. However, subjects with a primary neuropathologic diagnosis of dementia with Lewy bodies (DLB) were excluded because these people were judged to have clinical symptoms primarily due to non-AD neurodegeneration and would confound the analysis.
Clinical symptoms were categorized using the Clinical Dementia Rating (CDR) global score.12 The CDR is an instrument that grades subjects' cognitive and functional abilities. The clinician, incorporating input from an informant who knows the subject well, evaluates the subject's performance in 6 domains: memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. Impairment in each of the 6 domains is evaluated by the clinician as none (0), questionable or very mild (0.5), mild (1), moderate (2), and severe (3). An algorithm combines the scores from the individual items to give a global score.13 Subjects with a CDR global score of 0 at their last clinical assessment were considered to have normal cognition, and thus comprised the “asymptomatic group.” Subjects with a score of 0.5 or higher were considered to exhibit clinical characteristics consistent with MCI due to AD or AD dementia and comprised the “symptomatic group.” To best correlate clinical symptoms and neuropathologic features, the analytic sample was limited to subjects who died within 1 year of the last UDS clinical assessment.
Several characteristics were considered as possible sources of differences among asymptomatic and symptomatic subjects. Demographics included subject age (at last clinical assessment), sex, race, and duration of education. Clinical characteristics included history of depression, family history of dementia, APOE ε4 allele status, and the modified Hachinski Ischemic Score.14 Finally, the neuropathologic features assessed included the NIA-AA neurofibrillary tangles (B score) and neuritic plaques (C score), as well as presence of vascular pathology, presence of one or more microinfarcts, and the presence of Lewy bodies.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained from all participants. Research using the NACC database was approved by the University of Washington Institutional Review Board.
Statistical analysis.
The relationship between each characteristic and cognitive status (asymptomatic vs symptomatic) was first evaluated individually using logistic regression with generalized estimating equations, a popular method used to account for the clustering of subjects within a unit, which in this case was the Alzheimer's Disease Center. The regression models were run with an independent correlation structure and robust standard errors.
Characteristics found to be significant at the 0.10 α level in the bivariate analyses were included in a full multivariable model. To address the possible effects of missing data, additional multivariable models were run excluding APOE status and excluding both APOE and Hachinski Ischemic Score. All analyses were performed using R 2.14.2 “geeglm” package.15
RESULTS
Between September 2005 and September 2012, 1,775 UDS subjects had died and undergone autopsy. Of those, 1,436 had at least some diffuse or neuritic plaques (339 excluded because of lack of amyloid pathology). After excluding subjects with >1 year between death and last UDS visit, the analytic sample came to include 906 subjects. Of these, 82 (9%) were asymptomatic and 824 (91%) were symptomatic.
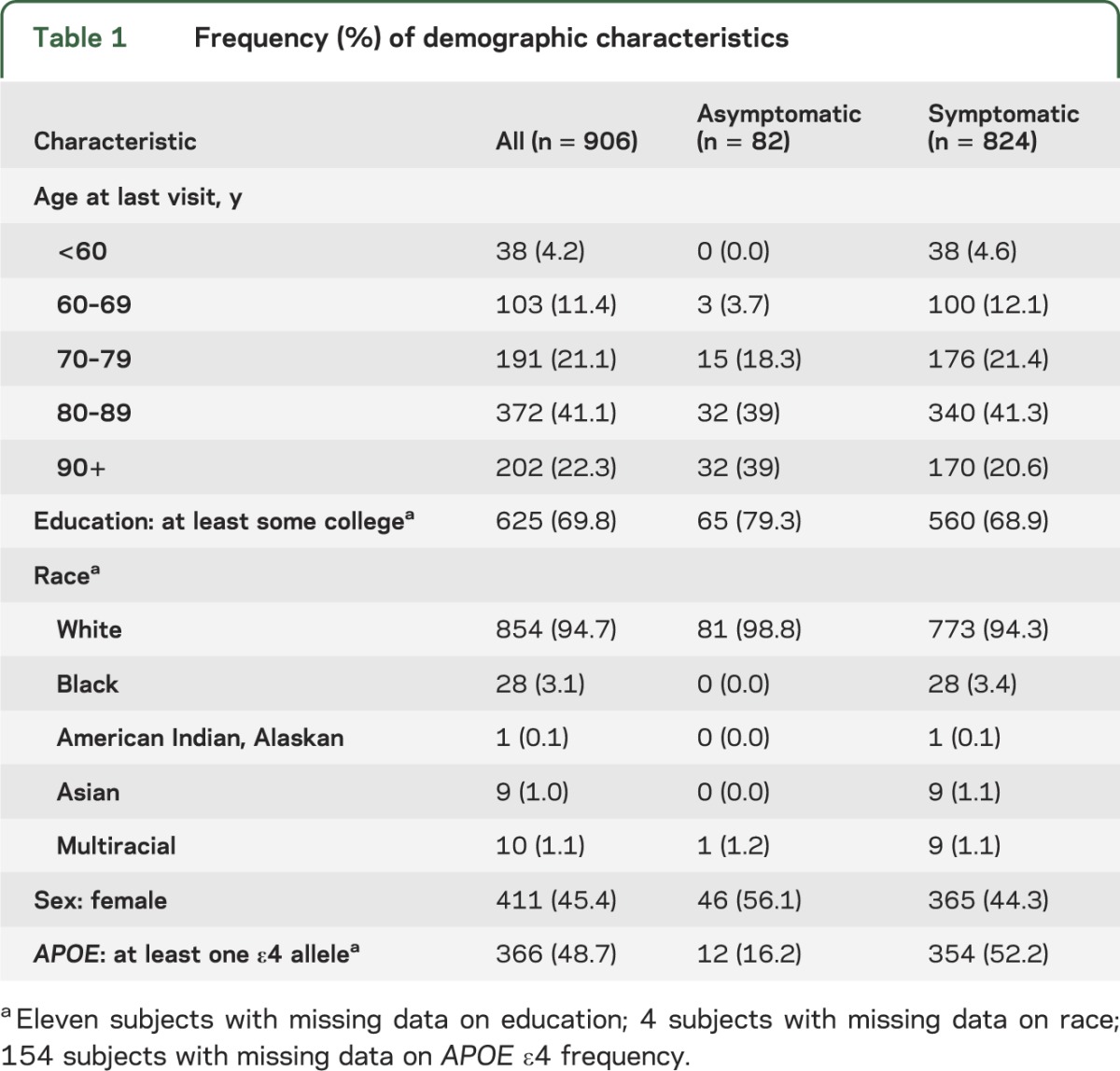
Characteristics of the asymptomatic and symptomatic subjects and the sample as a whole are displayed in table 1. Most subjects were white and had some college education. Mean age at last clinical assessment was higher for asymptomatic (86.2 ± 8.2 [SD] years) than symptomatic subjects (81.3 ± 11.2 years), and asymptomatic subjects had a lower frequency of carrying at least one APOE ε4 allele.
Table 1.
Frequency (%) of demographic characteristics
The figure displays the distribution of B and C scores among the symptomatic and asymptomatic participants. It should be noted that the NIA-AA staging scheme compresses 7 Braak stages into 4 “B” stages (0–3), which improves interrater reliability.7 The majority of symptomatic participants were at the high end of B and C scores, with B score of 3 (Braak stage V–VI) and frequent CERAD neuritic plaques. The asymptomatic subjects tended to have B score of 1 to 2 (Braak stage I–IV) but a wide range of neuritic plaque frequencies. There were, however, a few asymptomatic subjects with high B and C scores, as well as symptomatic subjects with low B and C scores.
Figure. Distribution of B and C scores among asymptomatic and symptomatic subjects with neuropathologic Alzheimer disease.

CERAD = Consortium to Establish a Registry for Alzheimer's Disease.
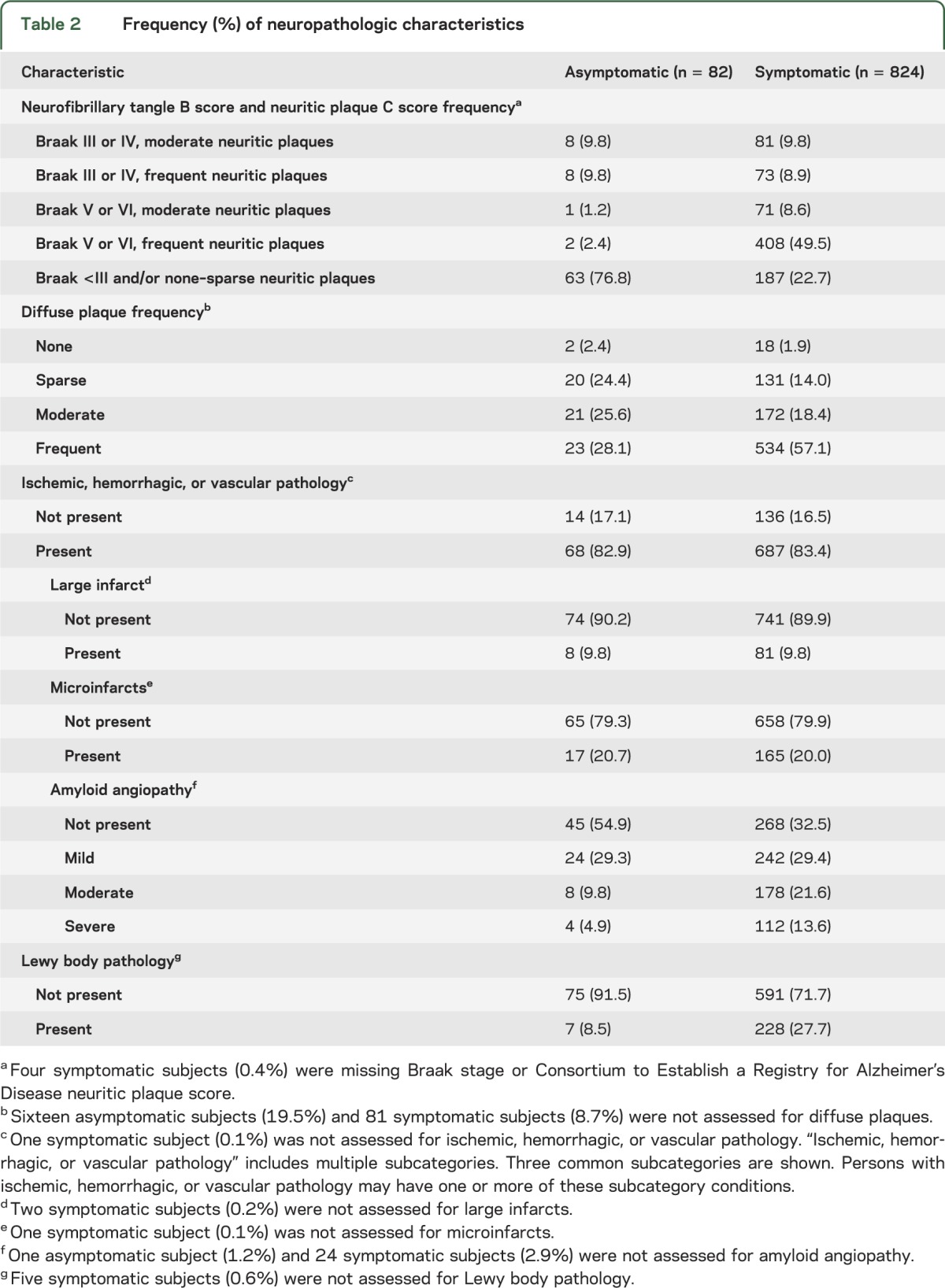
The frequencies of several neuropathologic features are described in table 2. As expected, having both the highest B score (3/3) and highest C score (3/3) was much more common in symptomatic than asymptomatic participants (49.5% vs 2.4%). A similar pattern was shown for diffuse plaques where symptomatic participants had a higher proportion of frequent diffuse plaques compared with asymptomatic subjects. There was minimal difference in the frequency of various cerebrovascular pathologies.
Table 2.
Frequency (%) of neuropathologic characteristics
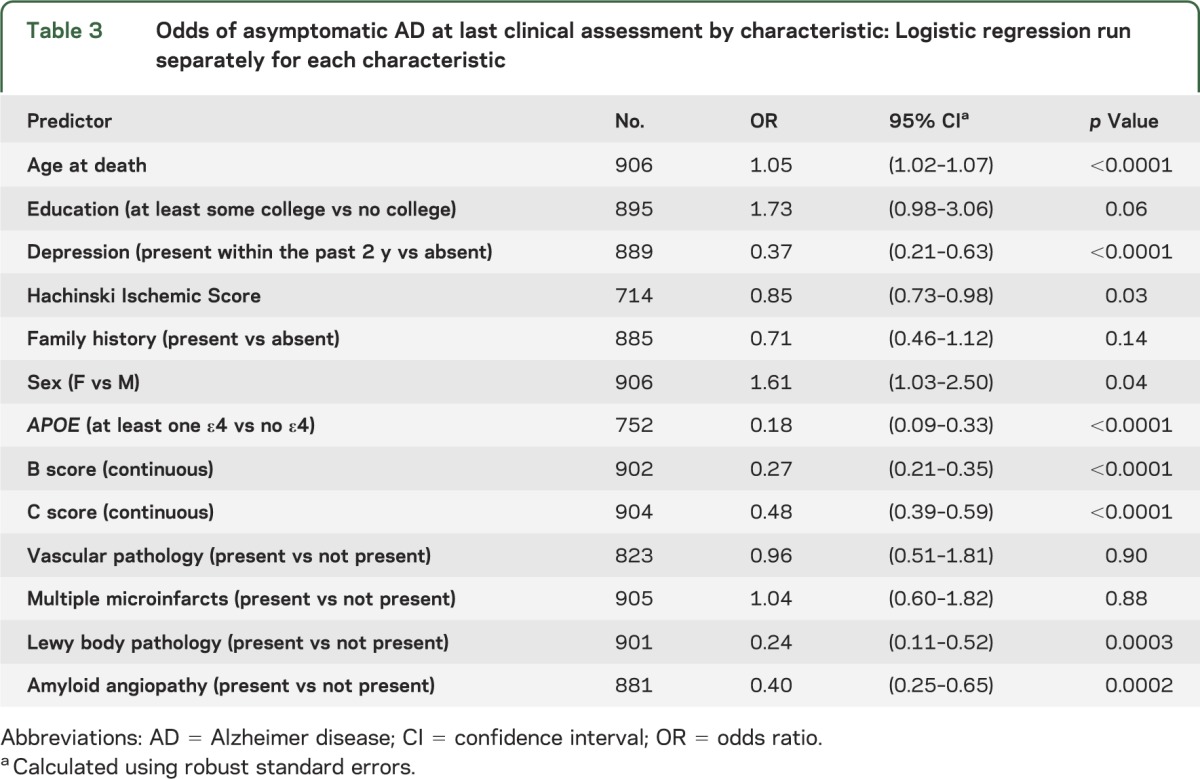
Several demographic and clinical characteristics were significantly associated with cognitive status in the bivariate analyses. The odds of being asymptomatic were increased for subjects who were older, had more education, had lower Hachinski Ischemic Score, did not have a recent history of depression, did not have an APOE ε4 allele, had lower B and C scores, and did not have Lewy body pathology (table 3). Race could not be modeled because of extremely small cell frequencies; all but one of the nonwhite subjects were symptomatic.
Table 3.
Odds of asymptomatic AD at last clinical assessment by characteristic: Logistic regression run separately for each characteristic
A full model (table 4) was fit using all of the predictors found to be significant in the bivariate models. In this model, the odds of being asymptomatic were significantly increased with older age, lower Hachinski Ischemic Score, no APOE ε4 allele, and lower B score. The association with age was such that the odds of being asymptomatic for a person aged 75 years was 75% higher than for a person aged 65. The effect of B score was quite marked; the odds of being asymptomatic more than tripled with each one stage decrease in B (neurofibrillary tangle) score.
Table 4.
Multivariable logistic regression models for odds of asymptomatic AD
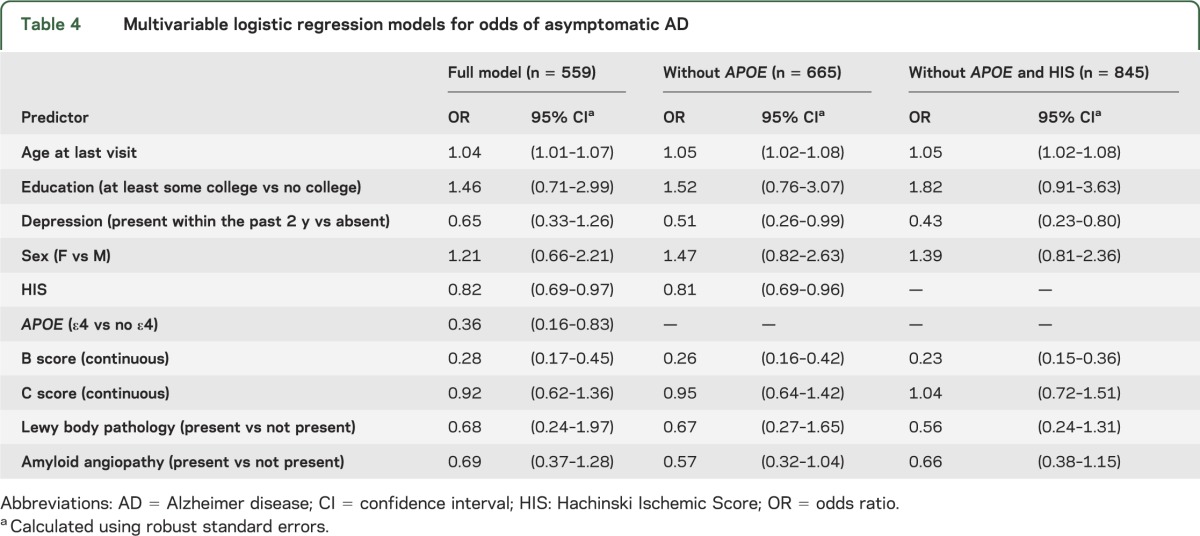
Because performing logistic regression required a complete case sample and much of the missing data were on APOE ε4 allele status and Hachinski Ischemic Score, the multivariable models were repeated excluding APOE and excluding both APOE and Hachinski Ischemic Score. Results, shown in table 4, were nearly identical to those in the full model, but depression became significant. In both models, the odds of being asymptomatic approximately doubled when the subject did not have a recent history of depression.
DISCUSSION
We sought to identify characteristics that were associated with symptoms among persons with underlying neuropathologic features of AD, as defined by the 2012 NIA-AA criteria. Our analysis revealed significant associations with several antemortem characteristics: compared with symptomatic subjects, asymptomatic subjects were older and had lower frequency of 1 or 2 APOE ε4 alleles, no history of depression, and fewer conditions consistent with vascular dementia.
There was a strong role for pathology in determining extent of clinical presentation. In bivariate analyses, neurofibrillary tangle (B) scores, neuritic plaque (C) scores, Lewy bodies, and amyloid angiopathy were all associated with increased odds of being symptomatic. However, in the multivariable model, only neurofibrillary tangle (B) score was associated with symptoms. The odds of being symptomatic increased 3-fold with every 1-point increase in neurofibrillary tangle (B) score. Nonetheless, the effect was incomplete as several asymptomatic subjects had pronounced pathology. Although highly correlated with being asymptomatic on bivariate analysis, neuritic plaque frequency became insignificant in the full model. In previous clinicopathologic and imaging studies, neurofibrillary tangles were similarly found to be more closely associated with cognitive decline than neuritic plaques.16–21
AD often occurs along with other neurodegenerative processes. A recent autopsy study of the oldest old found an association between presence of infarct pathology and symptoms.22 However, presence of microinfarcts was not associated with symptoms in our study. This might be explained by differences in the study samples, especially in terms of age,17 or differences in pathologic interpretation of lesions.5 Likewise, cerebral amyloid angiopathy (CAA) has been variably reported to be associated with increased risk of cognitive decline.23,24 The current study showed no independent effect of CAA once the extent of AD neuropathologic change was adjusted for, as shown previously.25
Interestingly, the presence of clinical vascular features on Hachinski Ischemic Score was significantly associated with symptoms whereas cerebrovascular pathology at autopsy was not. This difference could be attributable to the fact that vascular pathology was recorded as present vs absent. Within the category “present,” there may have been a range of severities and the simplification of absent vs present may have obscured a true relationship. Likewise, the Hachinski Ischemic Score captures features, such as emotional incontinence and somatic complaints, that may not necessarily represent a stroke on pathologic examination.
Depression was also associated with symptoms. Several studies have shown a relationship between depressive symptoms and clinical expression of AD.26−29 There is also evidence that depression could be associated with the pathology of the disease itself.30 However, it may not be possible, in the data available for the current study, to separate preexisting depression from symptoms of AD. We do not have data on lifetime experience with depression before development of symptoms (or for comparable time periods for asymptomatic persons).
The role of APOE was also interesting, in that it was strongly associated with symptoms even after adjusting for extent of underlying neuropathology. The residual effect of APOE after adjustment for neurofibrillary tangle (B) score and neuritic plaque (C) score may be attributable to the inability to adjust for Aβ (Thal phase) score in our data, or there may be another effect for which we are not able to account.31 For example, the presence of an APOE ε4 allele may be associated with pathology not captured here, such as greater levels of Aβ oligomers, which in turn have been linked to dementia symptoms after adjustment for amyloid plaque pathology.32
The association of older age with being asymptomatic seems counterintuitive, given increased AD incidence with increased age. This finding may be related to a somewhat increased severity for early-onset AD. It might also be attributable to a healthy survivor effect, with asymptomatic persons having a longer life expectancy compared with the symptomatic. Finally, this finding might be attributable to selection bias regarding those who enroll as normal controls.
On bivariate analysis, no college education and male sex were borderline significant predictors of symptoms. Although these variables were not significant in the multivariable models, the effect sizes were fairly large, suggesting that there might be an association that our study was underpowered to detect. Several previous studies have shown evidence of protective effect of increased cognitive reserve in AD2,33−35 and differences in presentation by sex, although not consistently.36–39
Before drawing conclusions from the data, the limitations of the study must be addressed. First, retrospectively fitting preexisting UDS data to the 2012 NIA-AA criteria has shortcomings. We were not able to derive an Aβ plaque (Thal phase) score. Using the surrogate of any diffuse plaques was a reasonable proxy for Thal phase ≥1, and thus a reasonable inclusion criterion. Nonetheless, it might have led to underassessment of cases that would have, in reality, met NIA-AA criteria. For example, some of the older autopsies used silver staining methods, thioflavin T, or Congo red, less-sensitive methods for detecting diffuse Aβ plaques than immunohistochemistry. Unfortunately, the staining methods are not available in the NACC neuropathology database, so we were unable to account for this protocol variation. Consequently, variation in staining methods between centers could have led to the exclusion of some subjects with AD neuropathologic (Aβ) changes not detected by silver and other stains. Moreover, based on prior AD classification schemes, there was likely a tendency to not perform special stains for diffuse plaques in asymptomatic subjects. This is indicated in our data by the fact that 19.5% of asymptomatic subjects did not have diffuse plaques assessed, compared with 8.7% of symptomatic subjects (table 2). However, the additional inclusion of subjects based on neuritic plaque (C) score ≥1 likely included most subjects with AD neuropathology who would have been misclassified because of lack of assessment of diffuse plaques or use of silver or other stains alone. Thus, we believe that with our inclusion criteria, most of the subjects in the NACC neuropathology database who had AD neuropathology were captured and that there was very little chance of misclassification of subjects without AD neuropathologic changes. Still, we were not able to stratify by Aβ plaque (A) Thal phase in the multivariable analysis, subsequently underassessing extent of Aβ deposition.
Second, there were considerable numbers of missing values for Hachinski Ischemic Score and APOE, which decreased the sample size. We performed sensitivity analyses excluding these variables, thus increasing the sample size. The findings from all 3 analyses were similar, supporting the likelihood that these are valid and robust findings.
Third, there are possibly biases in the nature of UDS subjects, especially among those who agree to autopsy, which limit our ability to draw inferences. For example, more highly educated subjects are more likely to enroll as normal controls, limiting our ability to draw inferences about the effect of education and cognitive reserve. In addition, there was only one nonwhite subject in the control group, prohibiting us from making inferences about the effect of race.
Fourth, there may have been confounding based on presence of Lewy bodies. Although Lewy body pathology is a common comorbidity in AD, to exclude the confounding influence of a second neurodegenerative disease, we excluded cases with a diagnosis of DLB. However, cases with incidental Lewy bodies were included. It is possible that some of these cases may now be recognized as a distinct entity called AD with amygdala Lewy bodies.40
Fifth, persons were classified as symptomatic vs asymptomatic. Dichotomization of an inherently continuous measure such as cognition is not ideal. However, we believe that use of the CDR as an outcome measure provides an objective and clinically relevant differentiation of people with and without recognizable cognitive symptoms.
Despite these limitations, these data allow us to draw reasonable conclusions regarding factors associated with clinical presentation of DAT in persons with AD neuropathology. There is obviously a very strong effect of neuropathology, with a 3-times-higher risk of becoming symptomatic for each point increase in neurofibrillary tangle (B) score. However, the effect is incomplete, with several asymptomatic subjects having very pronounced pathology. With persons with primary diagnosis of DLB excluded, there is no additional effect of the presence of Lewy bodies on likelihood of symptoms. There is no additional, independent effect of the presence of CAA after adjusting for extent of underlying AD neuropathology. Other factors that had a role in expression of symptoms included age and depression. The role of APOE is interesting, in that it was strongly associated even after adjusting for extent of underlying AD neuropathology. Future studies could add to this work by including a more diverse subject sample, especially for asymptomatic controls, and utilizing serial images to better assess the extent of cerebrovascular pathology at the time of cognitive assessment.
GLOSSARY
- AA
Alzheimer’s Association
- Aβ
β-amyloid
- AD
Alzheimer disease
- CAA
cerebral amyloid angiopathy
- CDR
Clinical Dementia Rating
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- CI
confidence interval
- DAT
dementia of the Alzheimer type
- DLB
dementia with Lewy bodies
- MCI
mild cognitive impairment
- NACC
National Alzheimer's Coordinating Center
- NIA
National Institute on Aging
- OR
odds ratio
- UDS
Uniform Data Set
AUTHOR CONTRIBUTIONS
S.E. Monsell contributed to the study concept and design, statistical analysis, interpretation of data, and drafting the manuscript. C. Mock contributed to the study concept and design, interpretation of data, and drafting the manuscript. C.M. Roe, N. Ghoshal, and J.C. Morris contributed to the study concept and design and editing of the manuscript for content. N.J. Cairns contributed to the study concept and design, interpretation of data, and editing of the manuscript for content. W. Kukull contributed to the study concept and design and editing of the manuscript for content.
STUDY FUNDING
The NACC database is supported by NIA grant UO1 AG016976.
DISCLOSURE
S.E. Monsell, C. Mock, and C.M. Roe report no disclosures. N. Ghoshal has participated or is currently participating in clinical trials of antidementia drugs sponsored by Elan/Janssen, Eli Lilly and Company, Wyeth, Pfizer, Novartis, and Bristol-Myers Squibb. J.C. Morris has participated or is currently participating in clinical trials of antidementia drugs sponsored by Janssen Alzheimer Immunotherapy Program and Pfizer. J.C. Morris has served as a consultant or has received speaking honoraria for Avid Radiopharmaceuticals, Eisai, Esteve, Janssen Alzheimer Immunotherapy Program, GlaxoSmithKline, Novartis, and Pfizer. N.J. Cairns and W. Kukull report no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roe CM, Xiong C, Miller JP, Morris JC. Education and Alzheimer disease without dementia: support for the cognitive reserve hypothesis. Neurology 2007;68:223–228 [DOI] [PubMed] [Google Scholar]
- 3.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 2009;68:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 2009;30:1026–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486 [DOI] [PubMed] [Google Scholar]
- 6.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997;18:S1–S2 [PubMed] [Google Scholar]
- 7.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord 2007;21:249–258 [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800 [DOI] [PubMed] [Google Scholar]
- 12.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414 [DOI] [PubMed] [Google Scholar]
- 13.Washington University School of Medicine Global Clinical Dementia Rating (CDR) based on CDR box scores [online]. Available at: http://www.biostat.wustl.edu/~adrc/cdrpgm/index.html. Accessed September 1, 2005
- 14.Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A. Pathological verification of ischemic score in differentiation of dementias. Ann Neurol 1980;7:486–488 [DOI] [PubMed] [Google Scholar]
- 15.R Development Core Team R: a language and environment for statistical computing. R Foundation for Statistical Computing [online]. Available at: http://www.R-project.org. Accessed March 1, 2012
- 16.Han SD, Gruhl J, Beckett L, et al. Beta amyloid, tau, neuroimaging, and cognition: sequence modeling of biomarkers for Alzheimer's disease. Brain Imaging Behav 2012;6:610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 2012;71:362–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 2007;66:1136–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992;42:631–639 [DOI] [PubMed] [Google Scholar]
- 20.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology 1992;42:1681–1688 [DOI] [PubMed] [Google Scholar]
- 21.Ghoshal N, Garcia-Sierra F, Wuu J, et al. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp Neurol 2002;177:475–493 [DOI] [PubMed] [Google Scholar]
- 22.James BD, Bennett DA, Boyle PA, Leurgans S, Schneider JA. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA 2012;307:1798–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011;37:75–93 [DOI] [PubMed] [Google Scholar]
- 24.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011;69:320–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 2010;20:66–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson RS, Barnes LL, Mendes de Leon CF, et al. Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology 2002;59:364–370 [DOI] [PubMed] [Google Scholar]
- 27.Green RC, Cupples LA, Kurz A, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol 2003;60:753–759 [DOI] [PubMed] [Google Scholar]
- 28.Barnes DE, Yaffe K, Byers AL, McCormick M, Schaefer C, Whitmer RA. Midlife vs late-life depressive symptoms and risk of dementia: differential effects for Alzheimer disease and vascular dementia. Arch Gen Psychiatry 2012;69:493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganguli M, Du Y, Dodge HH, Ratcliff GG, Chang CC. Depressive symptoms and cognitive decline in late life: a prospective epidemiological study. Arch Gen Psychiatry 2006;63:153–160 [DOI] [PubMed] [Google Scholar]
- 30.Rapp MA, Schnaider-Beeri M, Purohit DP, Perl DP, Haroutunian V, Sano M. Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry 2008;16:168–174 [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron 2009;63:287–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koffie RM, Hashimoto T, Tai HC, et al. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain 2012;135:2155–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray AD, Staff RT, McNeil CJ, et al. The balance between cognitive reserve and brain imaging biomarkers of cerebrovascular and Alzheimer's diseases. Brain 2011;134:3687–3696 [DOI] [PubMed] [Google Scholar]
- 34.Querbes O, Aubry F, Pariente J, et al. Early diagnosis of Alzheimer's disease using cortical thickness: impact of cognitive reserve. Brain 2009;132:2036–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortimer JA, Borenstein AR, Gosche KM, Snowdon DA. Very early detection of Alzheimer neuropathology and the role of brain reserve in modifying its clinical expression. J Geriatr Psychiatry Neurol 2005;18:218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts RO, Geda YE, Knopman DS, et al. The incidence of MCI differs by subtype and is higher in men: the Mayo Clinic Study of Aging. Neurology 2012;78:342–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hebert LE, Scherr PA, McCann JJ, Beckett LA, Evans DA. Is the risk of developing Alzheimer's disease greater for women than for men? Am J Epidemiol 2001;153:132–136 [DOI] [PubMed] [Google Scholar]
- 38.Yip AG, Brayne C, Matthews FE. Risk factors for incident dementia in England and Wales: the Medical Research Council Cognitive Function and Ageing Study—a population-based nested case-control study. Age Ageing 2006;35:154–160 [DOI] [PubMed] [Google Scholar]
- 39.Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry 2005;62:685–691 [DOI] [PubMed] [Google Scholar]
- 40.Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 2006;65:685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]