

Abstract
Objective:
To describe the clinical and neurophysiologic phenotype of a family with hereditary sensory and autonomic neuropathy type 1 (HSANI) due to a novel mutation in SPTLC2 and to characterize the biochemical properties of this mutation.
Methods:
We screened 107 patients with HSAN who were negative for other genetic causes for mutations in SPTLC2. The biochemical properties of a new mutation were characterized in cell-free and cell-based activity assays.
Results:
A novel mutation (A182P) was found in 2 subjects of a single family. The phenotype of the 2 subjects was an ulcero-mutilating sensory-predominant neuropathy as described previously for patients with HSANI, but with prominent motor involvement and earlier disease onset in the first decade of life. Affected patients had elevated levels of plasma 1-deoxysphingolipids (1-deoxySLs). Biochemically, the A182P mutation was associated with a reduced canonical activity but an increased alternative activity with alanine, which results in largely increased 1-deoxySL levels, supporting their pathogenicity.
Conclusion:
This study confirms that mutations in SPTLC2 are associated with increased deoxySL formation causing HSANI.
Hereditary sensory and autonomic neuropathy type 1 (HSANI) is an autosomal dominant (AD) sensory neuropathy complicated by ulcerations and amputations, with variable autonomic and motor involvement.1 To date, mutations in 5 genes have been reported to cause AD HSANI.2–7
Mutations in the enzyme serine palmitoyltransferase (SPT) cause HSANI.2,3 SPT is a heteromeric enzyme composed of 3 subunits (SPTLC1–3) located at the outer membrane of the endoplasmic reticulum.8,9 It catalyzes the first and rate-limiting step in de novo sphingolipid synthesis: the condensation of l-serine and palmitoyl-coenzyme A. Mutations in SPTLC1 account for 12% of patients with HSAN.10 More recently, mutations in SPTLC2 were found to cause HSANI with a similar phenotype.4 SPTLC1 and SPTLC2 mutations generally cause a shift in the substrate specificity of SPT leading to the alternative use of l-alanine and l-glycine over its canonical substrate l-serine.11,12 This forms an atypical category of 1-deoxysphingolipids (1-deoxySLs) lacking the C1 hydroxyl group, which impedes their conversion into complex sphingolipids but also their canonical degradation. Elevated 1-deoxySLs were found in the plasma and lymphoblasts of patients with HSANI11 and plasma and tissues of transgenic HSANI mice.13 The 1-deoxySLs were shown to be neurotoxic, blocking neurite formation in cultured neurons.11
Herein, we report a novel HSANI mutation in SPTLC2 associated with elevated plasma 1-deoxySLs. In vitro and cell culture studies show that this mutant has reduced canonical activity but forms high levels of 1-deoxySLs.
METHODS
Standard protocol approvals, registrations, and patient consents.
Ethical approval for this study was obtained from the Joint Medical and Ethics Committee at the National Hospital for Neurology and Neurosurgery. Written informed consent was obtained from all patients.
Patients.
One hundred seven patients with HSAN were selected from our inherited neuropathy database, including patients seen in the National Hospital for Neurology and Neurosurgery peripheral neuropathy clinics as well as patients whose DNA was referred from other hospitals for diagnostic and research testing. Most patients presented with distal progressive sensory loss, with or without ulcero-mutilating complications or autonomic dysfunction. Because of the overlap between Charcot-Marie-Tooth type 2B and HSANI, we included patients with motor involvement; however, sensory features were predominant. Diagnosis was based on clinical phenotype in addition to neurophysiology. All patients were negative for mutations in SPTLC1 and most were also negative for mutations in RAB7, NGFB, FAM134B, and NTRK1. Three hundred fifty-eight British control chromosomes were screened for control.
Patient assessment.
Patients found to have mutations underwent detailed clinical and neurophysiologic assessments including the Charcot-Marie-Tooth Neuropathy Score 2.14 Nerve conduction studies were performed using standard technique.
Genetic sequencing.
All 12 exons and flanking introns of SPTLC2 were amplified using Roche PCR reagents. Sequence reactions were performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and resolved on an ABI 3730xl Sequencer. For primers and primer conditions, see table e-1 on the Neurology® Web site at www.neurology.org. Sequence variants were confirmed by repeat sequencing.
PolyPhen2 (http://genetics.bwh.harvard.edu/pph/), SIFT (http://blocks.fhcrc.org/sift/SIFT.html), and aGVGD (http://agvgd.iarc.fr/) were used to predict the effect of the mutation on protein function.
Cloning.
The A182P was introduced into the SPTLC2 cDNA by site-directed mutagenesis as described previously11 (see table e-2 for primers). All constructs were verified by sequencing.
Stable expression of SPTLC2wt and the A182P mutation in HEK293 cells.
HEK293 cells (ATCC, Manassas, VA) were stably transfected with the empty vector (control), SPTLC2 wild type (wt), or the A182P mutant as described previously.11 Expression of the constructs was confirmed by reverse transcriptase–PCR and immune blotting (figure e-1).
Metabolic labeling assay.
Transfected HEK293 cells were cultured in Dulbecco’s modified Eagle medium for 3 days before medium was changed to Dulbecco’s modified Eagle medium without l-serine (Genaxxon BioScience, Ulm, Germany). After 2 hours of preincubation, isotope-labeled (2,3,3-d3, 15N) l-serine (1 mM) and (2,3,3,3)-d4–labeled l-alanine (5 mM) (Cambridge Isotope Laboratories, Inc., Andover, MA) were added to the cells. In some cases, cells were treated with Fumonisin B1 (FB1) (35 μM) as described. Cells were harvested after 24 hours, counted (Z2 Coulter Counter; Beckman Coulter, Brea, CA), and pelleted (800g, 5 minutes at 4°C). Pellets were stored at −20°C.
SPT in vitro activity assay and lipid-base extraction.
SPT in vitro activity was measured as described previously.15 In modification to the original protocol, 64 mM l-serine or 320 mM l-alanine (Sigma, St. Louis, MO) was used.
Acid-base extraction and analysis of sphingoid bases.
MetOH 500 µL including 200 pmol internal standard (d7-sphinganine and d7-sphingosine; Avanti Polar Lipids, Alabaster, AL) were added to 100 µL of plasma or frozen cell pellets resuspended in 100 µL phosphate-buffered saline. Lipid extraction was performed for 1 hour at 37°C with constant agitation at 1,000 rpm (Thermomixer Comfort; Eppendorf, Hamburg, Germany). Precipitated protein was removed by centrifugation (5 minutes at 16,000g). After transferring the supernatant into a new tube, lipids were extracted and analyzed by liquid chromatography–mass spectrometry as described earlier.11
Statistics.
One-way analysis of variance was used for statistical analysis. Significance was verified by Bonferroni multiple correction. Unless otherwise stated, p values <0.001 were considered significant. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
RESULTS
Genetics.
Sequencing of SPTLC2 in 107 index patients with HSANI revealed a novel mutation in one family. c.544G>C, A182P, was found in the index case (figure 1, III-2); this mutation was not present in 358 control chromosomes. One other affected member of this family who was tested carried the mutation (figure 1, IV-2).
Figure 1. Pedigree of HSANI family with SPTLC2-A182P mutation.
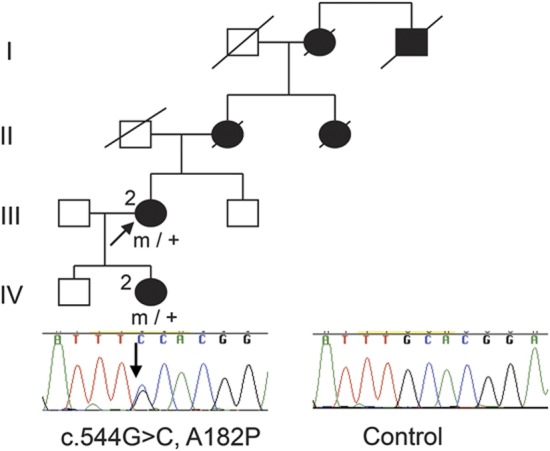
HSANI = hereditary sensory and autonomic neuropathy type 1; square = male; circle = female; diagonal line = deceased; filled symbol = affected; m/+ = heterozygous for mutation; arrow = proband; electropherogram of affected patient and control.
The A182P mutation affects a highly evolutionary conserved amino acid; PolyPhen2 and SIFT both predict that this change would be damaging, whereas aGVGD classifies the variant as class C25, where C65 is most likely to interfere with function and C0 less likely to interfere with function.
Clinical details.
The family had AD inheritance (figure 1). Clinical features are documented in table 1. Both patients had onset in the first decade, presenting with reduced sensation in the feet, with later development of motor weakness. Sensory loss occurred in a glove and stocking distribution; pinprick perception was affected to a greater extent than vibration perception, similar to patients with SPTLC1 mutations.16 Patient III-2 had painful tingling in the hands whereas IV-2 had no history of pain. Sensory complications including ulcers and accidental burns occurred in both patients, and severe wasting and weakness were present (figure e-2). There were no symptoms of hearing loss or autonomic dysfunction.
Table 1.
Clinical details

Neurophysiology.
Table 2 describes the neurophysiology of the patients with HSANI. Although this was predominantly an axonal neuropathy, motor conduction velocities in the upper limbs were slow (<38 m/s), even in patient III-2 who had reasonable upper limb motor amplitudes, a finding that we have previously documented in our HSANI families with SPTLC1 mutations.16
Table 2.
Nerve conduction studies
Plasma sphingoid and deoxysphingoid base levels.
Total sphinganine (SA) or sphingosine (SO) levels showed no difference between SPTLC2-A182P patients and healthy controls, whereas the 1-deoxySL levels were elevated in the plasma of the two A182P carriers. Higher 1-deoxySL plasma levels were found in the more severely affected patient, IV-2 (figure 2).
Figure 2. Sphingoid (A) and deoxysphingoid (B) base levels.
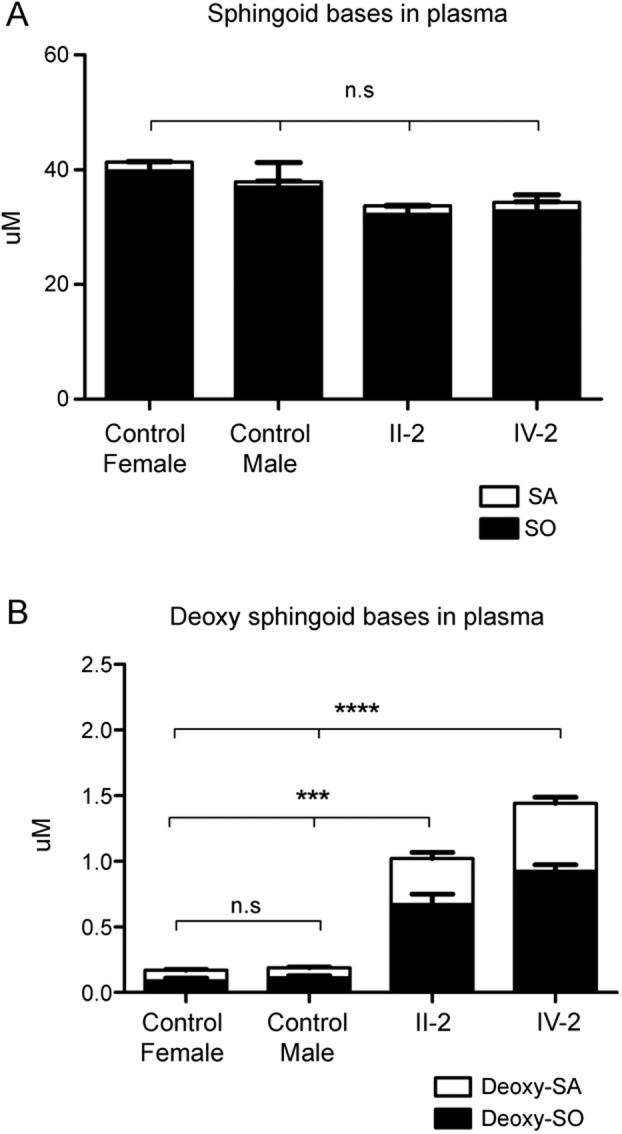
1-Deoxysphingolipids in plasma of patients and healthy controls. Significantly elevated 1-deoxysphingolipid levels were found for patients carrying the SPTLC2-A182P mutation. Data are shown as means, with error bars representing SDs. SDs were calculated using 1-way analysis of variance with Bonferroni multiple correction. n.s. = not significant; SA = sphinganine; SO = sphingosine. ***p < 0.001; ****p < 0.0001.
Effect of the SPTLC2-A182P mutation on SPT activity.
Most HSANI mutations in SPTLC1 and SPTLC2 are associated with reduced canonical activity and increased 1-deoxySL formation.2–4,17 The A182P mutant showed a 65% reduced in vitro activity with l-serine and a 3.5-fold increased activity with l-alanine (figure 3, A and B). These results were confirmed by a metabolic labeling assay using isotope-labeled (d3, 15N) l-serine (1 mM) and (d4) l-alanine (5 mM), which leads to the formation of isotope-labeled (M+3)SA and (M+3)deoxySA because one deuterium is lost during the condensation reaction.18 The assay was performed in the presence or absence of FB1, an inhibitor for ceramide synthase. The inhibition of ceramide synthase leads to the accumulation of SA over time, which is a measure for cellular SPT activity. In the presence of FB1, the SPTLC2wt cells showed a 2- to 3-fold higher accumulation of (M+3)SA and (M+3)deoxySA compared with controls (figure 3, C and D). In the A182P cells, the formation of (M+3)SA was reduced 2- to 3-fold (figure 3C). (M+3)deoxySA formation was 15-fold higher in A182P than in SPTLC2wt cells (figure 3D). In the absence of FB1, the sphingoid bases are mostly metabolized into ceramides and complex sphingolipids, which results in a predominant release of (M+3)SO after acid hydrolysis (figure 3C). In contrast, deoxySLs were predominantly present as (M+3)deoxySA (figure 3D). Interestingly, in SPTLC2wt cells, the total sum of isotope-labeled sphingoid bases was 50% higher in FB1-treated than in nontreated cells. In the absence of FB1, the activity with serine was the same in A182P and SPTLC2wt cells (figure 3C). No isotope-labeled 1-deoxySLs were found in control or SPTLC2wt cells when FB1 was absent. However, labeled 1-deoxySLs were formed in A182P cells, although total levels were about 8-fold lower compared with FB1-treated A182P cells (figure 3D). Because FB1 showed no effect on SPT activity in vitro (data not shown), we assume that the stimulatory effect of FB1 on SPT activity is indirect and probably associated with a recently described metabolic feedback regulation.19–21
Figure 3. SPT activity in vitro (A, B) and in HEK293 cells (C, D).

In vitro SPT activity with l-serine (A) and l-alanine (B) measured in 1 mg total protein lysate extracted from HEK293 cells expressing wt or mutant SPTLC2. (A) The A182P mutation leads to a significant decrease in keto-SA compared with the SPTLC2wt-expressing control (p < 0.001). (B) The A182P mutation leads to a significant increase in keto-deoxySA compared with the SPTLC2wt-expressing control (p < 0.0001). (C) SPT activity measured in HEK293 cells. SPT activity in the SPTLC2wt is increased in the presence of FB1, whereas this increase is lost for the A182P mutant (p < 0.0001). Without FB1, SPT activity is not different between mutant and SPTLC2wt. (D) Generation of deoxySLs in HEK293 cells. The A182P mutation leads to a significant increase in deoxySL formation compared with SPTLC2wt cells (p < 0.0001). In the presence of FB1, deoxySLs are also generated by SPTLC2wt cells (p < 0.001), but the formation was significantly higher in the A182P cells (p < 0.0001). Data are shown as means, with error bars representing SDs. SDs were calculated using 1-way analysis of variance with Bonferroni multiple correction. FB1 = Fumonisin B1; deoxy-SA = deoxysphinganine; deoxy-SO = deoxysphingosine; deoxySL = deoxysphingolipid; SA = sphinganine; SO = sphingosine; SPT = serine palmitoyltransferase; wt = wild-type.
No differences in the spatial distribution were seen between SPTLC2 and the A182P mutant. Immunohistochemistry showed in both cases a colocalization with the endoplasmic reticulum marker calnexin (figure e-3).
DISCUSSION
Mutations in SPTLC1 are a well-established cause of HSANI.2,3,16 An initial study of 12 families with HSANI did not find any patients with mutations in SPTLC2.22 However, because of the known association of SPT with HSANI, SPTLC2 remained a functional candidate and a later study established that mutations in SPTLC2 were also associated with HSANI.4 Our report provides genetic and functional data demonstrating that a novel A182P mutation found in one family is pathogenic, and gives detailed clinical and neurophysiologic descriptions of the phenotype. The age at onset in this family was in the first decade, younger than is typically seen in patients with HSANI. To date, 3 mutations in SPTLC2 have been described (V359M, G382V, and I504F).4 One of these was associated with early onset in the first decade and other atypical features such as motor conduction velocities in the demyelinating range, suggesting a wide phenotypic spectrum of HSANI.
Elevated blood 1-deoxySL levels were confirmed in both A182P carriers; higher levels were found in the more severely affected daughter than the mother. In vitro, the A182P mutant showed significantly reduced activity with l-serine and increased activity with l-alanine. This was confirmed by 2 metabolic labeling assays.
The combined genetic, clinical, and functional data confirm the association of this novel SPTLC2 mutation with HSANI. It further supports the concept that a pathologic 1-deoxySL formation underlies the pathogenesis of HSANI, indicating that HSANI, in contrast to most other inherited neuropathies, is a metabolic disorder. The 1-deoxySLs are hereby relevant HSANI biomarkers to validate the functional consequences of genetic SPTLC1 and SPTLC2 variants.
Supplementary Material
GLOSSARY
- AD
autosomal dominant
- 1-deoxySL
1-deoxysphingolipid
- FB1
Fumonisin B1
- HSANI
hereditary sensory and autonomic neuropathy type 1
- SA
sphinganine
- SO
sphingosine
- SPT
serine palmitoyltransferase
- wt
wild type
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Sinéad Murphy and Daniela Ernst drafted the manuscript and acquired, analyzed, and interpreted the data. Yu Wei, Matilde Laurà, Yo-Tsen Liu, James Polke, Julian Blake, John Winer, and Henry Houlden acquired, analyzed, and interpreted the data and revised the manuscript. Thorsten Hornemann and Mary Reilly were involved with the study concept, interpreted the data, revised the manuscript, and supervised the study.
STUDY FUNDING
Professor Reilly is grateful to the Medical Research Council and the Muscular Dystrophy Campaign and S. Murphy, M. Laurà, and M. Reilly are grateful to the National Institute of Neurological Disorders and Stroke/Office of Rare Diseases (ORD) (1U54NS065712-01) for their support. T. Hornemann and D. Ernst are grateful to the Gebert Rüf Foundation (GRS-047/09), the Center of Integrated Human Physiology (ZIHP), and “radiz”—Rare Disease Initiative Zurich, Clinical Research Priority Program for Rare Diseases, University of Zurich. This work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
DISCLOSURE
S. Murphy received a postdoctoral training fellowship from the Inherited Neuropathy Consortium Rare Disease Clinical Research Consortium, supported by the NIH National Institute of Neurological Disorders and Stroke/ORD (1U54NS065712-01). D. Ernst received funding from the Gebert Rüf Foundation (GRS-047/09). Y. Wei reports no disclosures. M. Laurà received grant funding from National Institute of Neurological Disorders and Stroke/ORD (1U54NS065712-01). Y.-T. Liu, J. Polke, J. Blake, J. Winer, and H. Houlden report no disclosures. T. Hornemann received funding from the Gebert Rüf Foundation (GRS-047/09). M. Reilly received grant funding from National Institute of Neurological Disorders and Stroke/ORD (1U54NS065712-01). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Houlden H, Blake J, Reilly MM. Hereditary sensory neuropathies. Curr Opin Neurol 2004;17:569–577 [DOI] [PubMed] [Google Scholar]
- 2.Bejaoui K, Wu C, Scheffler MD, et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet 2001;27:261–262 [DOI] [PubMed] [Google Scholar]
- 3.Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 2001;27:309–312 [DOI] [PubMed] [Google Scholar]
- 4.Rotthier A, Auer-Grumbach M, Janssens K, et al. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet 2010;87:513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verhoeven K, De Jonghe P, Coen K, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 2003;72:722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guelly C, Zhu P-P, Leonardis L, et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am J Hum Genet 2011;88:99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein CJ, Botuyan MV, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011;43:595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 2003;1632:16–30 [DOI] [PubMed] [Google Scholar]
- 9.Hornemann T, Richard S, Rutti MF, Wei Y, von Eckardstein A. Cloning and initial characterisation of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem 2006;281:37275–37281 [DOI] [PubMed] [Google Scholar]
- 10.Davidson GL, Murphy SM, Polke JM, et al. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol 2012;259:1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penno A, Reilly MM, Houlden H, et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 2010;285:11178–11187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zitomer NC, Mitchell T, Voss KA, et al. Ceramide synthase inhibition by Fumonisin B1 causes accumulation of 1-deoxysphinganine: a novel category of bioactive 1-deoxysphingoid bases and 1-deoxydihydroceramides biosynthesized by mammalian cell lines and animals. J Biol Chem 2009;284:4786–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eichler FS, Hornemann T, McCampbell A, et al. Overexpression of the wild-type SPT1 subunit lowers deoxysphingolipid levels and rescues the phenotype of HSAN1. J Neurosci 2009;29:14646–14651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy SM, Hermann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011;16:191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rütti MF, Richard S, Penno A, von Eckardstein A, Hornemann T. An improved method to determine serine palmitoyltransferase activity. J Lipid Res 2009;50:1237–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Houlden H, King R, Blake J, et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain 2006;129:411–425 [DOI] [PubMed] [Google Scholar]
- 17.Verhoeven K, Coen K, De Vriendt E, et al. SPTLC1 mutation in twin sisters with hereditary sensory neuropathy type I. Neurology 2004;62:1001–1002 [DOI] [PubMed] [Google Scholar]
- 18.Ikushiro H, Fujii S, Shiraiwa Y, Hayashi H. Acceleration of the substrate Calpha deproteination by an analogue of the second substrate palmitoyl-CoA in serine palmitoyltransferase. J Biol Chem 2008;283:7542–7553 [DOI] [PubMed] [Google Scholar]
- 19.Breslow DK, Collins SR, Bodenmiller B, et al. Orm family proteins mediate sphingolipid homeostasis. Nature 2010;463:1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S, Lone MA, Schneiter R, Chang A. Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc Natl Acad Sci USA 2010;107:5851–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siow DL, Wattenberg BW. Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. J Biol Chem 2012;287:40198–40204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawkins JL, Brahmbhatt SB, Auer-Grumbach M, et al. Exclusion of serine palmitoyltransferase long chain base subunit 2 (SPTLC2) as a common cause for hereditary sensory neuropathy. Neuromuscul Disord 2002;12:656–658 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}