

Abstract
Members of the actin family of proteins exhibit different biochemical properties when ATP, ADP-Pi, ADP, or no nucleotide is bound. We used molecular dynamics simulations to study the effect of nucleotides on the behavior of actin and actin-related protein 3 (Arp3). In all of the actin simulations, the nucleotide cleft stayed closed, as in most crystal structures. ADP was much more mobile within the cleft than ATP, despite the fact that both nucleotides adopt identical conformations in actin crystal structures. The nucleotide cleft of Arp3 opened in most simulations with ATP, ADP, and no bound nucleotide. Deletion of a C-terminal region of Arp3 that extends beyond the conserved actin sequence reduced the tendency of the Arp3 cleft to open. When the Arp3 cleft opened, we observed multiple instances of partial release of the nucleotide. Cleft opening in Arp3 also allowed us to observe correlated movements of the phosphate clamp, cleft mouth, and barbed-end groove, providing a way for changes in the nucleotide state to be relayed to other parts of Arp3. The DNase binding loop of actin was highly flexible regardless of the nucleotide state. The conformation of Ser14/Thr14 in the P1 loop was sensitive to the presence of the γ-phosphate, but other changes observed in crystal structures were not correlated with the nucleotide state on nanosecond timescales. The divalent cation occupied three positions in the nucleotide cleft, one of which was not previously observed in actin or Arp2/3 complex structures. In sum, these simulations show that subtle differences in structures of actin family proteins have profound effects on their nucleotide-driven behavior.
Keywords: actin, Arp3, nucleotide, dynamics, simulation
Introduction
Actin filaments form cytoskeletal and contractile structures vital to cellular function.1 Actin monomers bind ATP in a deep cleft and hydrolyze it shortly (k=0.3 s−1) after incorporation into filaments.2 The ADP-Pi intermediate slowly releases the γ-phosphate,3 leaving nonexchangeable ADP bound to most subunits in filaments. Bound nucleotides are not required for polymerization, but nucleotide-free actin denatures rapidly4 and each nucleotide state of monomeric and filamentous actin has different polymerization properties.5 The nucleotide bound to actin monomers and filaments also influences the affinities for binding proteins, including actin depolymerizing factor/cofilin,6 β-thymosins,7 and profilin.8
Since the first crystal structure of actin was solved in 1990,9 much effort has been devoted to investigating how the bound nucleotide influences the conformation and activity of the protein. Crystal structures and electron microscopy have revealed different conformations of the DNase binding loop (His40–Asp51) and changes in the width of the nucleotide binding cleft. However, the conformations of ATP–actin and ADP–actin are remarkably similar in most actin crystal structures, so we lack a consistent basis for the observed differences in biochemical assays.
Crystallization of actin monomers requires point mutations, covalent modification, or association with other proteins, and the resulting structures tend to vary based on modification strategies or crystallization conditions. For example, in crystals of actin with tetramethylrhodamine coupled to Cys374, the DNase binding loop is folded into an α-helix with bound ADP10 but has no secondary structure with bound ATP11 (see Fig. 1a). However, the DNase binding loop is disordered in crystal structures of both ATP–actin and ADP–actin with two point mutations in subdomain 4 to prevent polymerization.12 Variations in the nucleotide binding cleft also depend on crystallization conditions. In most crystal structures of actin, the nucleotide binding cleft is closed around either ATP or ADP.13–18 So far, the cleft is open only in certain crystals of ATP–β-actin bound to profilin, where the P1 and P2 loops were separated by an additional 3 Å from closed conformations.14 However, the cleft was closed in crystals of the same complex grown in different conditions.15 On larger length scales, reconstructions of electron micrographs of budding yeast actin filaments show the nucleotide cleft open in ADP filaments and closed in ADP filaments in beryllium fluoride (to mimic ADP-Pi).16
Fig. 1.

Arp3 and actin are homologous proteins that bind adenosine nucleotides. (a) Cα trace of overlaid Arp3 (cyan) and actin (red) showing ATP (yellow) bound to the cleft between the four subdomains (labeled 1–4). The DNase binding loop (DBL) and selected elements of secondary structure are indicated. (b) Cα trace of Arp3 indicating terminology of essential structural features. Red Cα trace shows regions that were disordered in crystal structures and modeled for the simulations. We defined five distances (A–D) to monitor changes in the structures. Distance A (black) defines the width of the subdomain 3/4 notch, a pocket between subdomains 3 and 4 that binds the adenine ring and ribose. Distances B1 (red) and B2 (green) define the width of the phosphate clamp [see (c)]. The phosphate clamp is composed of the P1 and P2 loops, β-strands with hairpin turns that line each side of the nucleotide cleft and directly contact the phosphates. Distance C (blue) measures the width across the cleft mouth, the region between subdomains 2 and 4 near the top of the nucleotide binding cleft. Distance D (orange) defines the width of the barbed-end groove, the region between subdomains 1 and 3 that is opposite to the cleft mouth. The Cα atoms from actin or Arp3 used to define the distances are as follows: A, Glu214/229–Met305/327; B1, Ser14/Thr14–Gly158/173; B2, Gly15/15–Asp157/172; C, Gly59/67–Glu207/222; and D, Leu163/Thr148–Phe352/Phe390. (c) Detail of ATP binding cleft of Arp3 (PDB accession code 1TYQ) showing hydrogen bonds between the P loops and the phosphates. The residues that contact the nucleotide are identical or similar in actin and Arp3, and the mode of interaction is conserved. (d) Cleft distances in actin and Arp3 from crystal structures. B2 is used to classify each structure as open (B2>7.0 Å), intermediate (6.0 Å<B2>7.0 Å), or closed (B2<6.0 Å).
The actin-related protein (Arp) 2/3 complex is an assembly of seven proteins that nucleates branched actin filaments in response to extracellular signals.17,18 The two subunits in the complex, Arp2 and Arp3, are closely related to actin (45% and 36% sequence identity to actin, respectively), have the same fold, and bind ATP. A crystal structure of the nucleotide-free crystals of Arp2/3 showed Arp3 in an open conformation and Arp2 with subdomains 1 and 2 so flexible that no electron density was present in the maps.19 Soaking these crystals in ATP caused the nucleotide cleft of Arp3 to close, while soaking them in ADP did not, giving weight to the possibility of an open conformation of ADP–actin.20 ATP bound subdomains 3 and 4 of Arp2, but the disordered subdomains 1 and 2 did not close around the nucleotide unless the crystal was chemically cross-linked with glutaraldehyde.21
Simulations of actin dynamics have run the gamut from density functional calculations of the active site to coarse-grained filamentous actin networks.22–29 All-atom simulations on nanosecond timescales have been helpful in complementing structural experiments. Most relevant to this study are molecular dynamics (MD) simulations of monomeric actin up to 50 ns in explicit water, where the nucleotide binding cleft of actin stayed closed when bound to both ATP and ADP.29 A simulation of actin–profilin showed that the open nucleotide binding cleft as observed in crystal structures14 was unstable when profilin was replaced with water.25
Here, we used MD simulations to study conformations of actin and Arp3 in a variety of nucleotide-bound states on the nanosecond timescale. These simulations have given us insights into several aspects of the effect of nucleotides on actin family proteins, including differences in the overall flexibility of actin and Arp3, responses of residues lining the nucleotide cleft, relay of structural changes outward from the cleft, pathways for partial release of the nucleotide, and the effect of the divalent cation on protein dynamics.
Results
Overview of simulations
We simulated the dynamics of actin and Arp3 for 8 and 5 ns, respectively, at 37 °C and 1 atm with bound ATP or ADP or without nucleotide (apo). In some cases, the nucleotide was changed at the onset of the simulation, for example, swapping ADP into an ATP crystal structure prior to solvation (ATP→ADP). This section and Table 1 provide an overview of the results for 12 simulations. The following sections use all of the simulations to draw conclusions about the dynamics of actin and Arp3.
Table 1.
Simulation results
| Simulationa | Actin |
Arp3 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATP→ATP | ADP→ADP | ATP→ADP | ATP→APO | ATP→ATP | ADP→ADP | APO→APO | ATP→APO | APO→ATP | Ca2+ | Chimera | Cold | |
| Starting PDB code | 1NWK | 1J6Z | 1NWK | 1NWK | 1TYQ | 1U2V | 1K8K | 1TYQ | 1K8K | 1TYQ | 1TYQb | 1TYQ |
| Simulation time (ns) | 8 | 8 | 8 | 8 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Simulation temperature (K) | 310 | 310 | 310 | 310 | 310 | 310 | 310 | 310 | 310 | 310 | 310 | 277 |
| Phosphate clamp (B1,B2) | ||||||||||||
| Start (crystal) | Closed | Closed | Closed | Closed | Intermediate | Open | Open | Intermediate | Open | Intermediate | Intermediate | Intermediate |
| Simulation | Stable | Stable | Stable | Distorts | Opens | Opens | Opens | Opens | Opens | Opens | Opensc | Closes |
| Cleft mouth (C) | ||||||||||||
| Start | Closed | Closed | Closed | Closed | Closed | Open | Open | Closed | Open | Closed | Closed | Closed |
| Simulation | Closes | Stable | Closes | Stable | Opens | Opens | Opens | Opens | Opens | Opens | Stable | Stable |
| Val159/174 | ||||||||||||
| Start | Down | Down | Down | Down | Up | Up | Up | Up | Up | Up | Up | Up |
| Simulation | Up | Down | Down | Down | Up | Down | Up | Up | Upd | Down | Up | Up |
| Sensor loop | ||||||||||||
| Start | Up | Down | Up | Up | Up | Up | Up | Up | Up | Up | Up | Up |
| Simulation | Up | Down | Up | –e | Up | Up | Fluctuated | Fluctuated | Up | Up | Up | Up |
| Ser14/Thr14 | ||||||||||||
| Start | Out | In | Out | Out | Out | Out | Out | Out | Out | Out | Out | Out |
| Simulation | Out | In | In | –e | Out | Out | Fluctuated | Fluctuated | Out | Out | Out | Out |
| DNase loop | ||||||||||||
| Start | Random coil | α-helix | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil |
| Simulation | Random coil | π-helix/random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil | Random coil |
| Average Cα RMS fluctuation (Å) | 0.98 | 1.09 | 0.99 | 1.12 | 1.34 | 1.40 | 1.42 | 1.48 | 1.39 | 1.37 | 1.21 | 1.01 |
| Average nucleotide RMS fluctuation (Å) | 0.40 | 0.52 | 0.49 | – | 0.71 | 0.84 | – | – | 0.70 | 0.52 | 0.50 | 0.29 |
Simulation nomenclature indicates nucleotide state in starting crystal structure and during simulation (e.g., ATP→ADP indicates that the nucleotide was removed from the ATP-containing crystal structure and replaced with ADP at the onset of the simulation). For simulations in which the nucleotide state in the simulation was not changed from the starting crystal structure, a shortened nomenclature is used in the text (e.g., the ATP→ATP actin simulation simply becomes ATP–actin).
Arp3–actin chimera is residues 370–375 of actin appended to the C-terminus of the 1–407 fragment of Arp3 from 1TYQ.
The phosphate clamp opened during the last 0.5 ns in this simulation.
The down position was occupied briefly for two brief periods during the last 1 ns of this simulation.
Distortion of the phosphate clamp and sensor loop occurs in this simulation; therefore, this parameter was not compared with other simulations.
The overall conformations of both ATP–actin and ADP–actin changed little during 8 ns of simulation time (Supplemental Figs. SF1A and SF1B). The P1 and P2 loops of actin remained tightly clamped around both nucleotides, but the conformation of ADP itself changed within the closed nucleotide cleft (details below). In the ATP–actin simulation, the initially unstructured DNase binding loop (His40–Asp51) remained unstructured and was the most mobile part of the protein. The DNase binding loop of the ADP–actin structure started as an eight-residue α-helix and relaxed to a more loosely coiled five-residue π-helix. When we replaced ATP with ADP at t=0, followed by solvation, helix αG (Glu224–Ser235) underwent large displacements but the DNase binding loop did not fold into an α-helix during 8 ns of simulation time. After replacing ATP with water (ATP→APO) at t = 0, the P1 loop collapsed into the area previously occupied by ATP but the cleft did not open, as previously predicted.11 Despite this distortion of the phosphate clamp, the overall dynamics of apo–actin were similar to the dynamics of ATP–actin and ADP–actin (not shown).
The backbone of Arp3 was more flexible than that of actin in our simulations. In the ATP–Arp3 and ADP–Arp3 simulations, the nucleotide binding cleft opened and underwent large oscillating motions, allowing the nucleotide to jiggle in the cleft and be partially released. The most dynamic parts of Arp3 were the DNase binding loop, the β12/β13 loop, and the αK/β15 insert (Fig. 1a). These features also had the highest B-factors or were disordered in crystal structures.19,21,30 When ATP was added to nucleotide-free Arp3, the cleft did not close, as observed in Arp2/3 complex crystal structures, but opened in a fashion similar to the other Arp3 simulations.
Actin favors a closed cleft, whereas Arp3 favors an open cleft
The nucleotide binding cleft of actin remained closed throughout the simulations, whereas the cleft of Arp3 opened consistently. In both proteins, the bound nucleotide had subtle effects on overall conformations on the timescales studied here. For all actin simulations, B2, the width of the phosphate clamp (Fig. 1b and c), stayed close to the starting distance of about 5 Å, with a standard deviation of less than 0.25 Å (Fig. 2a). In all but two of the Arp3 simulations, the clamp opened to a relatively stable distance of about 9 Å (~11 Å for the ADP–Arp3 simulation, see below), 1 Å greater than that observed in any Arp2/3 complex crystal structure (Table 1). The Arp3 phosphate clamp also fluctuated more than the actin phosphate clamp (Fig. 2a). The cleft mouths of actin and Arp3 followed the same trends as the clamp (Fig. 2b); while the width of the cleft mouths of both actin and Arp3 fluctuated more than the phosphate clamp, the mouth stayed closed in all actin simulations but opened in Arp3 simulations. The exceptions were a simulation of ATP–Arp3 at 4 °C, where adding ATP to the cleft in an intermediate position caused it to close stably around the nucleotide for the entire 5-ns trajectory (Fig. 5c), and a simulation of an Arp3–actin chimera at 37 °C, in which the phosphate clamps were closed or intermediate throughout most of the simulation and the cleft mouth stayed closed, as discussed below.
Fig. 2.
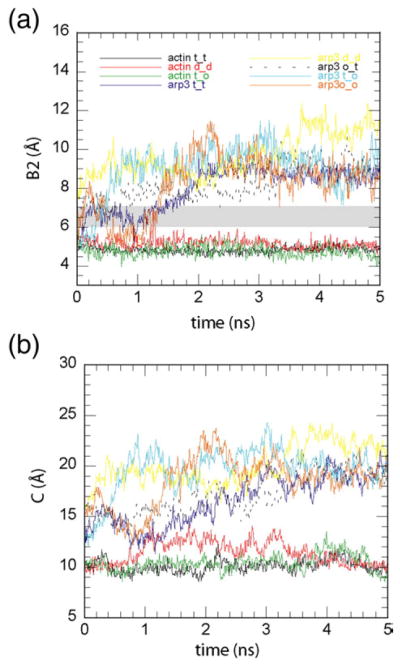
All nucleotide-bound states favor actin with a closed cleft and Arp3 with an open cleft. (a) Plot showing distance B2 as a function of simulation time. Shaded region marks the intermediate width of the clamp. Legend indicates which simulation corresponds to each trace, with t=ATP, d=ADP, o=APO, and an underscore replacing the arrow used in the simulation nomenclature from Table 1. The clamp remains closed (or intermediate) during the entire 8 ns (up to 5 ns shown here) for all actin simulations. The clamp opens in all Arp3 simulations, regardless of the nucleotide binding state. (b) Plot showing distance C as a function of simulation time. The cleft mouth stays closed for actin but opens in all Arp3 simulations. Color coding is the same as for (a).
Fig. 5.

The conserved C-terminal extension of Arp3 contributes to the tendency of the nucleotide binding cleft to open. (a) Surface representation of the starting structure for the ATP–Arp3 simulation with ATP (yellow) and the C-termini of actin (magenta) and Arp3 (cyan) in stick format. Rotation about the x-axis by 90° (right panel) shows the interaction of the C-terminal extension of Arp3 with the barbed-end groove. (b) Close-up showing interactions of the C-terminal extension of Arp3 with the barbed-end groove. Distance D is indicated by an orange dashed line. (c) Plot showing the width of the phosphate clamp (B2) and the cleft mouth (C) as a function of simulation time for the ATP–Arp3 simulation (red), the Arp3–chimera simulation (blue), and the 4 °C ATP–Arp3 simulation (black=Arp3–cold). Both the phosphate clamp and the cleft mouth showed a lower tendency to open in the Arp3–chimera simulation. The cleft stayed tightly closed when the simulation temperature was decreased to 4 °C (ATP–cold) even when the C-terminus of Arp3 was present. (d) Plot showing the width of the cleft mouth (C) and the barbed-end groove (D) as a function of simulation time for the ATP–Arp3 simulation (red) and the Arp3–chimera simulation (blue). In the ATP–Arp3 simulation, the barbed-end groove stayed near 14 Å for the majority of the simulation and the nucleotide cleft mouth opened dramatically to about 20 Å. In the ATP–chimera simulation, the barbed-end groove opened, stabilizing near 17 Å for 3 ns, and the cleft mouth remained closed. (e) Plot of barbed-end groove versus cleft mouth width for crystal structures of actin and Arp3.
ADP is less stable than ATP in the actin nucleotide binding cleft
The ATP and the phosphate clamp were remarkably stable during 8 ns of simulation of ATP–actin. The starting and final conformations of ATP itself and those of the phosphate clamp were nearly identical (Fig. 3a). Of five hydrogen bonds between the γ-phosphate (nonbridging oxygen atoms) and phosphate clamp, three were maintained throughout more than 90% of the simulation. The hydrogen bond between the γ-phosphate and the backbone amide of Val159 was lost after about 2 ns (Fig. 3b) and may have contributed to the upward flip of Val159 (Fig. 3a). We consider the significance of this conformational change below. The hydrogen bond between the backbone amide of Asp157 and the γ-phosphate was maintained through 67% of the simulation trajectory.
Fig. 3.

ATP is more stable than ADP in the actin nucleotide cleft. (a) Stereo figure showing the nucleotide binding cleft of the final frame of the 8-ns ATP–actin simulation (cyan) overlaid onto the starting structure (PDB accession code 1NWK, yellow). ATP and calcium are shown in purple (t=0) or magenta (t=8 ns). Hydrogen bonds to ATP in the starting structure are shown as dotted lines. Black dotted lines indicate hydrogen bonds that are also present in the final frame (but not shown here), whereas the red dotted line to Val159 indicates the hydrogen bond not present in the final frame. (b) Time course of hydrogen-bonding distances between atoms in the P1 (Gly14, green line) as well as P2 (Gly158, blue line, and Val159, red line) loops and atoms of the γ-phosphate of ATP. Both loops are hydrogen bonded to the γ-phosphate throughout most of the simulation (blue and green lines). The hydrogen bond between the backbone amide of Gly158 and the γ-phosphate (blue line) is lost for brief periods during the simulation. The hydrogen bond between Val159 and the γ-phosphate (red) is lost after about 2 ns. (c) Stereo figure showing the nucleotide binding cleft of the final frame of the 8-ns ADP–actin simulation (cyan) overlaid onto the starting structure (PDB accession code 1J6Z, yellow). ADP and calcium are shown in purple (starting conformation) or magenta (final conformation). Hydrogen bonds to ADP in the starting structure are shown as dotted lines. (d) Plot of the nucleotide torsion angle O4′–C4′–C5′–O5′ as a function of simulation time for the ATP–actin (green triangles), ADP–actin (blue), and (ATP→ADP)–actin swap (red) simulations. Torsions over 180° are plotted as negative values. (e) RMS fluctuation plotted as a function of arbitrary atom number in the nucleotide for the ATP–actin (green) and ADP–actin simulations.
Removal of the γ-phosphate destabilized the nucleotide. In the ADP–actin simulation, ADP started in the “standard” conformation (i.e., the same conformation as ATP in the ATP–actin simulation but with the absence of the γ-phosphate) (Fig. 3c). However, the O4′–C4′–C5′–O5′ dihedral angle, which measures the position of the arm connecting the phosphate chain to the ribose, underwent multiple torsional flips (Fig. 3d) before stabilizing in a new position with a torsion angle near 180°. We refer to this position as the “bent” conformation, because it was observed in the crystal structure of ADP–Arp2/3 complex in which ADP adopts a unique bent conformation in an open nucleotide cleft.20 Shortly after the ribose–phosphate adopted the bent conformation, the calcium ion moved approximately 2 Å toward the P2 loop side of the cleft. In this position, the calcium contacted Asp154 directly and the phosphates pointed directly down into the cleft. This conformation was maintained through the last 4 ns of the simulation. The rearrangements of ADP resulted in higher RMS fluctuations of the atoms in the nucleotide (Fig. 3e). These rearrangements were not expected based on the similarity of ATP and ADP in actin crystal structures. The movement of the calcium was also unexpected, as discussed below. We observed similar fluctuations of the O4′–C4′–C5′–O5′ dihedral angle and the calcium position in the simulation where we swapped ADP into ATP–actin at t=0, providing further evidence that ADP is less stable in the actin cleft than ATP.
Partial release of ATP and ADP in Arp3 simulations suggests multiple pathways for nucleotide dissociation and association
Opening of the nucleotide cleft in Arp3 simulations resulted in partial release of the nucleotide. Three major sets of contacts must be lost for nucleotide to dissociate from actin or Arp3. The notch between subdomains 3 and 4 provides a hydrophobic binding site for the adenine ring and a complementary surface with two hydrogen-bond acceptors for the ribose ring. The P1 and P2 loops provide the second set of contacts, forming the phosphate clamp. The divalent cation is the third anchor for the nucleotide; it coordinates the phosphates and makes direct or water-mediated contacts to residues lining the cleft.
The protein–nucleotide contacts dissociated in a different order in three simulations of Arp3 with bound nucleotides. However, in all cases, the phosphates lost contact with the P2 loop before losing contact with the P1 loop and tracked with the P1 side of the clamp as it opened. These are consistent with the transient breaking of the hydrogen bonds between the γ-phosphate and the P2 loop in the ATP–actin simulation (Fig. 3b).
In the ATP–Arp3 simulation (Fig. 4a), the phosphates lost contact with the P2 loop at 100 ps but remained hydrogen bonded to P1 on the other side of the clamp. After a further 1.7 ns, the adenine ring and ribose slipped out of the subdomain 3/4 notch. The calcium and the P1 loop retained the nucleotide in the cleft for the rest of the simulation.
Fig. 4.

Multiple pathways for partial release of nucleotide from Arp3. Stereo figures showing Cα traces of the overlaid starting structures (cyan) and either 4.0-ns (b) or 4.5-ns (a and c) snapshots from Arp3 simulations (blue). Nucleotides and calcium ions are shown in yellow (start) and orange (simulation snapshot), and water molecules are shown as blue pluses. (a) ATP–Arp3 simulation, starting with 1TYQ. The adenine ring and ribose are released from the subdomain 3/4 notch, and water molecules fill the region previously occupied by the nucleotide. (b) ADP–Arp3 simulation, starting with 1U2V. The phosphates of ADP rearrange to form a new hydrogen-bonding network (dotted lines), and the adenine ring and ribose dissociate from the subdomain 3/4 notch. (c) APO→ATP Arp3 simulation, starting with 1K8K. The adenine and ribose rings stay bound to the subdomain 3/4 notch. The phosphates lose contact to both the P1 and P2 loops but remain anchored in the cleft by the calcium, which contacts Asp169 directly. Water molecules form a layer between the phosphates and the clamp as the clamp opens.
In the ADP–Arp3 simulation (Fig. 4b), ADP started in the bent conformation and did not contact the calcium. The adenine and ribose rings immediately slipped out of the subdomain 3/4 notch. The loss of contacts to the protein allowed the adenine end of the nucleotide to fluctuate dramatically throughout the remainder of the simulation, while the phosphates remained relatively stable. The average RMS fluctuation for atoms in the adenine ring was 0.82 Å in the simulation of ADP–Arp3, whereas it was only 0.36 Å in the ADP–actin simulation. The phosphates lost contact with the P2 loop but stayed hydrogen bonded to the P1 loop until the last 0.5 ns. For the last half nanosecond, the nucleotide was anchored to the cleft by calcium and through hydrogen bonds with the guanidinium group of Arg374 and van der Waals interactions with the aliphatic portion of its side chain.
In the simulation where ATP was added to nucleotide-free Arp3 before equilibration, the adenine ring and ribose stayed bound to the subdomain 3/4 notch (Fig. 4c). The phosphates lost contact with the P2 loop and tracked with the P1 loop as the clamp opened. At 3.3 ns, the contacts with the P1 loop were also lost but ATP remained anchored by the calcium ion, which was centered in the cleft and made only water-mediated contacts with the protein.
A conserved C-terminal extension of Arp3 contributes to the tendency of the nucleotide binding cleft to open
We compared structures of actin and Arp3 for features that might explain the greater flexibility of the cleft of Arp3. The interactions with the bound nucleotides and hinge points for the rigid body motion that opens the cleft are largely identical. However, the C-terminus of Arp3 is five residues longer than that of actin. This extension of conserved residues forms a random coil that interacts with the hydrophobic barbed-end groove between subdomains 1 and 3 (Fig. 5a and b). Starting with the ATP-bound Arp3 crystal structure [Protein Data Bank (PDB) accession code 1TYQ], we created an Arp3–actin chimera by deleting five residues from the C-terminus of Arp3 and changing six residues at the newly created C-terminus to the corresponding residues in actin.
During a 5-ns simulation of this Arp3–actin chimera bound to ATP, both the phosphate clamp and the nucleotide cleft mouth showed a lower tendency to open than ATP–Arp3 (Fig. 5c and d). The phosphate clamp of the chimera stayed at a closed or an intermediate width until 4.5 ns, and the nucleotide cleft mouth stayed closed for the entire trajectory (Fig. 5c). In contrast, the barbed-end groove opened to maintain a stable distance near 17 Å for 3 ns and was an average of 16.6 Å wide over the entire simulation (Fig. 5d). This is near the width expected based on crystal structures of actin and Arp3 with closed nucleotide clefts (Fig. 5e). In the ATP–Arp3 simulation, the width of the barbed-end groove decreased to maintain an average distance of 14.3 Å and the nucleotide cleft mouth opened dramatically to about 20 Å. In both simulations, the width of the nucleotide cleft mouth was inversely correlated with the width of the barbed-end groove.
Movements of the nucleotide cleft mouth and phosphate clamp are correlated
The P1 and P2 loops line either side of the nucleotide cleft at the interface of the large and small domains. This geometrical arrangement may allow changes in the width of the phosphate clamp to be relayed to the nucleotide cleft mouth and vice versa. Crystal structures of actin and Arp3 suggest that this is the case, since increases in the width of the phosphate clamp are accompanied by increases in the width of the cleft mouth (Fig. 1d).
Our simulations show that the clamp and cleft mouth are correlated in Arp3, since the metrics B1 and B2 open in tandem with distance C (Fig. 2a and b). In the actin simulations, the correlation is not clear because of the modest changes in these distances. Computing the dot product between two vectors (with the starting and ending positions of each Cα constituting one vector), a technique called covariation analysis, provides another method to determine which residues move in tandem.31 By this method, Arp3 has a higher degree of correlated movement than does actin due to the greater rigid body motions that occur in the Arp3 trajectory. The P1 and P2 loops of both proteins are correlated with multiple atoms in subdomains 2 and 4, respectively (Fig. 6a and b). Thus, although the correlation between the phosphate clamp and the nucleotide cleft mouth in Arp3 is obvious, it seems likely to exist in actin as well.
Fig. 6.

Arp3 shows more correlated residue movements than actin. (a and b) Covariation analyses of (a) ATP–actin and (b) ATP–Arp3. Ribbon diagrams of actin and Arp3 show the starting conformation and lines connecting Cα atoms with a correlation coefficient of 0.70 or greater. (c and d) Porcupine plots of the contribution of the first eigenvector in the principal component analysis to the dynamics of (c) ATP–actin and (d) ATP–Arp3. Orientations are the same as in (a) and (b). Cα atoms are shown as spheres and represent the average position in the trajectory. Covariation and principal component analyses were carried out with the Dynatraj server.32
To understand the nature of correlated motions in actin and Arp3, we carried out principal component analysis on both proteins.33 “Porcupine” plots of the first eigenvector for the Arp3 simulations show that the main dynamic mode of Arp3 is rotation of each domain about axes pointing out of the page and centered either in subdomain 2 or in subdomain 4 (Fig. 6d). The nucleotide cleft motion is amplified moving from the center of the cleft to the cleft mouth, which is reflected in the large increases in distance C in the Arp3 simulations. The principal dynamic modes of actin appear less concerted due to the absence of large-scale rigid body motions in the actin simulations (Fig. 6c). In the ATP–actin and the ATP→ADP actin simulations, for instance, modest fluctuations of the phosphate clamps seem to be unconnected to the rigid body motions that close the cleft mouth by an additional 2 or 3 Å relative to the starting conformation (Fig. 2b).
The α-helix in the DNase binding loop is unstable in all nucleotide binding states tested
In the crystal structures of rhodamine-labeled actin, the DNase binding loop formed an α-helix when ADP was bound but was disordered when ATP was bound.10,11 These led to the hypothesis that the biochemical differences between ADP–actin and ATP–actin are at least partially due to folding of the DNase binding loop into an α-helix after the dissociation of the γ-phosphate. In our ADP–actin simulation, the DNase binding loop started with eight residues in an α-helical conformation (Fig. 7a). The helix partially unfolded at 1.7 ns, losing one turn (Fig. 7a). At 2.5 ns, the loop unfolded further to form a π-helix, in which the backbone torsion angles were helical, but the backbone hydrogen-bonding arrangement switched from i+4 to i+5 (Fig. 7b). No secondary structure formed in the DNase binding loop in either the ATP–actin or the ATP→ADP swap simulations over 8 ns (Fig. 7a).
Fig. 7.
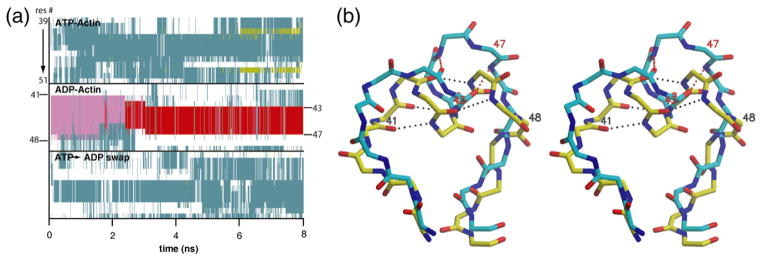
The α-helix in the DNase binding loop of ADP–actin is unstable. (a) Secondary structure of residues Arg39–Asp51 for ATP–actin (top), ADP–actin (middle), and the ATP→ADP actin swap (bottom) as a function of simulation time: α-helix in magenta, π-helix in red, random coil in white, turn in cyan, extended conformation in yellow. (b) Stereo figure of the overlaid DNase binding loops from the starting (yellow) and 5-ns (cyan) snapshots from the ADP–actin simulation showing hydrogen bonds that define the secondary structure. The hydrogen bonds at the end of each helix exhibit nonideal geometry.
The conformation of Ser14/Thr14 is sensitive to the nucleotide binding state but is not coupled to the conformation of the sensor loop on nanosecond timescales
Comparison of crystal structures of ATP–actin and ADP–actin shows that the absence of the γ-phosphate allows the P1 loop to move slightly inward and the hydroxyl of Ser14/Thr14 to rotate into the nucleotide cleft.10–12 These changes are accompanied by flipping of the backbone carbonyl of Glu72/Arg79 in the adjacent “sensor loop.” The sensor loop flip was proposed to promote folding of the DNase binding loop into an α-helix.10 Our MD simulations show that the conformation of Ser14/Thr14 is linked to the nucleotide state but that rotation of Ser14/Thr14 is not correlated with the sensor loop flip on nanosecond timescales.
In simulations of ATP–actin, Ser14 was stable in the “out” conformation and the sensor loop stayed in the “up” conformation (Fig. 8a), but upon replacement of ATP with ADP, Ser14 immediately rotated into the “down” conformation in the nucleotide cleft to occupy the position previously occupied by the γ-phosphate. However, the sensor loop did not respond by flipping into the down position.
Fig. 8.

Conformational changes of residues in and surrounding the nucleotide binding cleft. (a) Monitoring the position of Ser14/Thr14 and the sensor loop in actin and Arp3. χ1 of Ser14/Thr14 (solid lines) is plotted as a function of simulation time to monitor whether this residue is flipped into (“in”) or away from (“out”) the γ-phosphate binding site. The ψ angle of Glu72/Arg79 (dotted lines) is plotted as a function of simulation time to monitor whether its carboxyl group is flipped up (“sensor up”) or down (“sensor down”). Legend abbreviations are the same as those in Fig. 2. (b) Monitoring the position of the valine in the P1 loop. Plot of the ψ angle of Gly158/173 in the ATP–actin (red) and ADP–Arp3 (blue) simulations as a function of time. The actin simulation is truncated at 5 ns. The table on the right indicates the range of angles observed in actin and Arp3 crystal structures and the corresponding position of Val159/174 in these structures.
In simulations of Arp3 with bound ATP or ADP, both Thr14 and the sensor loop were stable in their starting conformations. However, in simulations without a bound nucleotide, both Thr14 and the sensor loop changed conformations, but these events were not tightly coupled in time (Fig. 8a). For example, in the apo–Arp3 simulation, the sensor loop flipped down permanently after 1.5 ns during a temporary 3-ns inward flip of Thr14. In the simulation where ATP was removed from Arp3, the sensor loop flipped down at 0.8 ns and Thr14 flipped inward 3.7 ns later.
These observations show that in the absence of nucleotide, Thr14 and the sensor loop transiently adopt both of their alternative conformations observed in crystal structures on the nanosecond timescale but their conformations are not tightly coupled. It is unknown whether the conformations of Ser14/Thr14 and the sensor loop are coupled on longer timescales. The greater overall flexibility and the wider nucleotide cleft of Arp3—especially in the absence of nucleotide—may be more permissive of these changes than actin, allowing us to observe them in 5-ns simulations.
The position of Val159/174 is not correlated with the nucleotide binding state on nanosecond timescales
The conformation of Val159/174 in the P2 loop is proposed to be sensitive to the presence of the γ-phosphate.16 This valine is found in either an “up” or a “down” conformation in Arp3 and actin crystal structures.13,21 With the side chain in the down position, the backbone amide of this residue can hydrogen bond to the γ-phosphate, but in the up position, it cannot. The up position of the valine may open up a path for phosphate to dissociate from the ADP-Pi intermediate. We observed both conformations of the valine in our simulations, but they were not correlated with the nucleotide state.
We used the Ψ angle of Gly173 to monitor the position of Val159/174 during the simulations (Fig. 8b). These valines changed very little relative to their starting positions in 9 of the 12 total simulations (Table 1). However, in the ATP–actin simulation, Val159 started in the down position and flipped up after 3 ns (Figs. 3a and 7b), about 1 ns after the γ-phosphate lost contact with the Val159 backbone amide. In ADP–Arp3, Val174 started in the up position but flipped down during the simulation. In the simulation where we added ATP to nucleotide-free Arp3, the valine occupied the down position twice briefly around 4.5 ns.
Calcium occupies multiple positions common to the nucleotide clefts of Arp3 and actin that influence nucleotide dynamics
In crystal structures of actin, a bound calcium exhibits heptagonal coordination to the β- and γphosphates and five water molecules, rather than contacting the protein directly. In our preliminary simulations of actin and Arp3, the calcium immediately moved to hydrogen bond to a conserved aspartate (Asp154/169) deep in the nucleotide binding cleft after constraints were released on the nucleotide and calcium. As described in Simulation Methods, we lowered the charge on the calcium in our production runs to reduce the electrostatic attraction to residues in the nucleotide cleft and more closely reflect the behavior in the crystal structures.
In our simulations, calcium occupied three stable positions in actin and Arp3 (Fig. 9a). In all but one of the crystal structures of actin or the Arp2/3 complex, calcium occupies position A, where it coordinates a shell of water molecules hydrogen bonded to three conserved residues that line the nucleotide cleft: Asp154/169, Gln137/144, and Asp11/11. Only in the simulations presented here was calcium found in position B, directly contacting the backbone carbonyl of Ser155/170, the side chain carboxylate of Asp154/169, or both. Contact with Ser155/170 requires a torsional flip of the peptide backbone that has not been observed in crystal structures. In the open calcium–ADP Arp2/3 complex structure (PDB accession code 1U2V), calcium occupied position C, contacting Asp11 directly but not within hydrogen-bonding distance of the phosphates.20
Fig. 9.
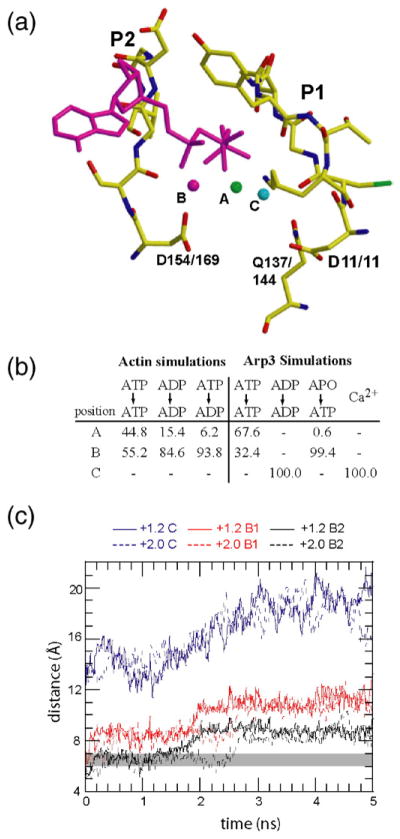
Calcium occupies multiple positions in the actin and Arp3 simulations. (a) Stick representation showing the phosphate clamp, ATP, and the three calcium binding sites observed in the crystal structures and simulations. Shown here is the phosphate clamp from the ATP–Arp3 simulation at 0.5 ns. In this simulation, the calcium (magenta, labeled B) occupies a secondary position in which it directly contacts the side chain of Asp169, the carbonyl of Ser170, or both. The primary site (green, labeled A), occupied in nearly all actin and Arp3 crystal structures, is centered in the cleft and makes only water-mediated contacts to the protein. In the tertiary site (cyan, labeled C), the calcium binds directly to Asp11. (b) Percentage of the time calcium occupies each of the binding sites during the simulations. (c) Plot of nucleotide cleft distances B1, B2, and C as a function of simulation time for (APO→ATP)–Arp3 simulations in which the calcium is given a +2.0 or +1.2 charge.
The position of the calcium influenced the position of the nucleotide in our simulations. In the actin simulations, the bound calcium vacillated between positions A and B, populating the sites about equally with ATP bound but favoring the B site with ADP bound (Fig. 9b). Movement of calcium into the B position drew the phosphates deeper into the nucleotide cleft, resulting in the temporary loss of contact between the γ-phosphate and the P2 loop in the ATP–actin simulation and the major rearrangement of ADP in the ADP–actin simulation (Fig. 3c). In the Arp3 simulations, the position of the calcium influenced the pathway of partial nucleotide release. For instance, in the ATP–Arp3 simulation, calcium first occupied the B position but later moved into the A position to maintain contacts to the phosphates as the clamp opened and the phosphates tracked with the P1 loop (Fig. 4a). In the ADP–Arp3 simulation, calcium occupied only the C position, where it contacted Asp11 from the P1 loop (Fig. 4b). In this position, it could not link the phosphates to the P2 side of the nucleotide cleft. This may contribute to the greater opening of the phosphate clamp and cleft mouth in the ADP–Arp3 simulation as compared with the other Arp3 simulations. To determine whether our standard reduced charge on the calcium favored opening of the nucleotide cleft in Arp3, we ran a 5-ns simulation of ATP–Arp3 with the calcium charge set at +2.0. In this simulation, both the phosphate clamp and the nucleotide cleft mouth opened with a trajectory very similar to the simulation in which the charge was +1.2 (Fig. 9c).
Discussion
ATP–actin and ADP–actin both favor a closed nucleotide cleft at 37 °C
One proposed structural difference to account for the distinct biochemical properties of ADP–actin and ATP–actin is opening of the nucleotide cleft upon dissociation of the γ-phosphate. Reconstructions of electron micrographs of budding yeast actin filaments show the nucleotide cleft open in ADP filaments and closed in ADP filaments in beryllium fluoride (to mimic ADP-Pi).16 Further evidence for an open ADP–actin state comes from crystal structures of the Arp2/3 complex, in which ADP–Arp3 adopts an open conformation.20 However, an ATP-bound profilin–actin complex is the only crystal structure of actin with an open cleft.14 All ADP-bound actin crystal structures are closed, suggesting that the closed state of ADP–actin monomers is the most energetically stable. In the simulations presented here, we show that both ATP–actin monomers and ADP–actin monomers remain closed around the nucleotide for the entirety of the 8-ns simulations in a constant NPT ensemble at energies of 37 °C and 1 atm. The actin cleft also stayed closed around ADP for at least 50 ns at slightly lower temperature in another MD study.29
In contrast, both ATP–Arp3 and ADP–Arp3 opened dramatically in 5-ns simulations, showing that motions necessary to open the Arp3 nucleotide cleft are accessible on the timescales we investigated. We note that the domain motions that open the cleft are very similar to those inferred from comparisons between closed crystal structures of actin and the open profilin–actin structure.15 The Arp3 example suggests that these MD simulations should have revealed open conformations of monomeric ADP–actin if they exist. The Arp3–actin chimera simulation showed how allosteric interactions can influence cleft opening. Further MD simulations and biophysical experiments are necessary to determine if similar allosteric interactions between adjacent subunits may affect the nucleotide cleft conformation in actin filaments.
Structural basis for differences between actin and Arp3
Vertebrate actins and Arp3 are 36% identical and have the same overall fold. The nucleotide binding clefts in the two proteins are identical, and residues that contact ATP in actin and Arp3 are conserved. Consequently, it is surprising that the proteins differ dramatically in at least three ways. The affinity of ATP for Arp3 (Kd =1.3 μM)34 is 1000-fold lower than that for actin (Kd =1.2 nM).35 Once incorporated into a filament, actin hydrolyzes ATP, whereas significant ATP hydrolysis by Arp3 has not been detected.36–38 Finally, in simulations here and elsewhere,24,29 actin remains tightly closed around its bound ATP or ADP, while here the nucleotide cleft of Arp3 opened with or without bound nucleotide.
Our simulations revealed a plausible mechanism to explain these differences: the C-terminal extension of Arp3 acts like an intrinsic profilin that binds the barbed-end groove and promotes the open conformation of the nucleotide cleft. Profilin, a small protein that binds to the barbed-end groove of actin,15 can stabilize an open conformation14 and binds with higher affinity to nucleotide-free actin than ATP–actin or ADP–actin.8 MD simulations have shown that removal of profilin from the open actin–profilin complex results in the closure of the nucleotide binding cleft of actin on nanosecond timescales.25
Three lines of evidence support the hypothesis that the C-terminus of Arp3 acts like an intrinsic profilin. First, among the nine currently available crystal structures of the Arp2/3 complex, the 5 C-terminal residues of Arp3 that extend beyond the C-terminus of actin show a greater tendency to be ordered when the nucleotide cleft of Arp3 is open than when it is closed.19–21 In the three Arp3 structures with open nucleotide binding clefts, an average of 3.4 of these 5 residues is ordered, while in the three structures with closed clefts, an average of 0.3 of these 5 residues is ordered. This difference suggests that stable interactions of the C-terminal extension with the groove between subdomains 1 and 3 facilitate closure of the barbed-end groove and promote the rigid body motion that opens the nucleotide binding cleft. Second, Arp3 lacking the C-terminal extension showed a dramatic reduction in its tendency to open (Fig. 5c). Finally, both the residues in the C-terminus and those in the barbed-end groove that contact it are well conserved in Arp3 from diverse species.
The observed opening of the cleft of ATP–Arp3 during the simulations was unexpected based on crystal structures of the Arp2/3 complex, where the nucleotide binding cleft of Arp3 is open in all of the nucleotide-free crystal structures but closed or intermediate in all three ATP-bound structures. The temperature offers one explanation for this difference, since the cleft of ATP–Arp3 did not open during a simulation at 4 °C, the temperature for growth of Arp2/3 complex crystals.
Nucleotide-free actin does not open but the phosphate clamp collapses within 8 ns
Actin in solution is unstable without a nucleotide,4 and therefore a crystal structure does not exist. The apo–Arp3, in contrast, is stable and adopts an open conformation in crystals of nucleotide-free Arp2/3 complex.19,21 These observations, coupled with the fact that the cleft must open to bind or release nucleotide without significant steric clash, led to the proposal that apo–actin opens in the absence of nucleotide.11 This open state is expected to be on the path to unfolding and destabilization of the protein. However, after removing ATP from actin, the cleft stayed closed in our simulation. Shortly (0.2 ns) after removing ATP, the P1 loop collapsed inward into the space previously occupied by the nucleotide. The carbonyl of Ser14 from the P1 loop formed a hydrogen bond with the backbone amide of Asp157 that was stable for the remainder of the simulation. Despite this distortion, the Cα atoms of apo–actin showed only slightly higher average RMS fluctuation values as compared with other actin simulations (~1.1 versus 1.0 Å, see Table 1), indicating that expected denaturation did not occur in the time course of our simulation. This is not surprising given that the rate constant for the unfolding of nucleotide-free actin is 0.2 s−1.4 Previously, it was noted that the second-order rate constant for Mg–ATP association with apo–actin is not diffusion limited,35 and this may be so because apo–actin only transiently populates open conformational states.
The collapse of P1 in the absence of nucleotide raises questions about the mechanism of nucleotide exchange. It seems unlikely that after release of ADP the P1 loop would collapse, since this conformational change would inhibit subsequent ATP binding. One explanation is that the P1 loop does not collapse when profilin is bound and the cleft is open. We did not explore such a possibility here.
Relaying structural change from the phosphate clamp to the barbed-end groove and the mouth of the nucleotide cleft
Because the γ-phosphate of ATP interacts directly with the P1 and P2 loops in both actin and Arp3, it stands to reason that these loops may initiate conformational changes upon ATP hydrolysis and phosphate dissociation, providing one way for the bound nucleotide to influence the conformation of other parts of the protein. Here we have shown that changes in the phosphate clamp are relayed to both the barbed-end groove and the cleft mouth in Arp3. When the phosphate clamp opened in our Arp3 simulations, the P1 and P2 loops acted as levers, amplifying the opening motion from the base of the nucleotide cleft (near the axis for rigid body motion, see Fig. 6d) toward the cleft mouth. Opening the nucleotide cleft decreases the width of the barbed-end groove. Changing the width of the barbed-end groove alters the binding surface and could influence interactions with actin-binding proteins, thus providing a basis for the effect of nucleotides on their binding affinity.
While our simulations showed that changes in the phosphate clamp can be relayed to the barbed-end groove and the cleft mouth, we found no evidence that the clamp opens in response to the loss of the γ-phosphate in actin or Arp3. The clamp stayed closed in our simulations of monomeric actin, regardless of the nucleotide binding state. It remains to be seen whether allosteric interactions within the filament or between actin and its binding partners alter the clamp’s sensitivity to the γ-phosphate. The phosphate clamp opened in all but one of the Arp3 simulations at 37 °C, including the ATP–Arp3 simulation. In contrast, crystal structures of the Arp2/3 complex have shown that Arp3 is sensitive to the presence of the γ-phosphate at 4 °C. When apo–Arp2/3 crystals were soaked with nucleotides, ATP binding caused the clamp to close but ADP binding did not.20 However, a recent cocrystal structure of the ADP–Arp2/3 complex (also at 4 °C) showed the cleft of Arp3 closed around ADP.21 Together, these observations suggest a low energetic barrier between the closed and open conformations of Arp3. Much work is needed to determine how the low energetic barrier between these conformations is related to the kinetic and thermodynamic details of Arp2/3 complex activation.
Partial release of the nucleotide in Arp3 simulations suggests multiple potential pathways for dissociation and association of nucleotides
The opening of the nucleotide cleft in the Arp3 simulations allowed the nucleotide to dissociate partially. Three sets of interactions anchor the nucleotide to the cleft, and the order in which these interactions are lost is different in each of the three simulations. This suggests that the binding energy is distributed relatively equally among functional groups of the nucleotide. The dissociation of the adenine and ribose rings from the subdomain 3/4 notch in the ATP–Arp3 and ADP–Arp3 simulations was unexpected, because in crystal structures of the Arp2/3 complex, subdomains 1 and 2 of the Arp2 subunit are completely disordered and nucleotide binds solely through interactions with subdomains 3 and 4, suggesting that much of the binding energy is derived from the interactions with the subdomain 3/4 notch.20 While the binding affinity of adenosine for actin or any of the actin-related proteins has not been measured, the presence of multiple low-energy pathways for release is consistent with the idea that the conformational change, which opens the nucleotide cleft, is the rate-limiting step for the release of the nucleotide.
Our simulations showed that ADP was more mobile in the nucleotide cleft of actin than ATP. ADP was not stable in the “standard” conformation observed in all ADP–actin crystal structures currently available. A torsional flip of the O4′–C4′–C5′–O5′ bond changed ADP to the “bent” conformation previously observed in the ADP–Arp2/3 complex crystal structure in which the nucleotide cleft of Arp3 is open. This conformation was proposed to be a “prerelease” ADP conformation.21 Our results suggest a similar potential role for the bent ADP conformation in actin.
The conformation of Ser14/Thr14 is sensitive to the bound nucleotide but is not coupled to the conformation of the sensor loop on nanosecond timescales
In crystal structures of actin with bound ADP or ATP, the loss of the γ-phosphate allows the P1 loop to move slightly inward and Ser14 to rotate inward toward the cleft.10,12 The P1 loop also moves inward in Arp3 in an ADP-bound Arp2/3 complex structure.21 These changes are accompanied by a flip of the backbone carbonyl of Glu72/Arg79, a residue in a short loop flanking the cleft, which contacts the P1 loop, termed the sensor loop (Fig. 1c). In the actin ATP→ADP swap simulation, Ser14 immediately rotated into the cleft, suggesting that the conformation of this residue is tightly linked to nucleotide state. However, the sensor loop did not flip, so the conformation of Ser14 is not linked tightly to the conformation of the sensor loop, at least on the timescales investigated here. In the nucleotide-free Arp3 simulations, Thr14 changed rotomers and the sensor loop flipped, but these changes were not correlated on the nanosecond timescale.
In the ADP–Arp3 simulation, Thr14 does not rotate into the cleft. Energetically, this change may not be favored unless the nucleotide cleft is closed. Consistent with this hypothesis, Thr14 is not rotated into the cleft in the ADP–Arp3 structure with an open nucleotide cleft,20 but the structure of the Arp2/3 complex cocrystallized with ADP showed Arp3 with a closed cleft and Thr14 rotated inward.21 Therefore, the relay of structural changes initiated by the rotomer state of Ser14/Thr14 may depend on a network of interactions among nucleotide phosphates, water, and protein that exists only when the nucleotide cleft is closed.
The secondary structure of the DNase binding loop is unstable, regardless of nucleotide binding state
In simulations where the DNase binding loop started in a random coil conformation, it remained a random coil regardless of the nucleotide binding state. In the ADP–actin simulation, the DNase loop helix partially unfolded from an α-helix to form a π-helix. The π-helix is rarely seen in crystal structures and is thought to be unstable under most circumstances.39 The observed instability of the DNase loop α-helix is consistent with crystal structures of nonpolymerizable point mutants of actin, which show that the DNase binding loop is disordered in both ADP- and ATP-bound states.12 The DNase loop α-helix was also unstable in MD simulations of ADP–actin by other investigators and partially unfolded into either a one-turn α-helix or a five-residue π-helix over 50 ns.29 The overall instability of the α-helix in these simulations agrees with our observations, but the partial unfolding was slower. This difference probably stems from the higher temperature used in our simulations (37 versus 27 °C).
The cumulative results of actin MD simulations and crystal structures from multiple labs are consistent with an analysis by Rould et al., which indicated that nucleotide-dependent folding of the DNase binding loop in the 1J6Z structure is caused by intermolecular propagation of the sensor loop flip through crystal packing interactions.12 This leaves open the question of why ADP–actin and ATP–actin exhibit distinct biochemical properties. The flip of the sensor loop upon loss of the γ-phosphate is the most consistent difference between ATP–actin and ADP–actin and Arps in crystal structures, but it is unclear how this change relays the nucleotide state to other parts of the molecule. Clearly, much is yet to be learned about the relationship between nucleotide binding and conformation in these proteins.
Val159/174 and back door phosphate release
Val159/174 in the P2 loop hydrogen bonds directly to the γ-phosphate. Mutational analyses of yeast actin showed that Val159 plays a role in linking the nucleotide state to large-scale conformational changes in actin subunits within filaments.16 This residue has been observed in both up and down positions in actin and Arp3.13,21 In the down position, its backbone amide hydrogen bonds to the γ-phosphate and its side chain blocks the “back door” for phosphate release as proposed.28 We previously hypothesized that as the γ-phosphate moves away from the β-phosphate after hydrolysis, it stays hydrogen bonded to Val159/174 as this residue flips up to open the back door.21 After the back door opens, the phosphate could exit without opening of the nucleotide cleft, a requirement necessitated by the observation that phosphate, but not nucleotide, can exchange in filamentous muscle actin. In two of our simulations, the valine switched between up and down conformations rapidly, suggesting a relatively low-energy barrier between the two positions. However, we expected that the valine would be more likely to occupy the down position in ATP-containing simulations in order to maintain the hydrogen bond to the γ-phosphate and to occupy the up position in the ADP simulations, where no γ-phosphate was present. While this is consistent with crystal structures, which show that the nucleotide state is not tightly linked to the conformation of this valine, it leaves an open question about how hydrolysis could favor the opening of the back door.
Calcium occupies three positions in the nucleotide clefts of actin and Arp3 that influence nucleotide dynamics
The identity of the divalent cation influences numerous aspects of actin function, including its critical concentration,40 affinity for ADP or ATP,41 and rate of ATP hydrolysis.2 While no biochemical information addresses the effect of divalent cations on Arp3 and Arp2, differences in crystal structures containing magnesium or calcium indicate that the identity of the divalent may also be critical in the Arp2/3 complex.21 In our initial simulations, the calcium tended to interact directly with negatively charged residues in the nucleotide cleft rather than indirectly through a sphere of water as seen in crystal structures of actin. After the charge on the calcium was adjusted, the simulations placed the calcium in three sites, which affected the position of the phosphates and their orientation relative to the phosphate clamps. Interestingly, the lower charge on the calcium did not affect the overall propensity of Arp3 to open, but the trajectories of divalent cation, nucleotide, and protein differed significantly in the simulations with Ca+1.2 and Ca+2.0. While speculation about how population of each divalent site in our simulations might influence biochemical properties is premature, we point out the importance of this detail in analyzing any actin simulation.
Implications for understanding Arp2/3 complex function
No ATPase activity has been detected for Arp3, but fluorescence resonance energy transfer experiments showed that ATP binding to Arp3 causes a conformational change.42 The opening and closing of the cleft of Arp3 may play a critical role in Arp2/3 complex function. Reconstructions of the Arp2/3 complex at filament branch junctions from electron micrographs show Arp3 in a closed conformation, similar to that observed in crystal structures of the Arp2/3 complex with bound ATP.43 The tendency of Arp3 to populate open states may play a role in keeping the Arp2/3 complex inactive until bound to nucleating promoting factors at a branch. Because the C-terminus favors cleft opening, it may be displaced from the barbed-end groove of Arp3 during activation of the Arp2/3 complex. Clearly, there is much to be learned about how the nucleotide states of Arp2 and Arp3 are involved in branching.
Simulation Methods
Coordinates of the crystal structure of rabbit skeletal muscle actin with bound adenosine-5′-imidotriphosphate and a rhodamine label on Cys374 were obtained from the PDB (accession code 1NWK).11 We removed rhodamine, surface calcium ions, and water molecules and converted the nonhydrolyzable adenosine-5′-imidotriphosphate to ATP. We modeled certain features from other crystal structures. We based the DNase binding loop on the open profilin–actin structure (PDB accession code 1HLU),14 the N-terminus on the DNase–actin crystal structure (PDB accession code 1ATN),44 and the C-terminus on the gelsolin–actin structure (PDB accession code 1ESV).45 The CHARMM topology file (version 27) was modified to accommodate the N-terminal acetyl–aspartate and the methylated histidine (His73). ADP–actin was prepared in a similar manner from the 1J6Z10 structure with the same sources for the N- and C-termini. Coordinates for Arp3 were taken from the apo–Arp2/3 crystal structure (PDB accession code 1K8K),19 the ADP-soaked crystals (PDB accession code 1U2V),20 or the ATP-soaked crystals (PDB accession code 1TYQ).20 The DNase binding loop of 1HLU was appended to the Arp3 structures after mutating residues to match Arp3 and adding the extra eight residues inserted into Arp3 in this region. The N- and C-termini of 1U2V were used to model missing residues at the termini of Arp3 in 1K8K and 1TYQ. Newly built portions of the starting models were refined using the refi_zone command in the software program O.
We used the Visual Molecular Dynamics (VMD) software package46 to add hydrogen atoms to actin and Arp3. Proteins were completely resolvated using a variation on the VMD solvate package. The solvate package creates a box of water of specified volume around the protein where the distance between oxygen atoms of neighboring water molecules is no less than 2.4 Å. After using this package to solvate the proteins studied here, we observed a gap or vacuum close to the protein and particularly few water molecules in the nucleotide binding cleft (Supplemental Fig. S2A). After close examination, it appeared that hydrogen atoms were being included in the distance calculation between water molecules and the protein. However, since the spacing among water molecules themselves appeared correct, we wrote a program to merge the protein structure with the box of water, removing water molecules within 2.4 Å of nonhydrogen atoms on the protein. The resulting positions of the water molecules close to the protein agreed qualitatively with crystal structures (Supplemental Fig. S2B). We used the VMD plug-in autoionize to add 100 mM NaCl to the existing solvent of about 30,000 explicit water molecules (TIP3 water model47). Atomistic interactions were calculated using the CHARMM force field (version 27).48 Electrostatics were calculated from particle mesh Ewald sums. Simulations were run with periodic boundary conditions in an isobaric–isothermal (constant NPT) ensemble using the NAMD software package.49
Actin and Arp3 were equilibrated in their solvent as follows: backbone atoms, nucleotide, and Ca2+ were fixed for 1 ps, while all other atoms were allowed to move. Next, only the nucleotide and Ca2+ were fixed for 1 ps; the system was heated to 37 °C, while the Cα atoms were fixed for 3 ps; the Langevin piston was then turned on for 5 ps; and, finally, the system was equilibrated at constant volume for 10 ps. It is widely known that the electrostatic potentials of the CHARMM force field have difficulty accurately modeling interactions of divalent cations. Our initial simulations showed that after constraints were released on the nucleotide and calcium, the calcium ion consistently moved to make a direct hydrogen bond to the conserved aspartate in the cleft (Asp154/169). In the actin crystal structures, the calcium has a complete shell of hydration and does not directly contact any of the residues in the nucleotide cleft.11,13 In these structures, the calcium is coordinated to the water in a heptagonal arrangement, with an average distance of 2.5 Å. We sought to approximate realistic behavior by lowering the charge on the calcium. Using the (APO →ATP)–Arp3 simulation as a model, we lowered the calcium charge in increments of 0.2 and then equilibrated, minimized, and heated the system before running 50-ps test simulations. Using this method, the highest charge at which the calcium did not make a direct contact to the aspartate after 50 ps was +1.2. We used this charge for all simulations that included calcium. Despite the lower charge, the calcium maintained a tendency to interact directly with Asp154/169 and Asp11/11.
Production runs of the Arp3 simulations were carried out on a San Diego Supercomputer Center DataStar IBM p690 cluster. One nanosecond of simulation time required 7 h on 128 processors. Actin simulations were run across eight processors at a rate of 1 ns per 2 days.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health through research grants GM066311 and GM026338 (to T.D.P.). P.D. and B.J.N. were supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Awards F32GM074504 and F32GM074374. We thank Ross C. Walker and the San Diego Supercomputer Center for providing the supercomputer time necessary for the completion of this project. We also thank William Jorgensen and Preston Moore for discussions about divalent cations.
Abbreviations used
- MD
molecular dynamics
- Arp
actin-related protein
- PDB
Protein Data Bank
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2007.11.068
Note added in proof: Cleavage of the DNase-loop of actin by ECP protease increases the rate of nucleotide exchange about two-fold and has been hypothesized to increase the population of the open conformation of actin in solution (Khaitlina, S.Y. & Strzelecka-Golaszewska, H. (2002). Biophys. J. 82, 321–334). To learn if this cleavage alters actin dynamics, we broke the backbone between Gly42 and Val43 at the end of the simulation of ATP-actin and extended the simulation. The nucleotide cleft stayed closed and the break in the backbone had little effect on the dynamics of actin over the additional 2 ns of simulation.
References
- 1.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 2.Blanchoin L, Pollard TD. Hydrolysis of ATP by polymerized actin depends on the bound divalent cation but not profilin. Biochemistry. 2002;41:597–602. doi: 10.1021/bi011214b. [DOI] [PubMed] [Google Scholar]
- 3.Carlier MF, Pantaloni D. Direct evidence for ADP-Pi–F-actin as the major intermediate in ATP–actin polymerization.Rate of dissociation of Pi from actin filaments. Biochemistry. 1986;25:7789–7792. doi: 10.1021/bi00372a001. [DOI] [PubMed] [Google Scholar]
- 4.Kinosian HJ, Selden LA, Gershman LC, Estes JE. Non-muscle actin filament elongation from complexes of profilin with nucleotide-free actin and divalent cation-free ATP–actin. Biochemistry. 2004;43:6253–6260. doi: 10.1021/bi036117s. [DOI] [PubMed] [Google Scholar]
- 5.Fujiwara I, Vavylonis D, Pollard TD. Polymerization kinetics of ADP- and ADP-Pi–actin determined by fluorescence microscopy. Proc Natl Acad Sci USA. 2007;104:8827–8832. doi: 10.1073/pnas.0702510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maciver SK, Zot HG, Pollard TD. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J Cell Biol. 1991;115:1611–1620. doi: 10.1083/jcb.115.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De La Cruz EM, Ostap EM, Brundage RA, Reddy KS, Sweeney HL, Safer D. Thymosin-beta(4) changes the conformation and dynamics of actin monomers. Biophys J. 2000;78:2516–2527. doi: 10.1016/S0006-3495(00)76797-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vinson VK, De La Cruz EM, Higgs HN, Pollard TD. Interactions of Acanthamoeba profilin with actin and nucleotides bound to actin. Biochemistry. 1998;37:10871–10880. doi: 10.1021/bi980093l. [DOI] [PubMed] [Google Scholar]
- 9.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin: DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 10.Otterbein LR, Graceffa P, Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 11.Graceffa P, Dominguez R. Crystal structure of monomeric actin in the ATP state. Structural basis of nucleotide-dependent actin dynamics. J Biol Chem. 2003;278:34172–34180. doi: 10.1074/jbc.M303689200. [DOI] [PubMed] [Google Scholar]
- 12.Rould MA, Wan Q, Joel PB, Lowey S, Trybus KM. Crystal structures of expressed non-polymerizable monomeric actin in the ADP and ATP states. J Biol Chem. 2006;281:31909–31919. doi: 10.1074/jbc.M601973200. [DOI] [PubMed] [Google Scholar]
- 13.Vorobiev S, Strokopytov B, Drubin DG, Frieden C, Ono S, Condeelis J, et al. The structure of nonvertebrate actin: implications for the ATP hydrolytic mechanism. Proc Natl Acad Sci USA. 2003;100:5760–5765. doi: 10.1073/pnas.0832273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chik JK, Lindberg U, Schutt CE. The structure of an open state of beta-actin at 2.65 Å resolution. J Mol Biol. 1996;263:607–623. doi: 10.1006/jmbi.1996.0602. [DOI] [PubMed] [Google Scholar]
- 15.Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U. The structure of crystalline profilin–beta-actin. Nature. 1993;365:810–816. doi: 10.1038/365810a0. [DOI] [PubMed] [Google Scholar]
- 16.Belmont LD, Orlova A, Drubin DG, Egelman EH. A change in actin conformation associated with filament instability after Pi release. Proc Natl Acad Sci USA. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006;7:713–726. doi: 10.1038/nrm2026. [DOI] [PubMed] [Google Scholar]
- 18.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 19.Robinson RC, Turbedsky K, Kaiser DA, Marchand JB, Higgs HN, Choe S, Pollard TD. Crystal structure of Arp2/3 complex. Science. 2001;294:1679–1684. doi: 10.1126/science.1066333. [DOI] [PubMed] [Google Scholar]
- 20.Nolen BJ, Littlefield RS, Pollard TD. Crystal structures of actin-related protein 2/3 complex with bound ATP or ADP. Proc Natl Acad Sci USA. 2004;101:15627–15632. doi: 10.1073/pnas.0407149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nolen BJ, Pollard TD. Insights into the influence of nucleotides on actin family proteins from seven structures of Arp2/3 complex. Mol Cell. 2007;26:449–457. doi: 10.1016/j.molcel.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akola J, Jones RO. Density functional calculations of ATP systems: 2. ATP hydrolysis at the active site of actin. J Phys Chem B. 2006;110:8121–8129. doi: 10.1021/jp054921d. [DOI] [PubMed] [Google Scholar]
- 23.Dalhaimer P, Discher DE, Lubensky TC. Crosslinked actin networks show liquid crystal elastomer behaviour, including soft-mode elasticity. Nature Phys. 2007;3:354–360. [Google Scholar]
- 24.Chu JW, Voth GA. Allostery of actin filaments: molecular dynamics simulations and coarse-grained analysis. Proc Natl Acad Sci USA. 2005;102:13111–13116. doi: 10.1073/pnas.0503732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minehardt TJ, Kollman PA, Cooke R, Pate E. The open nucleotide pocket of the profilin/actin x-ray structure is unstable and closes in the absence of profilin. Biophys J. 2006;90:2445–2449. doi: 10.1529/biophysj.105.072900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ming D, Kong Y, Wu Y, Ma J. Substructure synthesis method for simulating large molecular complexes. Proc Natl Acad Sci USA. 2003;100:104–109. doi: 10.1073/pnas.232588999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tirion MM, ben-Avraham D. Normal mode analysis of G-actin. J Mol Biol. 1993;230:186–195. doi: 10.1006/jmbi.1993.1135. [DOI] [PubMed] [Google Scholar]
- 28.Wriggers W, Schulten K. Investigating a back door mechanism of actin phosphate release by steered molecular dynamics. Proteins: Struct Funct Genet. 1999;35:262–273. [PubMed] [Google Scholar]
- 29.Zheng X, Diraviyam K, Sept D. Nucleotide effects on the structure and dynamics of actin. Biophys J. 2007;93:1277–1283. doi: 10.1529/biophysj.107.109215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nolen B, Ngo J, Chakrabarti S, Vu D, Adams JA, Ghosh G. Nucleotide-induced conformational changes in the Saccharomyces cerevisiae SR protein kinase, Sky1p, revealed by X-ray crystallography. Biochemistry. 2003;42:9575–9585. doi: 10.1021/bi0344331. [DOI] [PubMed] [Google Scholar]
- 31.Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins: Struct Funct Genet. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 32.Barrett CP, Hall BA, Noble ME. Dynamite: a simple way to gain insight into protein motions. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2280–2287. doi: 10.1107/S0907444904019171. [DOI] [PubMed] [Google Scholar]
- 33.Tai K, Shen T, Henchman RH, Bourne Y, Marchot P, McCammon JA. Mechanism of acetylcholinesterase inhibition by fasciculin: a 5-ns molecular dynamics simulation. J Am Chem Soc. 2002;124:6153–6161. doi: 10.1021/ja017310h. [DOI] [PubMed] [Google Scholar]
- 34.Dayel MJ, Holleran EA, Mullins RD. Arp2/3 complex requires hydrolyzable ATP for nucleation of new actin filaments. Proc Natl Acad Sci USA. 2001;98:14871–14876. doi: 10.1073/pnas.261419298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De La Cruz EM, Pollard TD. Nucleotide-free actin: stabilization by sucrose and nucleotide binding kinetics. Biochemistry. 1995;34:5452–5461. doi: 10.1021/bi00016a016. [DOI] [PubMed] [Google Scholar]
- 36.Dayel MJ, Mullins RD. Activation of Arp2/3 complex: addition of the first subunit of the new filament by a WASP protein triggers rapid ATP hydrolysis on Arp2. PLoS Biol. 2004;2:E91. doi: 10.1371/journal.pbio.0020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Clainche C, Pantaloni D, Carlier MF. ATP hydrolysis on actin-related protein 2/3 complex causes debranching of dendritic actin arrays. Proc Natl Acad Sci USA. 2003;100:6337–6342. doi: 10.1073/pnas.1130513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin AC, Welch MD, Drubin DG. Arp2/3 ATP hydrolysis-catalysed branch dissociation is critical for endocytic force generation. Nat Cell Biol. 2006;8:826–833. doi: 10.1038/ncb1443. [DOI] [PubMed] [Google Scholar]
- 39.Fodje MN, Al-Karadaghi S. Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng. 2002;15:353–358. doi: 10.1093/protein/15.5.353. [DOI] [PubMed] [Google Scholar]
- 40.Pollard TD. Rate constants for the reactions of ATP– and ADP–actin with the ends of actin filaments. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kinosian HJ, Selden LA, Estes JE, Gershman LC. Nucleotide binding to actin. Cation dependence of nucleotide dissociation and exchange rates. J Biol Chem. 1993;268:8683–8691. [PubMed] [Google Scholar]
- 42.Goley ED, Rodenbusch SE, Martin AC, Welch MD. Critical conformational changes in the Arp2/3 complex are induced by nucleotide and nucleation promoting factor. Mol Cell. 2004;16:269–279. doi: 10.1016/j.molcel.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 43.Rouiller I, Xu X-P, Amann KJ, Egile C, Nicastro D, Li R, et al. The structural basis of actin filament branching by Arp2/3 complex. J Cell Biol. 2008 doi: 10.1083/jcb.200709092. In the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabsch W, Holmes KC. The actin fold. FASEB J. 1995;9:167–174. doi: 10.1096/fasebj.9.2.7781919. [DOI] [PubMed] [Google Scholar]
- 45.Morton WM, Ayscough KR, McLaughlin PJ. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Biol. 2000;2:376–378. doi: 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- 46.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klien ML. Comparison of single potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 48.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM—a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 49.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.