

Abstract
The multi-faceted roles of calcium-independent phospholipase A2β (iPLA2β) in numerous cellular processes have been extensively examined through utilization of the iPLA2-selective inhibitor (E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one (BEL). Herein, we employed accurate mass/high resolution mass spectrometry to demonstrate that the active site serine (S465) and C651 of iPLA2β are covalently cross-linked during incubations with BEL demonstrating their close spatial proximity. This crosslink results in macroscopic alterations in enzyme molecular geometry evidenced by anomalous migration of the cross-linked enzyme by SDS-PAGE. Molecular models of iPLA2β constructed from the crystal structure of iPLA2α (patatin) indicate that the distance between S465 and C651 is approximately 10 Å within the active site of iPLA2β. Kinetic analysis of the formation of the 75 kDa iPLA2β-BEL species with the (R) and (S) enantiomers of BEL demonstrated that the reaction of (S)-BEL with iPLA2β was more rapid than for (R)-BEL paralleling the enantioselectivity for the inhibition of catalysis by each inhibitor with iPLA2β. Moreover, we demonstrate that the previously identified selective acylation of iPLA2β by oleoyl-CoA occurs at C651 thereby indicating the importance of active site architecture for acylation of this enzyme. Collectively, these results identify C651 as a highly reactive nucleophilic residue within the active site of iPLA2β which is thioesterified by BEL, acylated by oleoyl-CoA and located in close spatial proximity to the catalytic serine thereby providing important chemical insights on the mechanisms through which BEL inhibits iPLA2β and the topology of the active site.
Phospholipases A2 (PLA2s) catalyze the hydrolysis of the sn-2 carboxylic ester bond of glycerophospholipids to generate free fatty acids and lysophospholipids, which, together with their downstream metabolites, are involved in lipid metabolic and signaling pathways 1–5. Eukaryotic PLA2s are grouped into three main families: secretory, cytosolic, and calcium-independent phospholipases A2 6. Calcium-independent PLA2s (iPLA2s) or patatin-like phospholipases (PNPLA in HUGO nomenclature) are intracellular enzymes that do not require calcium ion for membrane association or catalysis and consist of nine family members (PNPLA1 through PNPLA9) also known as iPLA2 (β, γ, δ, ε, ζ, η, φ, ι κ) 7–9. Each of these calcium-independent PLA2s contain conserved nucleotide-binding (GXGXXG) and lipase (GXSXG) consensus motifs separated by a 10–50 amino acid spacer arm which collectively form a catalytic domain highly homologous to the plant phospholipase/lipase patatin. Previously published studies have demonstrated that calcium-independent phospholipase A2β (iPLA2β or PNPLA9) plays an important role in agonist-induced arachidonic acid release, receptor mediated signaling, insulin secretion, apoptosis, cellular proliferation and migration, vascular inflammation, and lysolipid production mediating calcium influx 10–20.
The mechanism-based inhibitor (E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one (BEL), was originally developed for studying the catalytic mechanism of chymotrypsin 21. In later studies, BEL was demonstrated to be a selective and potent mechanism-based inhibitor of iPLA2β 22, 23, and has been widely used to distinguish iPLA2 activities from those of other phospholipases A2 13, 24–30. Two plausible mechanisms for iPLA2 inhibition were considered based on the proposed mechanism for the inhibition of chymotrypsin by BEL 1, 21. In both mechanisms, the inactivation of the enzyme is initiated by nucleophilic attack of the serine hydroxyl on the lactone carbonyl thereby opening of the bromoenol lactone ring of BEL to generate a tethered α-bromomethyl ketone (Scheme 1, Step I) within the active site of the enzyme. This tethered α-bromomethylketone is a powerful alkylating agent that has several possible downstream fates. The α-bromomethyl ketone while tethered to the catalytic serine (S465 in iPLA2β) is subject to nucleophilic attack by one or more spatially adjacent nucleophilic residues (Step II). Covalent modification of these residues would impair the catalytic activity of the enzyme even after subsequent deacylation of S465 (Step III) due to steric hindrance at the active site. This mechanism is supported by multiple lines of evidence for iPLA2β, including kinetic assays and autoradiography 22, 23, but the alkylated residue(s) in iPLA2β were not identified. A second mechanistic possibility is that the enzyme undergoes deacylation immediately after hydrolyzing the bromoenol lactone ring of BEL followed by regeneration of the active site serine to generate a diffusible α-bromomethyl keto carboxylic acid (Step IV). This α-bromomethyl keto carboxylic acid could potentially alkylate any available cysteine residues (including those on the protein surface distal to the active site) that could also potentially result in inactivation of the enzyme. This mechanism was proposed based on the mass spectrometric observation that multiple cysteine residues were covalently modified by the keto carboxylic acid formed by iPLA2β after BEL treatment 31, but it fails to address the fact that the ring-opened diffusible BEL (i.e., the α-bromomethyl keto carboxylic acid) does not inhibit the activity of the enzyme when added exogenously in comparable concentrations in the presence of reducing agents 23. In early work, we calculated the capture ratio of the ring opened compound using a variety of methods to demonstrate that the majority of inhibition was mediated by covalently bound BEL at the active site in the presence of a reducing agent 22. The reactive α-haloketones that were released by hydrolysis of the acyl enzyme were rapidly inactivated by reducing agents such as DTT in the incubation medium in vitro or by GSH in vivo22.
Scheme 1. Proposed mechanisms for the inactivation of the iPLA2β by BEL.

I.. Nucleophilic attack of the carbonyl carbon of the bromoenol lactone ring of BEL by the catalytic serine (S465) of iPLA2β generates the electrophilic α-bromomethyl ketone.
Pathway A:
II. The α-bromomethyl ketone remains tethered to the active site through an acyl link-age. An appropriately positioned nucleophilic residue (within the active site) then attacks the α-carbon of the ring-opened BEL intermediate to eliminate bromide resulting in the stable alkylation of the neighboring nucleophilic residue in iPLA2β. Hydroxide ion or water can also be the attacking nucleophile (II′).
III. Following deacylation of the active site serine by iPLA2β catalyzed hydrolysis, the stable α-ketone remains covalently linked to the active site of the enzyme through the adjacent nucleophilic amino acid resulting in inhibition of enzyme activity by the active site.
Pathway B:
IV. The acyl-enzyme intermediate undergoes hydrolysis to release the α-bromomethyl ketone carboxylic acid.
V. The diffusible α-bromomethyl ketone carboxylic acid reacts with cysteine residues on the enzyme, resulting in the covalent modification of the enzyme at sites distant to the active site.
In this study, we analyzed BEL-inactivated iPLA2β by mass spectrometry, SDS-PAGE and auto-radiography. Remarkably, incubation of iPLA2β with BEL results in the generation of a rapidly migrating covalently modified species of iPLA2β on SDS-PAGE. Tandem mass spectrometric analyses of the rapidly migrating band demonstrated a BEL-bridged internal crosslink between the active site serine (S465) and C651 indicating an internally tethered protein with a decreased Stokes radius. Moreover, the BEL crosslink between S465 and C651 demonstrates the close spatial proximity of the active site in iPLA2β to C651. Importantly, C651 is between the IQ (residues 622–635) and “1-9-14” (residues 694–705) motifs of the calmodulin binding domain in iPLA2β previously identified 32. Substantiating these results was the marked enantioselectivity and kinetics of iPLA2β inhibition by (S)-BEL vs. (R)-BEL demonstrated through the extent of covalent modification of iPLA2β residues following treatment with each enantiomer. Collectively, these results provide important chemical insights into the mechanisms by which BEL inhibits iPLA2β and the interaction of the enzyme active site with C651 which is present in the calmodulinbinding domain of iPLA2β.
EXPERIMENTAL PROCEDURES
Materials
(E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one (BEL) was purchased from Cayman Chemical (Ann Arbor, MI). [3H]-BEL was synthesized and purified as previously described 33. Antibodies against iPLA2β epitopes were generated in rabbits prior to purification by immunoaffinity chromatography as described previously 32. Simply Blue™ Safe Stain and BenchMark™ Protein Ladder were purchased from Invitrogen (Carlsbad, CA). ECL™ Western Blotting Detection Reagents were from GE Healthcare Bio-Sciences Corp. (Piscataway, NJ). Sequence grade modified trypsin was from Promega (Madison, WI). Most solvents were purchased from Burdick & Jackson (Muskegon, MI). Nano-HPLC columns and pre-columns were obtained from Dionex (Sunnyvale, CA). Most other chemicals were obtained from Sigma (St. Louis, MO).
Recombinant His-tagged iPLA2β
Recombinant His-tagged iPLA2β was expressed and purified from Sf9 cells as described previously 34.
Measurement of iPLA2 β Catalytic Activity
Inhibition of the phospholipase activity of purified recombinant iPLA2β by BEL was measured through radiolabeled substrate assays (using 1-palmitoyl-2-[1-14C]arachidonoyl-sn-glycero-3-phosphocholine 35), real-time fluorescence assays (using 2-decanoyl-1-(O-(11-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pro-pionyl)amino)undecyl)-sn-glycero-3-phosphocholine 36), or by electrospray ionization mass spectrometry (using 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine 37) as previously described.
Separation of BEL Enantiomers
BEL enantiomers were separated by HPLC using a chiral column as described previously 35. Briefly, a Chirex column comprised of 3,5-dinitrobenzoyl-(R)-phenylglycine attached to a silica matrix was used as the stationary chiral phase. After equilibration with hexane/dichloroethane/ethanol (150:15:1), the BEL enantiomers were injected and eluted isocratically at a flow rate of 2 mL/min. The elution of BEL was monitored by UV absorbance at 280 nm. Peaks corresponding to (S)- or (R)-BEL retention times 19 and 21 min, respectively) were collected, dried under a nitrogen stream and stored at −20°C. The concentrations of (R)- and (S)-BEL were determined by its UV absorbance at 280 nm in acetonitrile (ε280 = 6130 cm−1 M−1)35.
Polyacrylamide Gel Electrophoresis of Covalent BEL- iPLA2β Adducts
Racemic BEL, (R)-BEL and (S)-BEL were stored as solids and redissolved in ethanol immediately before use. After dissolving in ethanol, BEL was injected into 25 mM imidazole buffer, pH 7.8 containing 1 mM DTT and 50 mM NaCl and incubated with purified iPLA2β for the indicated time periods at room temperature. The ratio of BEL to protein in the reaction varied from 0 to 10. The reactions were terminated by addition of 2 × SDS-PAGE loading buffer (0.125 M Tris, pH 6.8, 4 % SDS (w/v), 20 % (v/v) glycerol, 0.001 % bromphenol blue (w/v), 0.2 M DTT (DL-dithiothreitol)) and the mixture was boiled for 5 min. BEL-protein adducts were electrophoretically separated on 7% SDS-PAGE gels and stained with Coomassie Blue. The resultant gels were scanned with a Kodak Image Station 440 to quantify the protein band intensities using Kodak 1D software.
[3H]-BEL Labeling and Autoradiographic Analysis
Prior to use, [3H]-BEL was purified by HPLC on a reverse phase column (C18, 4.6 mm × 250 mm, Beckman ODS) using a mobile phase of 60% acetonitrile in water at a flow rate of 1 mL/min. The elution of BEL was monitored by UV absorbance at 280 nm. Chloroform and methanol were added to the collected BEL fraction to obtain a final ratio of 1:1:1 (chloroform : methanol : eluted fraction (v/v/v)). Optically pure [3H]-BEL was extracted into chloroform, dried under a nitrogen stream, and redissolved in ethanol. The concentration of BEL was determined by its UV absorbance at 280 nm in acetonitrile (ε280 = 6130 cm−1 M−1). Purified [3H]-BEL was incubated with iPLA2β in a solution of 25 mM imidazole, pH 7.8, 1 mM DTT and 50 mM NaCl at a molar ratio of 3:1 (BEL : protein) at room temperature for 5 min. The reaction was terminated by adding an equal volume of 2× SDS-PAGE buffer. Proteins were separated on 7% SDS-PAGE gels and visualized by Coomassie Blue staining. Following fixation with 40% methanol containing 10% acetic acid, the gels were incubated with a fluorographic reagent (Amplify, Amersham) for 30 min, dried under vacuum and exposed to film.
Covalent Acylation of iPLA2β with Oleoyl-CoA
Purified iPLA2β (5 μM) was incubated in the presence or absence of oleoyl-CoA (50 μM) for 60 min at 30°C. Protein was precipitated with CHCl3/CH3OH as previously described 38, dried using a Speed-Vac, and solubilized in 25 mM NH4HCO3 containing 0.1% RapiGest™ (Waters, Milford, MA). Trypsin was added to the samples at a ratio of 1:30 (w/w) followed by incubation at 37°C for two hours. After termination of the trypsinolysis by addition of trifluoroacetic acid (0.5% final concentration), the samples were further incubated at 37°C for 45 min and centrifuged at 13,000 rpm for 15 min. The supernatant was removed and the pellet was resuspended in 70% isopropanol by a combination of vortexing and sonication. Both the supernatant and the pellet fractions were analyzed by nano-LC/MS/MS. Processed samples were applied to a PepMap C4 precolumn (300 μm × 1 mm, Dionex, Sunnyvale, CA) using a Surveyor autosampler (ThermoFisher Scientific, Waltham, MA) and washed with 0.1% formic acid for 5 min. A nonlinear gradient from 90% mobile phase A (0.1% formic acid in water) and 10% mobile phase B (50% acetonitrile/50% isopropanol, 0.1% formic acid) to 10% A and 90% B was applied to elute the tryptic peptides from the precolumn onto a reverse-phase C4 analytical column (75 μm × 15 cm, Dionex, Sunnyvale, CA) at a flow rate of 250 nL/min. Peptides eluting from the C4 column were introduced into an LTQ-Orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA) using a TriVersa™ Nanomate system (Advion, Ithaca, NY) with a spray voltage of 1.7 kV. The mass spectrometer was operated in a data-dependent acquisition mode with dynamic exclusion enabled allowing the five most intense ion peaks from the full mass scan to undergo product ion mass analysis. The instrument was calibrated externally. Full mass scans were performed by the Orbitrap mass analyzer with a mass resolution of r = 30,000 at m/z 400 Thomson. The lock mass option was enabled to ensure a mass accuracy of < 5 ppm (utilizing the m/z value of the protonated diisooctyl phthalate (391.2843) as the lock mass). Product ion mass spectra were acquired either by the ion trap or the Orbitrap mass analyzer with a mass resolution of 15,000 at m/z 400 Thomson. For the product ion analyses, a normalized collision energy of 30% was applied, the activation time was set at 30 ms with the activation parameter q = 0.250, and precursor ions were isolated within the range of 2 to 4 Thomson. The acquired data was searched against a customized protein sequence database using both SEQUEST (Bioworks™, ThermoFisher Scientific, Waltham, MA) and MASCOT algorithms (Matrix Science, London, UK). Positive search results were manually confirmed.
Western Blotting
Proteins were separated on 7% SDS-PAGE gels and transferred to immobilon-P membranes in 10 mM CAPS buffer (pH 11) containing 10% methanol. Powdered milk (5% (w/v)) was used to block the membranes prior to incubation with primary antibody. Horseradish peroxidase-linked secondary antibody was used in combination with an ECL detection system to visualize immunoreactive bands.
In-gel Digestion of Proteins for Mass Spectrometric Analyses
Protein bands visualized with Coomassie Blue were excised from the gel, cut into small pieces (~ 1 mm3), and destained twice by vortexing for 10 min in 100 μL of 25 mM ammonium bicarbonate containing 50% acetonitrile. The gel pieces were dried utilizing a Speed Vac and then rehydrated in 25 mM ammonium bicarbonate, pH 8 containing trypsin (12.5 μg/mL) on ice for 45 min. Excess trypsin solution was then removed and a small amount of 25 mM ammonium bicarbonate, pH 8 was added to cover the gel pieces. Following overnight incubation at 37°C, 100 μL of 50% acetonitrile containing 5% formic acid were then added and the resultant tryptic peptides were extracted by vigorous vortexing and sonication. The extraction step was repeated once and the peptide extracts were combined and evaporated to ~ 10 μL using a Speed Vac. The peptide extract was diluted in 10% acetonitrile containing 0.1% formic acid before analysis by either MALDI-TOF or ESI-LC/MS/MS.
In-solution Digestion of Protein for Mass Spectrometric Analyses
Native or BEL-treated protein was precipitated by chloroform and methanol as described previously 38. Briefly, four volumes of methanol and one volume of chloroform were added to the protein solution. After vortexing, three volumes of water were then added and the mixture was centrifuged at 14,000 × g for 15 min. The upper phase was discarded and replaced with three volumes of methanol. Proteins were recovered as a pellet following centrifugation for 15 min at 14,000 × g, and re-constituted into 0.1% RapiGest™ in 25 mM NH4HCO3. Trypsin was added at a ratio of 1:30 (enzyme to protein, w/w) and the samples were incubated at 37°C for two hours. Trifluoroacetic acid was then added to a final concentration of 0.5%. The samples were further incubated at 37°C for 45 min and centrifuged at 13,000 rpm for 15 min. The supernatant was transferred to a new tube, and the pellet was resuspended into 70% isopropanol by vortexing and intermittent sonication. Both the supernatant and the pellet fractions were analyzed by mass spectrometry.
Analysis of the Intact Protein by MALDI-TOF
iPLA2β was incubated with BEL as described above. Reactions were terminated by addition of 0.1% TFA (final pH=2) followed by overnight dialysis (Slide-A-Lyzer MINI Units, MWCO 7000, Pierce, Rockford, IL) against one liter of water at 4°C. Samples were then mixed with a matrix solution (5 mg/ml α-CHCA in 50% acetonitrile and 0.1% TFA) and spotted onto a MALDI target plate. Mass spectra were acquired in the positive-ion linear mode on a 4700 Proteomics Analyzer (MALDI-TOF/TOF) with a Nd:YAG laser at a 200 Hz repetition rate. BSA was added as an internal standard for calibration.
ESI-LC/MS/MS Analysis
Tryptic peptides were loaded onto a PepMap C4 precolumn (300 μm × 1 mm, Dionex, Sunnyvale, CA) by a Surveyor autosampler (ThermoFisher Scientific, Waltham, MA). The precolumn was then washed with 0.1% formic acid for 5 min. A nonlinear gradient from 90% of mobile phase A (0.1% formic acid in water) and 10% of mobile phase B (50% acetonitrile/50% isopropanol, 0.1% formic acid) to 10% of A and 90% of B was applied to elute the peptides from the precolumn onto a reverse-phase C4 analytical column (75 μm × 15 cm, Dionex, Sunnyvale, CA) at a flow rate of 250 nL/min. The eluted peptides were introduced into an LTQ-Orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA) by a TriVersa™ Nanomate system (Advion, Ithaca, NY) at a constant voltage of 1.7 kV. The mass spectrometer was operated in a data-dependent acquisition mode with dynamic exclusion enabled, i.e. each full mass scan in the Orbitrap was followed by tandem mass scans (product ion analysis) of the five most intense peaks from the full mass spectrum in the ion trap. In selected cases, the most intense ion peak from the peptide fragmentation was selected for further fragmentation to provide additional sequence information (i.e., MS3 analyses). The instrument was calibrated externally following the manufacturer’s instructions. Full mass spectra were acquired in the Orbitrap mass analyzer with a target mass resolution of r = 30,000 at m/z 400 Th. MS2 spectra were acquired either in the ion trap or in the Orbitrap mass analyzer with a target mass resolution of r = 15,000 at m/z 400 Th. For MS2 and MS3 analyses, the normalized collision energy of 25% was applied, the activation time was set at 30 ms with an activation parameter q = 0.25, and precursor ions were isolated within the range of 2 and 4 Th. AGC target settings for FT full mass scan, FT MSn scan and ion trap MSn scan were 5 × 105, 2 × 105 and 1 × 104, respectively. The maximum injection time for MSn scan (FT and ion trap) and FT full mass scan were 1 s and 500 ms, respectively. The acquired data were searched against a customized protein sequence database using both SEQUEST and Mascot algorithms 39, 40. Positive search results were individually confirmed.
RESULTS
Incubation of BEL with iPLA2β Results in the Production of Two Resolvable Bands by SDS-PAGE that Each Contain BEL-linked Covalent Adducts
Intriguingly, analysis of iPLA2β incubated with racemic BEL by SDS-PAGE revealed the presence of two bands visualized by Coomassie Blue staining, whereas only one band was present in the untreated iPLA2β sample (Fig. 1A). The upper band of the BEL-treated enzyme migrated similarly as the native iPLA2β displaying a molecular weight of ~ 80 kDa. The lower band migrated more rapidly exhibiting an apparent molecular weight of 75 kDa (i.e., approximately 5 kDa less than the upper band).
Figure 1. SDS-PAGE autoradiographic analyses of BEL-treated iPLA2 β.
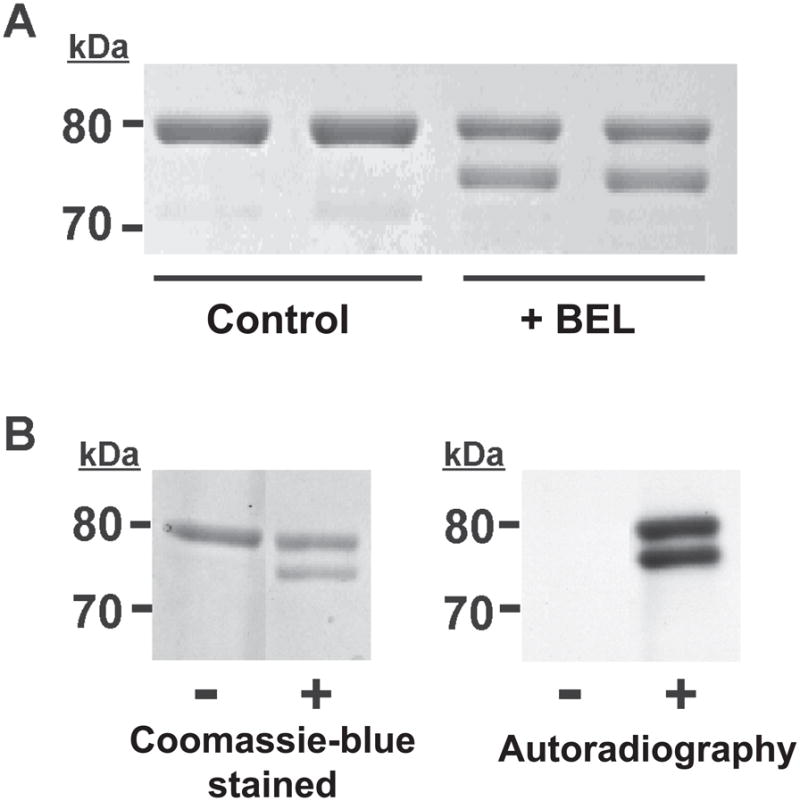
A, Resolution of BEL-treated iPLA2β into two bands by SDS-PAGE. iPLA2β was incubated with racemic BEL at 22°C for 10 min and electrophoresed on a 7% SDS-PAGE gel. The upper band has an apparent molecular weight similar to native iPLA2β (80 kDa) while the lower band has an apparent molecular weight of approximately 75 kDa. B, Radiolabeling of iPLA2β with racemic [3H]-BEL. iPLA2β was incubated with racemic [3H]-BEL at room temperature for 5 min followed by separation on a 7% SDS-PAGE gel stained with Coomassie Blue (left) followed by visualization of the radiolabeled bands by autoradiography (right). Two bands are present in the lane containing [3H]-BEL-treated iPLA2β indicating that both bands are modified by BEL. “−“ indicates protein treated with ethanol vehicle alone; “+” indicates protein treated with [3H]-BEL.
To determine whether one or both bands of the doublet observed by SDS-PAGE were covalently modified by BEL, we incubated iPLA2β with racemic [3H]-BEL, subjected the treated enzyme to SDS-PAGE, and analyzed the gel by autoradiography. An identical doublet pattern was observed after visualization by autoradiography as that present on the Coomassie blue-stained SDS-PAGE gel (Fig. 1B) indicating that [3H]-BEL was incorporated into both bands in similar amounts. Since the incorporation of radiolabeled BEL survives boiling under denaturing conditions and subsequent SDS gel electrophoresis, each band likely contains covalently bound BEL. These results suggest that the incubation of iPLA2β with BEL yields at least two covalently modified products which can be resolved by SDS-PAGE.
Western Blotting and MALDI-TOF Mass Spectrometry Demonstrate that the 75 kDa Band is Intact iPLA2β
Since SDS-PAGE and autoradiographic analyses showed that the 75 kDa band had an apparent molecular weight which was 5 kDa less than the native iPLA2β protein and was covalently modified by BEL, we postulated that the molecular weight difference could potentially result from cleavage of the iPLA2β protein backbone, perhaps through an unusual proteolytic or protein splicing event that occurred following reaction of the enzyme with BEL. To determine whether the 75 kDa band was the product of a BEL-mediated cleavage reaction, we performed Western blotting using antibodies directed against the N-terminus, middle and C-terminus of iPLA2β and measured the masses of the protein products by MALDI-TOF mass spectrometry. Western blotting demonstrated that the 75 kDa band contained the all identifiable epitopes present in the 80 kDa native protein (Fig. 2B). Thus, the de novo appearance of the 75 kDa band after incubation with BEL did not result from proteolytic cleavage and contains the C-terminus, the N-terminus as well as the internal residues 277–295 and 506–525.
Figure 2. Western blot analysis of BEL-treated iPLA2β utilizing antibodies directed against four different regions of the iPLA2β protein.
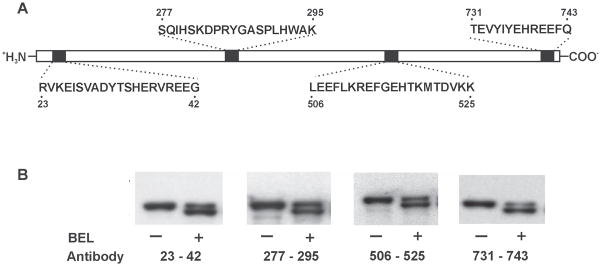
Epitopes for the four antibodies are indicated in Panel A. iPLA2β was incubated with (S)-BEL at 22°C for 3 min. Following separation on a 7% SDS-PAGE gel, the protein bands were transferred to Immobilon-P PVDF membranes and probed separately with the four different antibodies against iPLA2β in Panel B. Both native and BEL-treated iPLA2β are immunoreactive to all four antibodies. “−“ indicates the control sample and “+” indicates the BEL-treated sample.
To confirm the structural integrity of iPLA2β, racemic BEL-treated iPLA2β was directly analyzed by MALDI-TOF mass spectrometry in the linear mode. A single broad peak with a mass of 82,931 Th is observed in the mass spectrum of the native iPLA2β, which agrees well with its predicted mass (83,860 Th) (Fig. 3A). The peak is broad because the resolution of MALDI-TOF MS in the linear mode using this instrument is not sufficient to resolve the isotopologues of an 80 kDa protein or multiple protein species with small mass shifts caused by common protein modifications (e.g., deamidation, acetylation, phosphorylation, etc.). A peak with a similar shape and average mass is observed in the mass spectrum of the racemic BEL-treated iPLA2β (Fig. 3B). Close examination of these mass spectra revealed that the peak of BEL-treated iPLA2β is 318 Da greater in mass than the native protein, suggesting the covalent linkage of a single BEL-derived moiety to iPLA2β. However, since the observed peak represents a mixture of native and the BEL-modified iPLA2β and is broad due to the relatively low mass resolving power of the instrument for the small mass shift (318 Da) of the native protein, the observed mass shift does not necessarily reflect the exact mass difference caused by the modification. However, clearly, no peak with a mass ≈ 5 kDa less than the predicted peak is present in the mass spectrum. Since the mass resolution of MALDI-TOF MS in the linear mode should be sufficient to distinguish two molecular species with a low kDa mass difference, the absence of a peak with a 5 kDa lower mass suggests that the more rapidly migrating band is due to alterations in the conformation of BEL-treated iPLA2β that either modifies its interaction with the polyacrylamide matrix or changes its effective Stokes radius and does not result from cleavage of the peptide backbone of the protein.
Figure 3. MALDI-TOF mass spectrometric analyses of native and BEL-treated iPLA2β holoproteins.
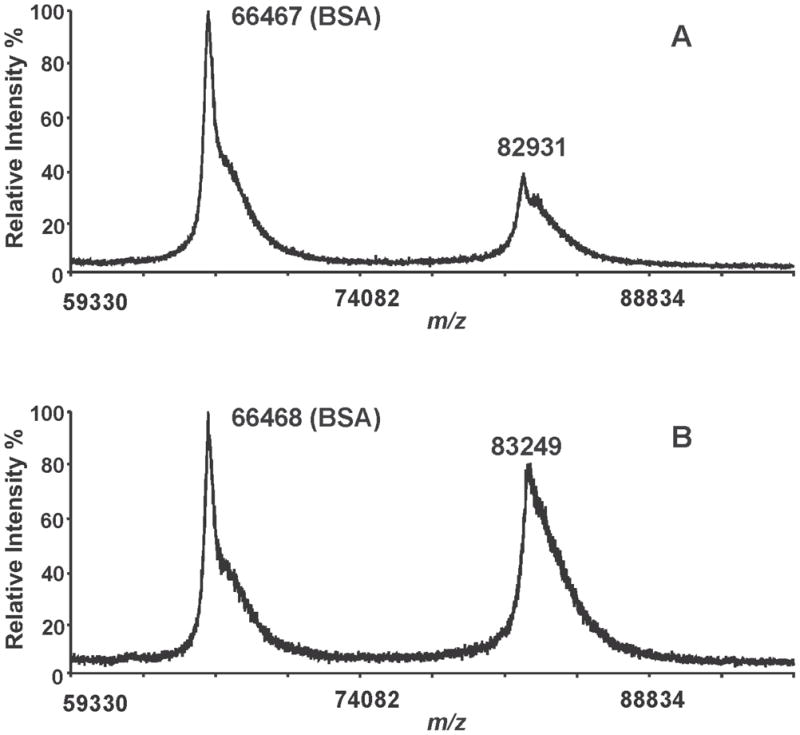
Native (A) and BEL-treated iPLA2β (B) were mixed with a MALDI matrix solution (5 mg of α-CHCA in 50% acetonitrile/0.1% TFA), and spotted onto a MALDI target plate. Bovine serum albumin was added as a molecular mass marker and the acquired mass spectra were calibrated using its monomeric and dimeric masses. A single peak corresponding to iPLA2β is observed in the BEL-treated sample and there is no indication of the presence of a peak possessing a 5 kDa mass shift. These results demonstrate that the two bands observed by SDS-PAGE of the BEL-treated iPLA2β have similar molecular weights.
Both S465 and C651 of iPLA2β Are Covalently Modified by BEL
To identify the residues in iPLA2β which are covalently modified by the mechanism-based suicide inhibitor BEL, we first incubated racemic BEL with purified recombinant iPLA2β and analyzed the tryptic peptides by LC-MS/MS. Importantly, these experiments were performed in the presence of DTT to scavenge the reactive α-bromomethyl ketone derivative of BEL which upon release from the catalytic serine may diffuse out of the active site and non-specifically modify distant iPLA2β nucleophilic residues. In the BEL-treated sample, two unique tryptic peptides were detected as triply-charged ions at m/z 880.4606 Th and 908.4530 Th, respectively. Their monoisotopic masses are 254.0939 Da greater than the expected values for the iPLA2β peptides 456DLFDWVAGTSTGGILALAILHSK478 (D456-K478) and 644SPQVPVTCVDVFRPSNPWELAK665 (S644-K665) at a mass accuracy of 3.5 ppm (Fig. 4, Panels A and D). Collision-induced dissociation (CID) of each peptide (generating MS2 spectra) revealed the presence of ions that closely correlated with those predicted for the unmodified tryptic peptides as well as the presence of unique peaks having masses 254 Da greater than anticipated.
Figure 4. Mass spectral analysis of the tryptic peptides of BEL-modified iPLA2β identifying S465 and C651 as sites of modification.

Purified recombinant iPLA2β was incubated with racemic BEL at a molar ratio of 1:1 at 22°C for 3 min. The reaction was terminated by addition of CHCl3/CH3OH, vortexing, and centrifugation to pellet the precipitated protein. The protein pellet was solubilized in buffer containing RapiGest™, digested with trypsin, and analyzed by LC/MS/MS as described in “Experimental Procedures”. A and D, Full mass spectra showing the presence of unique peaks in the BEL-treated sample in the mass range of m/z 878–887 and 905–915, respectively. B, CID mass spectrum (MS2) of the ion at m/z 880.36 (3+) which corresponds to the BEL-modified tryptic peptide, 456DLFDWVAGTSTGGILALAILHSK478. C, CID mass spectrum (MS3) of the ion at m/z 913. E, CID mass spectrum (MS2) of the ion at m/z 908.36 (3+) which corresponds to the BEL-modified tryptic peptide, 644SPQVPVTCVDVFRPSNPWELAK665. F, CID mass spectrum (MS3) of the ion at m/z 1156. MS2 and MS3 mass spectra in combination with high mass accuracy (< 5 ppm) identify that S465 and C651 are modified by an α-keto-substituted carboxylic acid leading to a mass increase of 254 kDa.
In the MS2 spectrum of the putative modified peptide D456-K478, all of the observed y ions (y15 – y21) exhibited increased masses (i.e., 254 Da greater than the expected mass values) in comparison to those anticipated for the unmodified tryptic peptide. In contrast, the masses of all observed b ions (b4 - b7, and b9) matched well with the expected values. These results indicate that the modified residue is located in the sequence of 465STGGILALAILHSK478 (Fig. 4B). To determine which residue within this sequence was covalently modified, MS3 experiments were performed. Fragmentation of the ion at m/z 933.01 Th (present in the MS2 spectrum and assigned as the doubly-charged y17 ion) demonstrated the anticipated masses in the MS3 spectrum indicating that the modification site is present in the sequence 462AGTS465 (Fig. 4C). Collectively, the MS2 and MS3 spectra identify S465, the only overlapping residue between the sequences 462AGTS465 and 465STGGILALAILHSK478, as the site of covalent modification by BEL.
Next, the ion at m/z 908.4530 Th representing the putative modified peptide S644-K665 was investigated further by MS2 and MS3 fragmentation studies. Analysis of the MS2 spectrum revealed the presence of y16 – y20 ions that each possessed a mass increase of 254 Da relative to the expected values for the unmodified peptide. These results indicate that the addition of BEL occurred in the sequence 650TCVDVFRPSNPWELAK665. Notably, the b3, b4, b6, b7, y11, y12 and y14 ions do not exhibit any shifts in mass. Collectively, these results demonstrate that C651 is the modified residue (Fig. 4). To substantiate this conclusion, we acquired the MS3 spectrum of the ion at m/z 1157.24 Th (Fig. 4). The b* and y ions observed in this MS3 spectrum unambiguously identify C651 as the site of modification. Collectively, LC-MS/MS analysis of the tryptic digest from BEL-treated iPLA2β demonstrated that both S465 and C651 were modified upon BEL treatment leading to a mass shift of 254 Da.
Our finding that C651 of iPLA2β is modified after treatment with BEL is in accordance with the results of Song et al., which demonstrated that several cysteine residues were modified by BEL in the absence of DTT and that C651 was the only modified cysteine residue in the presence of 1 mM DTT 31. In contrast to the present results, no modification of the lipase consensus sequence motif, GTSTG, was detected in those experiments in either the absence or presence of DTT.
S465 and C651 Are Covalently Linked by a Bidentate BEL Adduct
Since both S465 and C651 of iPLA2β are covalently modified by BEL and at least one product of the reaction of iPLA2β with BEL migrates more rapidly than the native protein on an SDS-PAGE gel, we hypothesized that S465 and C651 were cross-linked by BEL as a covalent bridge as proposed in the inhibition mechanism (Scheme 1, Pathway A). To investigate whether S465 and C651 were connected through a BEL-derived covalent adduct, we analyzed the tryptic digest of BEL-treated iPLA2β by LC-MS/MS specifically searching for the species corresponding to the cross-linked product of the S465-containing tryptic peptide (D456-K478, designated as peptide α) and the C651-containing tryptic peptide (S644-K665, designated as peptide β) through C16H12O2 (the formula of the BEL-derived product formed after the lactone of BEL has been hydrolyzed with subsequent elimination of the bromide). A candidate peptide was thus identified with a mass which matches that of the putative cross-linked species. This peptide was detected as an ion either with a +5 charge (the peak at m/z 1018.7330 Th) or a +6 charge (the peak at m/z 849.1084 Th) at a mass accuracy of < 3.5 ppm (Fig. 5A). The tandem mass spectra (MS2 spectra) of these two ions were then acquired in the Orbitrap mass analyzer at a resolution of 15,000 at m/z 400 Th to verify the peptide sequence and to determine which residues were covalently modified (Fig. 5B & 5C). Since the high mass accuracy product ion spectra were acquired in the Orbitrap, the charge states and the m/z values of the product ions were determined with high confidence levels ensured by the high resolution and high mass accuracy of the FT data. In the MS2 spectra, y and b ions corresponding to the fragments from peptide α (y3a through y6a, y8a, y9a, y11a through y13a ions and b3a through b9a ions) and peptide β (y11β through y14β ions, b4β, b6β and b7β ions) were observed, indicating that these two tryptic peptides are present in the putative cross-linked adduct. Peaks assigned as y14a through y20a ions all displayed a mass shift corresponding to the addition of peptide β and a fragment with a mass of 236. Correspondingly, y16β, and y18β through y20β ions all display a mass shift corresponding to the addition of peptide α and a fragment with a mass of 236 (Fig. 5B & 5C). Collectively, the assigned fragments from the MS2 mass spectra (Table I) demonstrate that S465 and C651 are cross-linked through a covalent derivative of BEL with the formula C16H12O2. The proposed structure of the cross-linked peptides and the identified fragments are illustrated in Fig. 5D. Based on the structure of the cross-linker, we estimate that the maximum distance between the sulfur of C651 and the oxygen of S465 is 9 Å.
Figure 5. Mass spectrometric evidence for the cross-linking of S465 and C651.

Trypsinized iPLA2β treated with (S)-BEL was analyzed by ESI-LC/MS/MS as described in “Experimental Procedures”. A, The survey full mass spectrum acquired using the Orbitrap mass analyzer with a resolution r = 30,000 at m/z 400 shows that the cross-linked peptide is detected as ions in two different charge states. The peaks at m/z 849.1113 and 1018.7330 represent the monoisotopic ions of the +6- or +5- charged cross-linked peptide, respectively. Each of the ions were fragmented by CID in the ion trap, and the fragments were analyzed in the Orbitrap mass analyzer with a resolution r = 15,000 at m/z 400. B, the tandem mass spectrum (MS2 spectrum) of the ion at m/z 1018.7330 Th. C, the tandem mass spectrum (MS2 spectrum) of the ion at m/z 849.1113 Th. D, the structure and the identified fragments of the cross-linked peptides. The S465-containing tryptic peptide (α) and C651-containing tryptic peptide (β) are cross-linked by a link-age between S465 and C651 via a BEL hydrolytic product.
Table 1.
Fragment Ions Identified in the Tandem Mass Spectra of the Cross-linked Peptide
| Fragments from Peptide α (456 – 478) | Fragments from Peptide β (644 – 665) | ||||
|---|---|---|---|---|---|
|
| |||||
| Ion Type | Charge State | m/z | Ion Type | Charge State | m/z |
| b3α | 1 | 376.1868 | b4β | 1 | 412.2192 |
| b4α | 1 | 491.2147 | b6β | 1 | 608.3403 |
| b5α | 1 | 677.2937 | b7β | 1 | 709.3886 |
| b6α | 1 | 776.3622 | b10β | 4 | 912.4721 |
| b7α | 1 | 847.3981 | y11β | 2 | 672.8599 |
| b8α | 1 | 904.4179 | y12β | 2 | 722.3905 |
| b9α | 1 | 1005.4678 | y13β | 2 | 779.9047 |
| y3α | 1 | 371.2044 | y14β | 2 | 829.4113 |
| y4α | 1 | 484.2885 | y16β | 4 | 1121.3323 |
| y5α | 1 | 597.3713 | y16β | 5 | 897.2646 |
| y6α | 1 | 668.4098 | y18β | 4 | 1170.3571 |
| y8α | 1 | 852.5314 | y18β | 5 | 936.4869 |
| y9α | 1 | 965.6143 | y19β | 4 | 1195.1229 |
| y11α | 1 | 1135.7209 | y20β | 5 | 981.9154 |
| y12α | 1 | 1192.7418 | |||
| y12α | 2 | 596.8738 | |||
| y13α | 1 | 1293.7811 | |||
| y13α | 2 | 647.3988 | |||
| y14α | 5 | 817.8368 | |||
| y16α | 4 | 1061.5631 | |||
| y17α | 4 | 1079.3209 | |||
| y17α | 5 | 863.6592 | |||
| y18α | 4 | 1104.0878 | |||
| y18α | 5 | 883.4738 | |||
| y19α | 4 | 1150.6109 | |||
| y19α | 5 | 920.6881 | |||
| y20α | 4 | 1179.6131 | |||
| y20α | 5 | 943.6931 | |||
| y21α | 5 | 973.1121 | |||
| y21α | 6 | 811.0901 | |||
Peptide α: 456DLFDWVAGTSTGGILALAILHSK478
Peptide β: 644SPQVPVTCVDVFRPSNPWELAK665
Differential Kinetics of Covalent Modification of S465 and C651 by (S)-BEL and (R)-BEL
Previously, it was demonstrated that BEL could be chromatographically resolved into its (S)- and (R)-enantiomers by chiral HPLC, and that (S)-BEL selectively inhibits iPLA2β activity in comparison to (R)-BEL (17). To investigate whether (S)-BEL may be selective in the covalent modification of S465 and C651, iPLA2β was treated with either (S)- or (R)-BEL, and analyzed by SDS-PAGE and ESI-LC/MS/MS. The results demonstrated that treatment of iPLA2β with either (S)- or (R)-BEL produced similar changes in iPLA2β electrophoretic behavior as racemic BEL and that both are able to modify S465 and C651.
To further scrutinize differences in enantioselectivity, we performed detailed kinetic analyses by incubating iPLA2β with (S)- or (R)-BEL for various time intervals and comparing the ratio of the two bands formed by Western analysis and correlating this to covalent alterations determined by mass spectrometry. As shown in Fig. 6A and 6B, incubations with (S)- BEL or (R)-BEL both result in a similar kinetic profile of the formation of the doublet pattern following SDS-PAGE. Notably, the intensity of the upper band decreases whereas the intensity of the lower band increases with incubation time. However, the rate at which the intensity of the lower band increases is markedly faster for (S)-BEL-treated iPLA2β than for the (R)-BEL-treated sample. For (S)-BEL, the intensity of the 75 kDa band was nearly equal to that of the 80 kDa band after only two minutes incubation (Fig. 6A), but for (R)-BEL, the intensity of the 75 kDa band increases at a slower rate. The 80 kDa band was still dominant after 60 min incubation in the presence of (R)-BEL (Fig. 6B). To quantitatively determine the time-dependence of enantioselective alterations, we measured the net intensity of each band by densitometry, normalized that value to the total net intensity of both bands in the same lane (relative intensity) and plotted the relative intensity of each band as a function of incubation time. As shown in Fig. 6C, the intensity of the 75 kDa band increased rapidly in the presence of (S)-BEL, reached a similar intensity as the 80 kDa band within two minutes, and remained at this level for the remaining incubation time examined. In contrast, for (R)-BEL, the intensity of the 75 kDa band increased slowly, and even after 60 min incubation represented only 35% of the total intensity present in the 80 kDa band.
Figure 6. Time course assay analysis of the reaction of (S)-BEL and (R)-BEL with iPLA2β as determined by SDS-PAGE and densitometry.
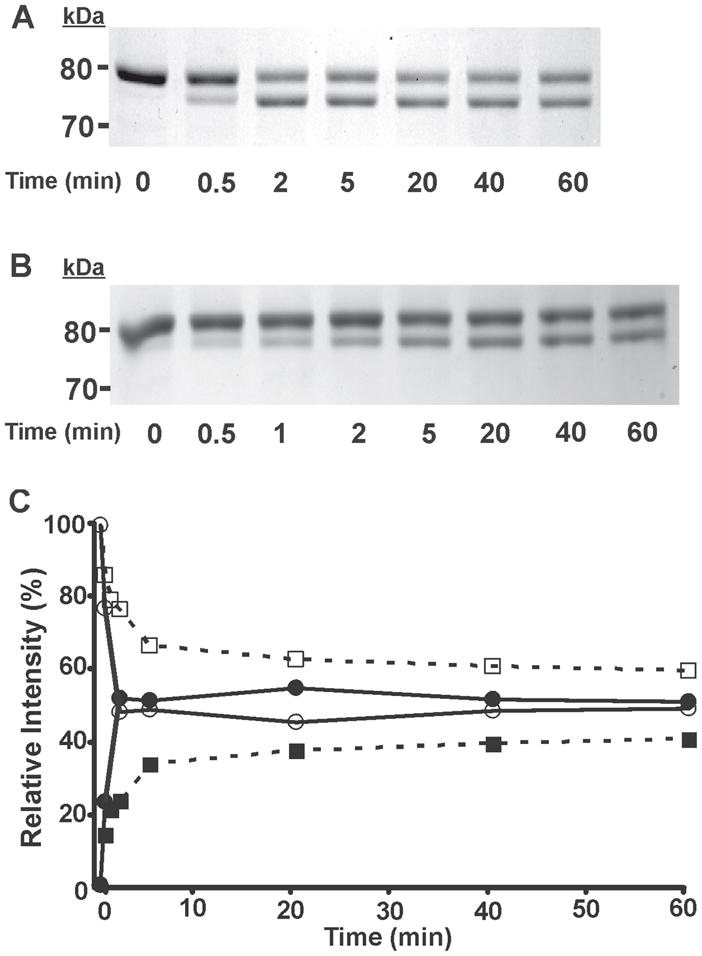
Purified recombinant iPLA2β was incubated with (S)-BEL (A) and (R)-BEL (B) at 22°C for various time intervals as indicated. The reactions were terminated by adding an equal amount of SDS-PAGE sample loading buffer (2×) and the iPLA2β adducts were separated on a 7% SDS-PAGE gel prior to visualization by Coomassie Blue staining. C, the net intensity of each protein band from A and B were measured utilizing a Kodak Image Station, normalized to the total intensity of the protein bands present in the same lane, and expressed as a percentage of the total intensity. The relative intensity of each protein band was then plotted against the incubation time. Solid circles, 75 kDa band of iPLA2β following incubation with (S)-BEL; Open circles, 80 kDa band of iPLA2β following incubation with (S)-BEL; Solid squares, 75 kDa band of iPLA2β following incubation with (R)-BEL, Open squares, 80 kDa band of iPLA2β following incubation with (R)-BEL. All experiments were performed in duplicate.
In addition, we also assessed the levels of the two modified tryptic peptides and the cross-linked peptide in (S)-BEL- and (R)-BEL-treated samples by mass spectrometry through a semi-quantitative method using the relative peak intensities of known peptides. In this method, the peak area of a target peptide is normalized against the average peak area of a group of representative iPLA2β peptides that serve as endogenous internal standards, and the resultant number (normalized relative abundance) represents the level of the target peptide in a sample. Comparison of the normalized relative abundance of a target peptide in different samples would provide insight into how different reaction conditions affect the level of this peptide. The normalized peak areas of the two modified peptides and the cross-linked peptide from (S)-BEL- or (R)-BEL-treated samples were calculated as described in “Experimental Procedures” and depicted in Fig. 7.
Figure 7. Comparison of the extent of iPLA2β modification by (S)-BEL and (R)-BEL.
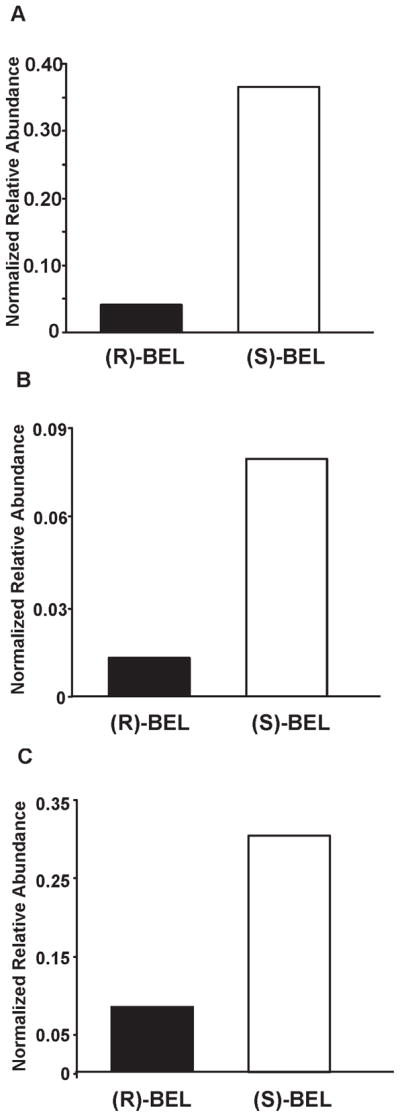
An equal amount of purified iPLA2β was incubated with either (S)-BEL or (R)-BEL at a molar ratio of 1:1 at 22°C for 3 min. The reaction was terminated by precipitating the protein with chloroform/methanol. The protein pellet was solubilized in buffer containing RapiGest™, digested with trypsin, and analyzed by LC/MS/MS as described in “Experimental Procedures”. The normalized relative abundance of a target peptide was calculated as described in “Experimental Procedures”. A, the normalized relative abundance of the BEL-modified S465-containing tryptic peptide (D456-K478) in (R)-BEL- or (S)-BEL-treated samples. B, the normalized relative abundance of the BEL modified C651-containing tryptic peptide (S644-K665) in (R)-BEL- or (S)-BEL-treated samples. C, the normalized relative abundance of the cross-linked peptide in (R)-BEL or (S)-BEL-treated samples. The results indicate that treatment with (S)-BEL results in a higher level of protein modification. By examining the changes in band intensity following SDS-PAGE in the time-course experiment and comparing the levels of modified S465 and C651 as determined by ESI-LC/MS/MS, we found that (S)-BEL is more efficient at covalent modification of iPLA2β than (R)-BEL. These observations are in good agreement with the fact that (S)-BEL is a selective inhibitor of iPLA2β, and demonstrate the enantioselectivity of (S)- vs. (R)-BEL in the kinetics of covalent modification of iPLA2β leading to inhibition of enzymic activity.
As shown in the figure, the normalized peak area of the adducted S465-containing peptide is ten times higher in the (S)-BEL-treated sample than in the (R)-BEL treated sample (Fig. 7A, 0.35 vs. 0.035). Similarly, a nine-fold difference was observed for the modified C651-containing peptide and four-fold difference for the cross-linked peptide from the (S)-BEL-treated sample relative to the (R)-BEL-treated iPLA2β (Fig. 7B, 0.009 vs. 0.001 and 0.30 vs. 0.08). This difference likely reflects the stereoelectronic characteristics and interactions of (S)-BEL and (R)-BEL at the active site and their differential ability to be catalytically cleaved to modify iPLA2β.
C651 is Acylated in vitro by Oleoyl-CoA
Previously, we demonstrated that purified recombinant iPLA2β possesses palmitoyl-CoA hydrolase activity 34 in addition to its well-characterized lysophospholipase and phospholipase A2 activities 41, 42. Importantly, the acyl-CoA thioesterase activity of iPLA2β is completely dependent upon the presence of the catalytic serine (S465) of the enzyme 34. Moreover, iPLA2β is covalently autoacylated in a highly substrate-specific manner (by oleoyl- but not palmitoyl-CoA), which occurs at a second site distinct from the hydrolytic lipase site (GXSXG) 34. Therefore, iPLA2β modulates multiple metabolic alterations resulting from the production of lipid 2nd messengers (e.g., arachidonic acid and lysolipids) or alternatively through hydrolysis or transacylation of saturated acyl-CoAs from specific cellular membrane compartments.
Since localization of the acylation site would provide insight into the mechanism of acyl-CoA hydrolysis and iPLA2β structure, we incubated iPLA2β in the presence and absence of oleoyl-CoA followed by precipitation, solubilization, and trypsinolysis of the protein prior to analysis of the resultant peptides by LC/MS/MS as described in “Experimental Procedures.” A peptide with a mass 264 Da greater than the expected mass of 644SPQVPVTCVDVFRPSNPWELAK665 (Fig. 8A) was identified indicative of the addition of an oleoylate moiety to the peptide. The CID mass spectrum (MS2) of this peptide revealed several fragments whose m/z values correspond well with the b3 and b4 ions predicted for the unmodified tryptic peptides as well as those whose m/z values exhibit a mass increase of 264 Th greater than the predicted values for the unmodified peptides (y15, y16, y18, y19 and y20 ions). The MS2 spectrum of the S644-K665 peptide confirms its sequence identity, but does not provide sufficient information for us to determine the modification site (Fig. 8B). Therefore, we performed MS3 analysis of the ion at m/z 1161.8 from the MS2 spectrum to identify the residue modified by oleic acid. Because the m/z values of all the y ions (y9 – y14) match the predicted values for the unmodified peptide while the ion at m/z 1161.8 represents a modified fragment with a mass increase of 264 Th greater than the control fragment, we concluded that C651 is modified by an oleate group (Fig. 8C). Collectively, these results identify C651 as a highly reactive nucleophilic residue which is in close spatial proximity with the catalytic serine of iPLA2β (S465) and is a target for both alkylation by the α-bromomethyl ketone of BEL as well as acylation by oleoyl-CoA.
Figure 8. Identification of Covalent Oleoylation of C651 in iPLA2β by Oleoyl-CoA.

A, Full scan mass spectrum (m/z 350 – 1600) at 112.4 min of the LC-MS/MS analysis of the tryptic digest from iPLA2β treated with oleoyl-CoA in vitro. A triply-charged ion of m/z 911.8425 was observed with a resolution power of 30000 at m/z 400. The calculated mass of this ion (2732.5041 Th) matches the mass of the oleoylated 644SPQVPVTCVDVFRPSNPWELAK665 with a mass accuracy of 3 ppm. B, the ion of m/z 911.8425 was selected for CID (collision-induced dissociation) fragmentation. The resulting fragments (y and b ions as labeled) confirm the sequence of the peptide as 644SPQVPVTCVDVFRPSNPWELAK665 and localize the modification site as C651. C, the ion of m/z 1161.8 (the base peak in panel B) from the fragment of 911.8425 was selected for further fragmentation by CID (MS3). The fragmentation pattern of this ion substantiates the identity of the oleoylated peptide and the modification site.
DISCUSSION
Since its initial discovery as a selective and potent mechanism-based inhibitor for iPLA2β, BEL has been widely used to explore the multiple biologic functions of iPLA2β-mediated lipid 2nd messenger generation, calcium signaling, and cellular growth, proliferation and differentiation mediated by this enzyme 2–5, 8, 10, 15, 35, 43, 44. Thus, a detailed understanding of the mechanism through which iPLA2β is inactivated by BEL, the covalent adducts which are formed, and the fate of the highly reactive α-bromomethylketone under different redox conditions is central to defining the specificity and selectivity of this inhibitor. Accordingly, we utilized high mass accuracy mass spectrometry in conjunction with complimentary protein analytic techniques to identify three distinct BEL adducts of iPLA2β including a covalently cross-linked product demonstrating the close spatial proximity of the catalytic active site serine (S465) to C651 through a BEL-tethered adduct linking these residues. Examination of the enantioselective mechanism-based inhibition of iPLA2β with (R)-BEL and (S)-BEL in the current study revealed that the kinetics of covalent modification of iPLA2β with each of the BEL enantiomers was consistent with our previous observations demonstrating an order of magnitude difference in the potency of (S)-BEL to inhibit iPLA2β catalytic activity vs. (R)-BEL 35. The nucleophilic reactivity of C651 within the active site was further established by the identification of this residue as a site of covalent fatty acylation of iPLA2β by oleoyl-CoA as we previously reported 34.
Although the mechanism of inhibition of iPLA2s by BEL has been proposed to be similar to that previously elaborated by Katzenellenbogen et al. for inhibition of chymotrypsin by haloenol lactone suicide substrate inhibitors 21, 45–48, important questions remained regarding the chemical identity of reaction intermediates and the modified residues of iPLA2β. Nucleophilic attack of the carbonyl of the BEL bromoenol lactone ring by the catalytic serine (S465) of iPLA2β results in the formation of a highly reactive α-bromomethyl ketone adduct (Step I of Scheme 1). This highly reactive intermediate can either 1) react with a nucleophilic residue within the active site to form a tethered intermediate (Step II of Scheme 1) which can undergo hydrolysis at S465 (Step III of Scheme 1); 2) undergo elimination of the bromide (with water as the attacking nucleophile)(Step II′ of Scheme 1); or 3) undergo hydrolysis to release the α-bromomethyl ketone carboxylic acid which diffuses out of the active site to react with distal cysteine residues on the surface of the enzyme (Step V of Scheme 1). In our early work, we calculated that the average enzyme turnover number for inactivation of cytosolic myocardial iPLA2 by BEL is approximately 10 mol of BEL for every mole of phospholipase A2 and that the stoichiometry of BEL to fully inhibited iPLA2β is approximately 1.2 to 1 (BEL:enzyme ratio) 22. These data indicate that a substantial fraction of the α-bromomethyl keto carboxylic acid derivative of BEL leaves the catalytic site and diffuses into the surrounding solution. Thus, strong nucleophilic agents such as DTT or glutathione are required to rapidly scavenge the released α-bromomethyl ketone derivative of BEL to prevent extensive non-specific modification of iPLA2 surface cysteine residues and/or the inhibition of other enzymes. In initial in vitro studies, we utilized DTT to rapidly inactivate the released α-bromomethyl keto carboxylic acid derivative of BEL which diffused out of the active site 23, 49. We specifically point out that DTT does not completely attenuate BEL inhibition of iPLA2β, presumably due to its inability to effectively interact with the α-bromomethyl keto carboxylic acid derivatives of BEL generated and tethered to the active site.
Previously, Song and coworkers 31 reported that most cysteine residues of iPLA2β were modified upon treatment with BEL in the absence of DTT and believed that neither the catalytic site, S465, nor any adjacent residues were modified. These observations led to their proposal that inactivation occurs by release of ring opened BEL by diffusion of the α-bromomethyl keto carboxylic acid through alkylating cysteine residues (mechanism B in Scheme 1) many of which are distal from the active site. Furthermore, Song et al. reported that C651 was the only cysteine residue modified by BEL in the presence of DTT (1 mM), but its alkylation by BEL was not sufficient to completely inactivate iPLA2β activity. From this, they concluded that DTT could protect the enzyme from being inactivated by BEL through decreasing cysteine alkylation at sites distant to the active site. However, the direct demonstration of BEL attached to the active site serine and a cross-linked product with C651 identify a different mechanism responsible for iPLA2β inactivation by BEL.
Specifically, the results of the current study clearly demonstrate that S465 is modified as a BEL adduct, leading to a mass shift of 254 Da. This is of particular relevance since site directed mutagenesis of S465 to alanine abolishes the phospholipase41 and acyl-CoA hydrolase36 activities of iPLA2β. In addition, we provide evidence that reaction of iPLA2β with BEL results in the formation of a tethered adduct between S465 and C651. The absence of the BEL modified S465 containing peptide or the S465-BEL-C651 species in the work of Song et al.31 may have been due to their loss during workup using different isolation conditions or due to the low specific activity (~ 600 pmol fatty acid•min−1•mg−1 protein) of iPLA2β used in their study vs. ~ 1 μmol fatty acid•min−1•mg−1 protein used in the current study. The BEL-modified S465 and the cross-linked S465-BEL-C651 species are stable acylenzyme intermediates in the inactivation pathway as evidenced by their identification by tandem mass spectrometry, MALDI-TOF and a doublet present on SDS-PAGE after high temperature denaturation. The stability of these intermediates may be attributed to a slow or disabled deacylation step as has been demonstrated from studies on serine proteases whose structural and catalytic features are well established and have been shown to exhibit a high degree of structural homology with the patatin family of phospholipases/lipases 50–53. In a study on the inactivation mechanism of chymotrypsin by haloenol lactones, the deacylation of the active site serine was demonstrated to be the rate-limiting step in the pathway, and the half life of a stable acyl enzyme could be as long as 12.5 hrs 46. An acyl-enzyme intermediate formed between porcine pancreatic elastase and its natural substrate, β-casomorphin-7, was detected by mass spectrometry, crystallized and characterized for greater understanding of the catalytic mechanism 54. These studies suggested that the deacylation of the acyl enzyme intermediate requires the participation of a “hydrolytic water” molecule, which is appropriately positioned and activated by a nearby residue to generate a hydroxide ion which attacks the carbonyl carbon of the ester linkage. Following the hydrolysis of the bromoenol lactone by S465, the ring opened product allows greater mobility and flexibility of the BEL adduct within the active site. The position of this tethered adduct may displace hydrolytic water molecules or block their entrance or exit from the active site. As a result of these conformational changes, the carbonyl carbon of the ester linkage may not be susceptible to hydrolysis. The observation of an acyl enzyme intermediate formed with oleoyl-CoA that is stable on a catalytic timescale suggests that intramolecular transacylation could influence iPLA2β substrate specificity, kinetics, intermolecular interactions and membrane localization each resulting in novel modes of enzyme regulation.
The results presented in the current study are therefore consistent with mechanism A (Scheme 1) (i.e., a large fraction is cross-linked internally while tethered) while other reactive sites are also modified without departure from the active site. Specifically, the BEL derivative remains tethered to the active site serine (S465) through an ester linkage after the hydrolysis of the lactone ring, and this tethered species is poised to alkylate an accessible nucleophile within the active site, forming an irreversibly inactivated enzyme (Scheme 1, Step II). This cross-linked intermediate can also undergo a subsequent deacylation step leading to regeneration of the active site serine (Scheme 1, Step III). The three modification products identified in the present study implicate the thiol of C651 (-SH) and water as the reactive nucleophiles. The reaction with the thiol generates a modified C651 and the cross-linked products (Scheme 1, Steps II and III) while the reaction with water facilitated by H-bonding leads to debromination of the α-bromomethyl ketone derivative of BEL which remains esterified to S465 as a stable adduct (Scheme 1, Step II′). Formation of any one of the three modified products results in an inactive enzyme.
The modification of C651 by BEL and the presence of a BEL adduct cross-linking C651 to S465 indicates that C651 is in close spatial proximity to the active site serine. Based upon the mass shift and the proposed inactivation mechanism, we were able to determine the structure of the cross-link and the maximum distance between C651 and S465 as approximately 9 Å. Considering its close spatial proximity to the active site serine and its strong nucleophilicty, this cysteine residue may play an important role in regulating iPLA2 activity. Moreover, C651 is localized between two calmodulin binding motifs in the primary sequence of iPLA2β, i.e. a “1-9-14” motif (622IRKGQGNKVKKLSI635) and an IQ motif (701IQYFRLNPQLGSDI714), indicating that it may have a role in regulating inhibition of phospholipase activity by calmodulin 32.
Previous studies have demonstrated that (S)-BEL is a more potent inhibitor of iPLA2β than (R)-BEL 35. To investigate potential correlations between their inhibitory potencies toward iPLA2β and their abilities to covalently modify the enzyme, we compared the levels of the modified peptides in (S)-BEL and (R)-BEL-treated iPLA2β samples through a mass spectrometric-based semi-quantitative approach. The results showed that the levels of the modified peptides are significantly higher in (S)-BEL-treated iPLA2β than in the (R)-BEL-treated enzyme (i.e., the BEL-modified S465-containing tryptic peptide, the BEL-modified C651-containing tryptic peptide, and the cross-linked peptide are ten, six, and four times more abundant, respectively). The data indicate that the high potency of (S)-BEL against iPLA2β positively correlates with its ability to generate stable BEL modified peptides. The ability of (S)-BEL and (R)-BEL to covalently modify iPLA2β likely reflects differences in their diastereotopic interactions with the enzyme active site, which would affect: 1) binding affinity of BEL or phospholipid substrate; 2) the rate of formation of the acylenzyme intermediate; 3) the dwell time of the hydrolyzed inhibitor in the active site; and 4) the rate of deacylation. Future studies utilizing molecular models of iPLA2β based upon the crystal structures of patatin-like enzymes in combination with computational simulations using BEL enantiomers as probes will likely provide further insight into the enantioselectivity of BEL for other members of the iPLA2 family of enzymes.
In conclusion, this study has evaluated two distinct mechanisms for the inactivation of iPLA2β by its mechanism-based inhibitor, BEL. The results from mass spectrometric and SDS-PAGE analyses support a mechanism by which a stable acyl-enzyme intermediate is formed (after the lactone of BEL is hydrolyzed) by the enzyme and subsequent reactions of this intermediate with nearby nucleophilic residues in the active site resulting in inhibition of iPLA2β activity. The identification of nearly 50% cross-linked peptides by gel electrophoresis not only demonstrates the existence of the acyl-enzyme intermediate, but also reveals the spatial proximity of C651 to the active site S465 and that this interaction accounts for a substantive portion of BEL-mediated inactivation. Moreover, the inhibitory effects of (S)-BEL and (R)-BEL on iPLA2β have been shown to correlate with their abilities to covalently modify the enzyme. The current studies facilitate our understanding of the catalytic mechanism of PLA2β, demonstrate the active site connectivity with C651 and confirm the utility of BEL in identifying iPLA2β-mediated alterations in lipid metabolism that can be substantiated by complimentary genetic loss of function experiments as we have previously demonstrated 17, 44, 55.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grants 5PO1HL57278 and RO1HL41250. R. W. G. has financial relationships with LipoSpectrum and Platomics.
ABBREVIATIONS
- BEL
(E)-6-(bromomethylene)-3-(1-naphthalenyl)-2H-tetrahydropyran-2-one
- α-CHCA
α-cyano-4-hydroxycinnamic acid
- DTT
dithiothreitol
- ESI LC-MS
electrospray ionization liquid chromatography-mass spectrometry
- iPLA2
calcium-independent phospholipase A2
- MALDI-TOF
matrix-assisted laser desorption/ionization time-of-flight
- TFA
trifluoroacetic acid
References
- 1.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the Group IV phospholipase A2 family. Prog Lipid Res. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Hooks SB, Cummings BS. Role of Ca2+-independent phospholipase A2 in cell growth and signaling. Biochem Pharmacol. 2008;76:1059–1067. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green JT, Orr SK, Bazinet RP. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J Lipid Res. 2008;49:939–944. doi: 10.1194/jlr.R700017-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Jenkins CM, Cedars A, Gross RW. Eicosanoid signalling pathways in the heart. Cardiovasc Res. 2009;82:240–249. doi: 10.1093/cvr/cvn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog Lipid Res. 2011;50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res. 2006;47:1940–1949. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Wilkins WP, 3rd, Barbour SE. Group VI phospholipases A2: homeostatic phospholipases with significant potential as targets for novel therapeutics. Curr Drug Targets. 2008;9:683–697. doi: 10.2174/138945008785132385. [DOI] [PubMed] [Google Scholar]
- 9.Kienesberger PC, Oberer M, Lass A, Zechner R. Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res. 2009;50(Suppl):S63–68. doi: 10.1194/jlr.R800082-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehman JJ, Brown KA, Ramanadham S, Turk J, Gross RW. Arachidonic acid release from aortic smooth muscle cells induced by [Arg8]vasopressin is largely mediated by calcium-independent phospholipase A2. J Biol Chem. 1993;268:20713–20716. [PubMed] [Google Scholar]
- 11.Gross RW, Ramanadham S, Kruszka KK, Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry. 1993;32:327–336. doi: 10.1021/bi00052a041. [DOI] [PubMed] [Google Scholar]
- 12.Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273:13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 13.Roshak AK, Capper EA, Stevenson C, Eichman C, Marshall LA. Human calcium-independent phospholipase A2 mediates lymphocyte proliferation. J Biol Chem. 2000;275:35692–35698. doi: 10.1074/jbc.M002273200. [DOI] [PubMed] [Google Scholar]
- 14.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2a inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J Biol Chem. 2000;275:18248–18258. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 15.Su X, Mancuso DJ, Bickel PE, Jenkins CM, Gross RW. Small interfering RNA knockdown of calcium-independent phospholipases A2 b or g inhibits the hormone-induced differentiation of 3T3-L1 preadipocytes. J Biol Chem. 2004;279:21740–21748. doi: 10.1074/jbc.M314166200. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X, Wang D, Zhao Z, Xiao Y, Sengupta S, Zhang R, Lauber K, Wesselborg S, Feng L, Rose TM, Shen Y, Zhang J, Prestwich G, Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–29368. doi: 10.1074/jbc.M513105200. [DOI] [PubMed] [Google Scholar]
- 17.Moon SH, Jenkins CM, Mancuso DJ, Turk J, Gross RW. Smooth muscle cell arachidonic acid release, migration, and proliferation are markedly attenuated in mice null for calcium-independent phospholipase A2b. J Biol Chem. 2008;283:33975–33987. doi: 10.1074/jbc.M805817200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Zhao Z, Wei G, Yan L, Wang D, Zhang H, Sandusky GE, Turk J, Xu Y. Group VIA phospholipase A2 in both host and tumor cells is involved in ovarian cancer development. FASEB J. 2010;24:4103–4116. doi: 10.1096/fj.10-161356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Xie Z, Zhao Q, Pang H, Turk J, Calderon L, Su W, Zhao G, Xu H, Gong MC, Guo Z. Smooth muscle-specific expression of calcium-independent phospholipase A2b (iPLA2b) participates in the initiation and early progression of vascular inflammation and neointima formation. J Biol Chem. 2012;287:24739–24753. doi: 10.1074/jbc.M112.340216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song H, Wohltmann M, Tan M, Bao S, Ladenson JH, Turk J. Group VIA PLA2 (iPLA2b) is activated upstream of p38 mitogen-activated protein kinase (MAPK) in pancreatic islet b-cell signaling. J Biol Chem. 2012;287:5528–5541. doi: 10.1074/jbc.M111.285114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniels SB, Cooney E, Sofia MJ, Chakravarty PK, Katzenellenbogen JA. Haloenol lactones. Potent enzyme-activated irreversible inhibitors for a-chymotrypsin. J Biol Chem. 1983;258:15046–15053. [PubMed] [Google Scholar]
- 22.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 23.Zupan LA, Weiss RH, Hazen SL, Parnas BL, Aston KW, Lennon PJ, Getman DP, Gross RW. Structural determinants of haloenol lactone-mediated suicide inhibition of canine myocardial calcium-independent phospholipase A2. J Med Chem. 1993;36:95–100. doi: 10.1021/jm00053a012. [DOI] [PubMed] [Google Scholar]
- 24.Alzola E, Perez-Etxebarria A, Kabre E, Fogarty DJ, Metioui M, Chaib N, Macarulla JM, Matute C, Dehaye JP, Marino A. Activation by P2X7 agonists of two phospholipases A2 (PLA2) in ductal cells of rat submandibular gland. Coupling of the calcium-independent PLA2 with kallikrein secretion. J Biol Chem. 1998;273:30208–30217. doi: 10.1074/jbc.273.46.30208. [DOI] [PubMed] [Google Scholar]
- 25.Akiba S, Mizunaga S, Kume K, Hayama M, Sato T. Involvement of group VI Ca2+-independent phospholipase A2 in protein kinase C-dependent arachidonic acid liberation in zymosan-stimulated macrophage-like P388D1 cells. J Biol Chem. 1999;274:19906–19912. doi: 10.1074/jbc.274.28.19906. [DOI] [PubMed] [Google Scholar]
- 26.McHowat J, Creer MH. Calcium-independent phospholipase A2 in isolated rabbit ventricular myocytes. Lipids. 1998;33:1203–1212. doi: 10.1007/s11745-998-0324-5. [DOI] [PubMed] [Google Scholar]
- 27.Carnevale KA, Cathcart MK. Calcium-independent phospholipase A2 is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J Immunol. 2001;167:3414–3421. doi: 10.4049/jimmunol.167.6.3414. [DOI] [PubMed] [Google Scholar]
- 28.Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006;58:591–620. doi: 10.1124/pr.58.3.7. [DOI] [PubMed] [Google Scholar]
- 29.Gaposchkin DP, Farber HW, Zoeller RA. On the importance of plasmalogen status in stimulated arachidonic acid release in the macrophage cell line RAW 264.7. Biochim Biophys Acta. 2008;1781:213–219. doi: 10.1016/j.bbalip.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Saab-Aoude S, Bron AM, Creuzot-Garcher CP, Bretillon L, Acar N. A mouse model of in vivo chemical inhibition of retinal calcium-independent phospholipase A2 (iPLA2) Biochimie. 2013;95:903–911. doi: 10.1016/j.biochi.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Song H, Ramanadham S, Bao S, Hsu FF, Turk J. A bromoenol lactone suicide substrate inactivates group VIA phospholipase A2 by generating a diffusible bromomethyl keto acid that alkylates cysteine thiols. Biochemistry. 2006;45:1061–1073. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins CM, Wolf MJ, Mancuso DJ, Gross RW. Identification of the calmodulin-binding domain of recombinant calcium-independent phospholipase A2b. implications for structure and function. J Biol Chem. 2001;276:7129–7135. doi: 10.1074/jbc.M010439200. [DOI] [PubMed] [Google Scholar]
- 33.Durley RC, Parnas BL, Weiss RH. Synthesis of high specific activity tritium labeled (E)-6-(bromoethylene)-tetrahydro-3-(1-naphthalenyl)-2H-pyran-2-one as a selective probe for calcium-independent phospholipase A2. J Label Comp Radiopharm. 1992;31:685–691. [Google Scholar]
- 34.Jenkins CM, Yan W, Mancuso DJ, Gross RW. Highly selective hydrolysis of fatty acyl-CoAs by calcium-independent phospholipase A2b : Enzyme autoacylation and acyl-CoA-mediated reversal of calmodulin inhibition of phospholipase A2 activity. J Biol Chem. 2006 doi: 10.1074/jbc.M511623200. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2) b, and not iPLA2g, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J Biol Chem. 2002;277:32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 36.Jenkins CM, Yan W, Mancuso DJ, Gross RW. Highly selective hydrolysis of fatty acyl-CoAs by calcium-independent phospholipase A2b. Enzyme autoacylation and acyl-CoA-mediated reversal of calmodulin inhibition of phospholipase A2 activity. J Biol Chem. 2006;281:15615–15624. doi: 10.1074/jbc.M511623200. [DOI] [PubMed] [Google Scholar]
- 37.Yan W, Jenkins CM, Han X, Mancuso DJ, Sims HF, Yang K, Gross RW. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2g: identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. J Biol Chem. 2005;280:26669–26679. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- 38.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 39.Eng JK, Mccormack AL, Yates JR. An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database. J Am Soc Mass Spectr. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 40.Sadygov RG, Cociorva D, Yates JR., 3rd Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- 41.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 42.Wolf MJ, Gross RW. Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J Biol Chem. 1996;271:30879–30885. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]
- 43.Mancuso DJ, Abendschein DR, Jenkins CM, Han X, Saffitz JE, Schuessler RB, Gross RW. Cardiac ischemia activates calcium-independent phospholipase A2b, precipitating ventricular tachyarrhythmias in transgenic mice: rescue of the lethal electrophysiologic phenotype by mechanism-based inhibition. J Biol Chem. 2003;278:22231–22236. doi: 10.1074/jbc.C300033200. [DOI] [PubMed] [Google Scholar]
- 44.Dietrich HH, Abendschein DR, Moon SH, Nayeb-Hashemi N, Mancuso DJ, Jenkins CM, Kaltenbronn KM, Blumer KJ, Turk J, Gross RW. Genetic ablation of calcium-independent phospholipase A2b causes hypercontractility and markedly attenuates endothelium-dependent relaxation to acetylcholine. Am J Physiol Heart Circ Physiol. 2010;298:H2208–2220. doi: 10.1152/ajpheart.00839.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakravarty PK, Krafft GA, Katzenellenbogen JA. Haloenol lactones: enzyme-activated irreversible inactivators for serine proteases. Inactivation of a-chymotrypsin. J Biol Chem. 1982;257:610–612. [PubMed] [Google Scholar]
- 46.Sofia MJ, Katzenellenbogen JA. Enol lactone inhibitors of serine proteases. The effect of regiochemistry on the inactivation behavior of phenyl-substituted (halomethylene)tetra- and -dihydrofuranones and (halomethylene)tetrahydropyranones toward a-chymotrypsin: stable acyl enzyme intermediate. J Med Chem. 1986;29:230–238. doi: 10.1021/jm00152a011. [DOI] [PubMed] [Google Scholar]
- 47.Boulanger WA, Katzenellenbogen JA. 5-(Halomethyl)-2-pyranones as irreversible inhibitors of a-chymotrypsin. J Med Chem. 1986;29:1483–1487. doi: 10.1021/jm00158a027. [DOI] [PubMed] [Google Scholar]
- 48.Daniels SB, Katzenellenbogen JA. Halo enol lactones: studies on the mechanism of inactivation of a-chymotrypsin. Biochemistry. 1986;25:1436–1444. doi: 10.1021/bi00354a037. [DOI] [PubMed] [Google Scholar]
- 49.Hazen SL, Gross RW. Human myocardial cytosolic Ca2+-independent phospholipase A2 is modulated by ATP. Concordant ATP-induced alterations in enzyme kinetics and mechanism-based inhibition. Biochem J. 1991;280(Pt 3):581–587. doi: 10.1042/bj2800581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trimble LA, Street IP, Perrier H, Tremblay NM, Weech PK, Bernstein MA. NMR structural studies of the tight complex between a trifluoromethyl ketone inhibitor and the 85-kDa human phospholipase A2. Biochemistry. 1993;32:12560–12565. doi: 10.1021/bi00210a002. [DOI] [PubMed] [Google Scholar]
- 51.Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999;97:349–360. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- 52.Rydel TJ, Williams JM, Krieger E, Moshiri F, Stallings WC, Brown SM, Pershing JC, Purcell JP, Alibhai MF. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry. 2003;42:6696–6708. doi: 10.1021/bi027156r. [DOI] [PubMed] [Google Scholar]
- 53.Bottoms CA, White TA, Tanner JJ. Exploring structurally conserved solvent sites in protein families. Proteins. 2006;64:404–421. doi: 10.1002/prot.21014. [DOI] [PubMed] [Google Scholar]
- 54.Wilmouth RC, Clifton IJ, Robinson CV, Roach PL, Aplin RT, Westwood NJ, Hajdu J, Schofield CJ. Structure of a specific acyl-enzyme complex formed between b-casomorphin-7 and porcine pancreatic elastase. Nat Struct Biol. 1997;4:456–462. doi: 10.1038/nsb0697-456. [DOI] [PubMed] [Google Scholar]
- 55.Moran JM, Buller RM, McHowat J, Turk J, Wohltmann M, Gross RW, Corbett JA. Genetic and pharmacologic evidence that calcium-independent phospholipase A2b regulates virus-induced inducible nitric-oxide synthase expression by macrophages. J Biol Chem. 2005;280:28162–28168. doi: 10.1074/jbc.M500013200. [DOI] [PubMed] [Google Scholar]