

Abstract
Progressive supranuclear palsy (PSP) is a relatively common neurodegenerative tauopathy clinically characterized by parkinsonism, axial rigidity, and supranuclear gaze palsy. Pathologic findings of PSP are neuronal loss, gliosis, and neurofibrillary tangles in basal ganglia, diencephalon, and brainstem; there is increasing recognition of clinicopathologic variants of PSP.1
Progressive supranuclear palsy (PSP) is a relatively common neurodegenerative tauopathy clinically characterized by parkinsonism, axial rigidity, and supranuclear gaze palsy. Pathologic findings of PSP are neuronal loss, gliosis, and neurofibrillary tangles in basal ganglia, diencephalon, and brainstem; there is increasing recognition of clinicopathologic variants of PSP.1
PSP is usually a sporadic condition; however, there are reports of familial aggregation in PSP.2 Recently, a genome-wide association study (GWAS) identified several genes associated with increased risk of PSP.3 This report describes clinical, pathologic, and genetic differences in familial and sporadic autopsy-proven PSP.
Methods.
We identified cases of autopsy-proven PSP with family history of neurodegenerative disease, including PSP, parkinsonism, or dementia from available medical records in the brain bank at Mayo Clinic in Jacksonville, FL. Cases considered to have a positive family history were those who had at least one first- or second-degree relative with parkinsonism or dementia. We excluded cases with pathogenic mutations in MAPT. All brains were acquired with appropriate ethical approval, and all autopsies were approved by legal next of kin. Available medical records were abstracted for clinical information in a consecutive series of cases from the period 1998 to 2008, including cardinal clinical features of PSP, such as parkinsonism and unexplained falls, as well as other notable neurologic findings. All cases had standardized neuropathologic evaluation as part of CurePSP brain bank, including assessment of neuronal and glial tau pathology with phospho-tau immunohistochemistry in 21 brain regions.4 Atypical PSP included cases with extensive cortical involvement or paucity of tau pathology in cardinal regions.1 For each case, DNA was extracted from frozen brain tissue and genotyped for APOE and MAPT. A subset of PSP cases (n = 375) was included in the CurePSP GWAS3 (tables e-1 and e-2 on the Neurology® Web site at www.neurology.org). MAPT H1/H2 was determined from a single nucleotide polymorphism, rs8070723. For comparison, genotypes were available on healthy, living control subjects collected from neurologically normal family members or caregivers of patients with Parkinson disease followed at Mayo Clinic Florida.
Student t test was used to compare continuous variables. Fisher exact test or χ2 test was used to compare group differences for categorical variables. Pathologic tau lesion scores were compared with the Mann-Whitney rank sum test. Statistical significance was considered for p values <0.05. Statistical analysis was performed with SigmaPlot version 11.0 (Systat Software Inc., San Jose, CA).
Results.
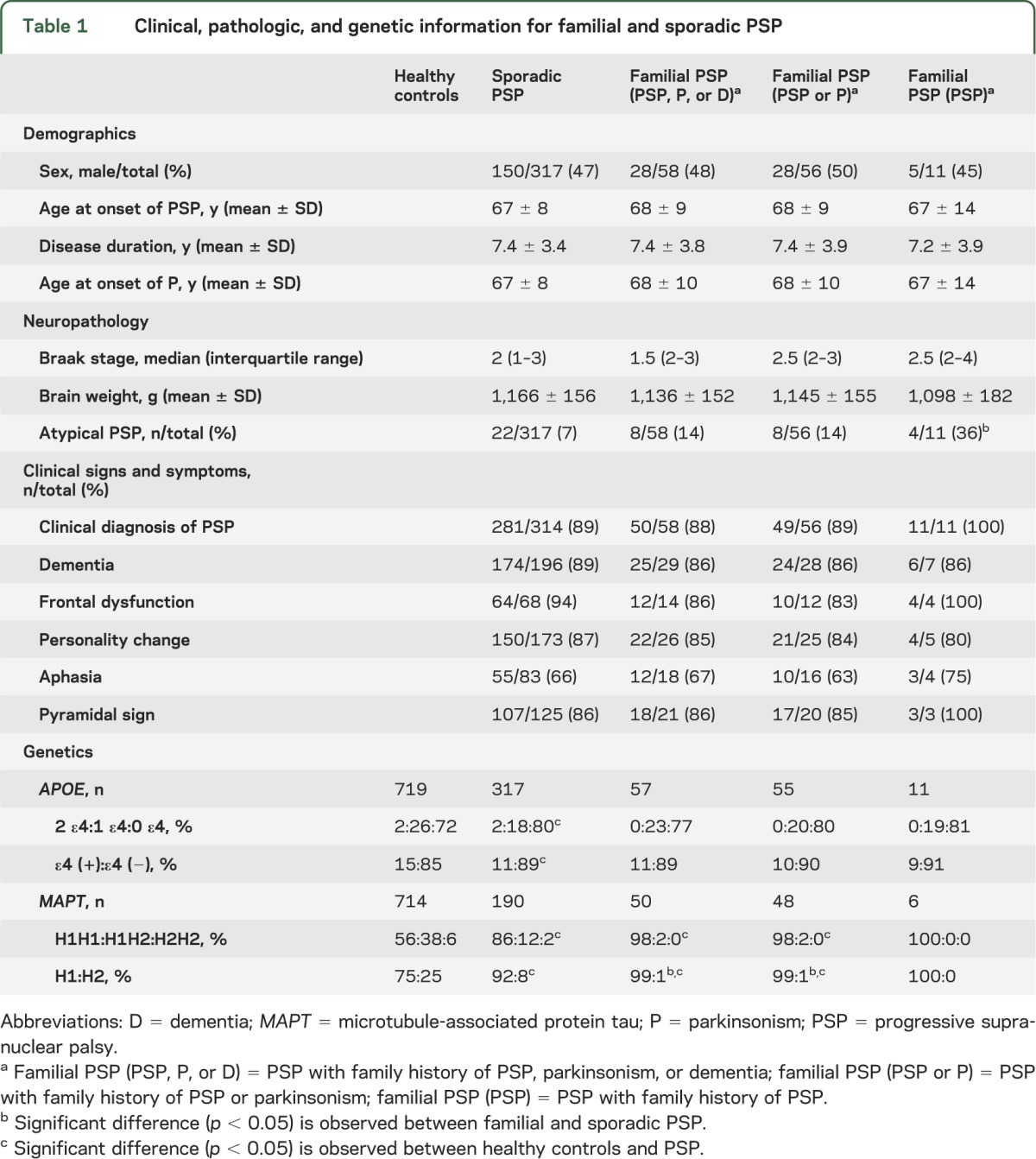
Of the 375 PSP cases, 58 (15%) had a family member with clinical history of PSP, parkinsonism, or dementia. Eleven cases (3%) had a family member with clinical history of PSP. Table 1 summarizes select demographic, clinical, pathologic, and genetic features of familial and sporadic PSP. Pathologically, familial PSP had more frequent atypical pathology compared with sporadic PSP (p = 0.004). In most regions, the severity of tau lesion scores was lower in familial than sporadic PSP, although there were no significant differences after adjustments for multiple comparisons (tables e-1 and e-2). Table e-3 provides genotype and allele frequencies for familial and sporadic PSP for genome-wide significant single nucleotide polymorphisms from the CurePSP GWAS.
Table 1.
Clinical, pathologic, and genetic information for familial and sporadic PSP
Discussion.
We found very few differences in pathologic and genetic characteristics between familial and sporadic PSP. Moreover, on a range of clinical features (table 1), familial and sporadic PSP did not differ regardless of whether one used stringent or liberal criteria for defining familial PSP. We defined familial PSP when probands had either a first- or second-degree relative with either parkinsonism or dementia; however, we acknowledge the difficulty of defining familial PSP based solely on clinical criteria, given the increasing recognition of clinical heterogeneity of pathologically confirmed PSP.1 In this study, we used different levels of stringency to overcome this weakness, by including dementia, parkinsonism, and the most stringent diagnosis, PSP, to define familial PSP.
Pathologically, we found that familial PSP tended to have less tau pathology compared with sporadic PSP. These results are contrary to those found in other familial neurodegenerative disorders, such as Alzheimer disease (AD); familial AD often has more severe pathology than sporadic AD.5 Moreover, familial tauopathies due to mutations in MAPT invariably have severe tau pathology.6 This unexpected finding may indicate that genetic factors other than MAPT may have an important role in familial PSP.
APOE ε4 allele frequency was significantly lower in PSP compared with controls, a finding that was noted in the CurePSP GWAS.3 In previous studies, presence of the ε4 allele has been associated with increased AD pathology in PSP.7 Familial PSP had a stronger association with the MAPT H1 haplotype than both controls and sporadic PSP. This finding suggests that MAPT H1 affects risk of familial PSP, but not necessarily the severity of tau pathology.
Although this report is based on a large series of autopsy-confirmed PSP, it is still underpowered to find small differences between familial and sporadic PSP, especially for genetic factors with small effect sizes, such as non-MAPT genes implicated in the CurePSP GWAS.3
Supplementary Material
Acknowledgment
The authors acknowledge the CurePSP Genetics Consortium led by Dr. Gerald Schellenberg, University of Pennsylvania, for genotyping data. The brain bank at Mayo Clinic in Jacksonville is supported by Cure PSP/Society for Progressive Supranuclear Palsy.
Footnotes
Supplemental data at www.neurology.org
Author contributions: Shinsuke Fujioka: drafting and revising the manuscript, analysis and interpretation of the data, final approval of the manuscript. Avi A. Algom: retrieving clinical data from brain bank database, final approval of manuscript. Melissa E. Murray: analysis or interpretation of data, final approval of manuscript. Audrey Strongosky: coordination of the study, final approval of manuscript. Alexandra I. Soto-Ortolaza and Rosa Rademakers: analysis or interpretation of data, final approval of manuscript. Owen A. Ross: analysis or interpretation of data, manuscript revision for intellectual content, final approval of manuscript. Zbigniew K. Wszolek: design and conceptualization of the study, final approval of manuscript. Dennis W. Dickson: neuropathologic analysis, design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript, final approval of manuscript.
Study funding: PSP patients included in this study were from brain donors to the PSP brain bank at Mayo Clinic in Jacksonville, which is supported by CurePSP/Society for Progressive Supranuclear Palsy and Mayo Clinic Florida Udall Center for Excellence in Parkinson Research (P50 NS072187-01S2).
Disclosure: S. Fujioka, A. Algom, M. Murray, A. Strongosky, and A. Soto-Ortolaza report no disclosures. R. Rademakers receives research support from the NIH (R01 NS065782, R01 AG02651, and P50 AG16574), the ALS Therapy Alliance, and the Consortium for Frontotemporal Degeneration Research, and serves on the medical advisory board of the Association for Frontotemporal Degeneration. O. Ross receives research support from the NIH (National Institute of Neurological Disorders and Stroke R01 NS078086 and P50NS072187) and the Michael J. Fox Foundation. Z. Wszolek receives research support from the NIH (R01 NS057567, 1RC2NS070276, and P50NS072187), Mayo Clinic Florida Research Committee CR program, and the Dystonia Medical Research Foundation, and holds and has contractual rights for receipt of future royalty payments from patents regarding: A novel polynucleotide involved in heritable Parkinson's disease. D. Dickson receives research support from the NIH (P50 AG016574, P50 NS072187, and P01 AG003949) and CurePSP/Society for Progressive Supranuclear Palsy. Go to Neurology.org for full disclosures.
References
- 1.Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010;23:394–400 [DOI] [PubMed] [Google Scholar]
- 2.Donker Kaat L, Boon AJ, Azmani A, et al. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 2009;73:98–105 [DOI] [PubMed] [Google Scholar]
- 3.Hoglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 2011;43:699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josephs KA, Mandrekar JN, Dickson DW. The relationship between histopathological features of progressive supranuclear palsy and disease duration. Parkinsonism Relat Disord 2006;12:109–112 [DOI] [PubMed] [Google Scholar]
- 5.Shepherd C, McCann H, Halliday GM. Variations in the neuropathology of familial Alzheimer's disease. Acta Neuropathol 2009;118:37–52 [DOI] [PubMed] [Google Scholar]
- 6.Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 1998;8:387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuboi Y, Josephs KA, Cookson N, Dickson DW. APOE E4 is a determinant for Alzheimer type pathology in progressive supranuclear palsy. Neurology 2003;60:240–245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.