

Abstract
Objectives:
We aimed to describe the clinical phenotype conferred by the intermediate-length huntingtin allele CAG repeat expansion in a population-based study.
Methods:
The Prospective Huntington At Risk Observational Study (PHAROS) enrolled adults at risk for Huntington disease (HD). They were assessed approximately every 9 months with the Unified Huntington’s Disease Rating Scale (UHDRS) by investigators unaware of participants' gene status. UHDRS scores were compared according to the Huntingtin gene CAG repeat number: expanded >36, intermediate 27–35, and nonexpanded controls <26.
Results:
Fifty (5.1%) of the 983 participants had an intermediate allele (IA). They were similar to controls on UHDRS motor, cognitive, and functional measures, but significantly worse behaviorally on apathy and suicidal ideation. On 5 of the 9 other behavioral items and on total behavior, the IA group's scores were worse than those of controls and expanded participants, who themselves scored significantly worse than controls on 6 behavioral measures. Retention rates at 4 years were 48% for the IA group compared to 58% and 60% for the expanded and control groups.
Conclusions:
In a cohort at risk for HD, the IA was associated with significant behavioral abnormalities but normal motor and cognition. This behavioral phenotype may represent a prodromal stage of HD, with the potential for subsequent clinical manifestations, or be part of a distinct phenotype conferred by pathology independent of the CAG expansion length.
Huntington disease (HD) is an autosomal dominant neurodegenerative disorder affecting motor, cognition, and behavior. It is caused by an unstable expansion of the cytosine-adenine-guanine (CAG) trinucleotide repeat within the huntingtin gene.1 The presence of 40 or more CAG repeats confers HD. At 36 to 39 repeats, there is reduced penetrance with a much later disease onset, if at all.2 A CAG expansion ranging from 27 to 35 CAG repeats is considered intermediate and is nonpathologic by definition,3 meaning that it has no direct phenotypic consequences. However, as an unstable or mutable normal allele, this intermediate allele (IA) has the small risk of expanding into the disease range upon germline transmission. Therefore, IA carriers, though unaffected themselves, may pass on the expanded version of the HD gene to cause manifest disease in their offspring.4 There have been reports of individuals with an IA developing late-onset HD,5 but population-based data are lacking. We sought to identify and describe the phenotype of individuals with the IA as a component of the Prospective Huntington At Risk Observational Study (PHAROS).6
METHODS
Study design and subjects.
PHAROS is an observational study involving 43 North American sites of the Huntington Study Group. Between July 1999 and January 2004, PHAROS enrolled 1,001 unaffected participants aged 26–55 years. The rationale behind the cohort size has been previously described.6 Subjects were at 50–50 risk for having inherited the HD repeat expansion by virtue of having an affected parent or sibling, but had chosen not to undergo predictive DNA testing. Individuals were excluded for severe depression or psychosis. Although de-identified examination of DNA was a component of the study, neither participants nor investigators were informed of individual results of gene analyses.
Clinical evaluation.
Approximately every 9 months, participants were clinically evaluated on all domains (motor, cognition, behavior, and functioning) of the Unified Huntington’s Disease Rating Scale (UHDRS).7
Data analyses.
Participants' UHDRS scores were examined according to (nonpredetermined) CAG expansion lengths as recommended by the American College of Medical Genetics3: expanded >36, intermediate 27–35, and nonexpanded controls <26. Analysis of variance using ranks was used to assess pairwise comparability at baseline among these 3 groups. Age- and sex-adjusted repeated measures analyses were also performed.
Genetic testing.
Blood samples were collected and the HD gene trinucleotide expansion length (CAGn) was measured at the DNA laboratory of the Molecular Neurogenetics Unit at Massachusetts General Hospital, as previously described.8
Standard protocol approvals, registrations, and patient consents.
The study protocol and consent procedure were approved by institutional review boards at all participating sites.
RESULTS
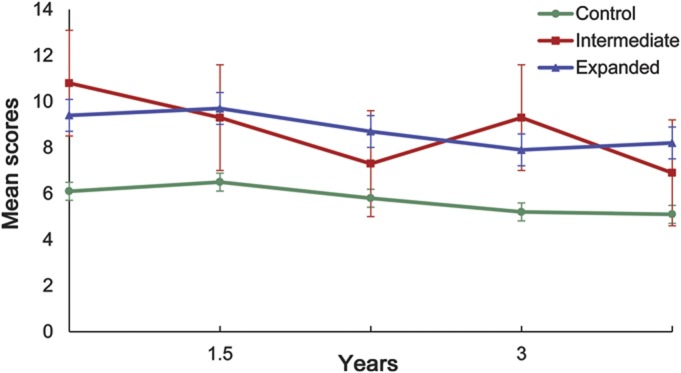
PHAROS enrolled 1,001 participants. Fourteen subjects with unequivocal HD motor features at baseline and 4 subjects missing CAG data were excluded from these analyses. Data from the remaining 983 participants were analyzed. Figure 1 shows the frequency distribution of the huntingtin alleles in this cohort. Baseline demographic and clinical characteristics are presented in tables 1 and 2. Both at baseline and over time, the IA group was found to be similar to the control group in regards to motor, cognition, and functioning. The mean UHDRS behavioral scores at baseline are presented in figure 2. Compared to the expanded and control groups, the IA group has higher (worse) baseline scores on all behavioral features except for depressed mood, anxiety, disruptive/aggressive behavior, and compulsive behavior. Despite this, the IA group is only significantly worse than the control group in regards to apathy (p = 0.0381) and suicidal thoughts (p = 0.0127). In contrast, the expanded group is significantly worse than controls with respect to baseline behavioral measures of irritability, anxiety, perseverative/obsessional thinking, low self-esteem/guilt, depressed mood, apathy, and total behavior score. Over the first 4 years of prospective evaluation, this pattern of behavioral abnormalities persists, but with a slight trend toward improved scores, as shown in figure 3. After 4 years, the retention rates were 46% for the intermediate group and 58% and 60% for the expanded and control groups.
Figure 1. Huntingtin allele frequency distribution in Prospective Huntington At Risk Observational Study cohort.

Table 1.
Baseline demographic information of PHAROS cohort by CAG expansion length
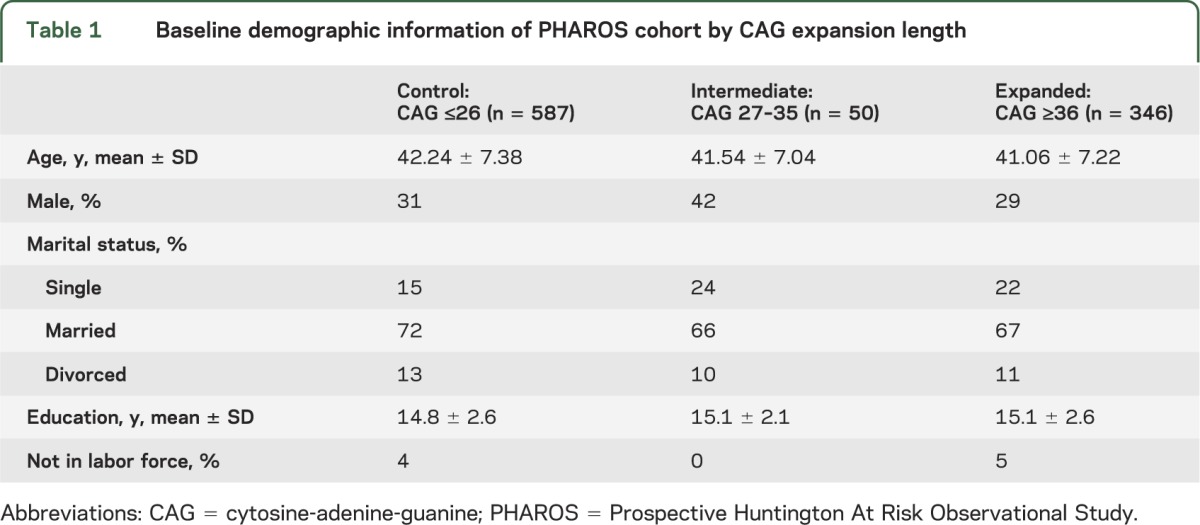
Table 2.
Baseline clinical characteristics of PHAROS cohort by CAG expansion length
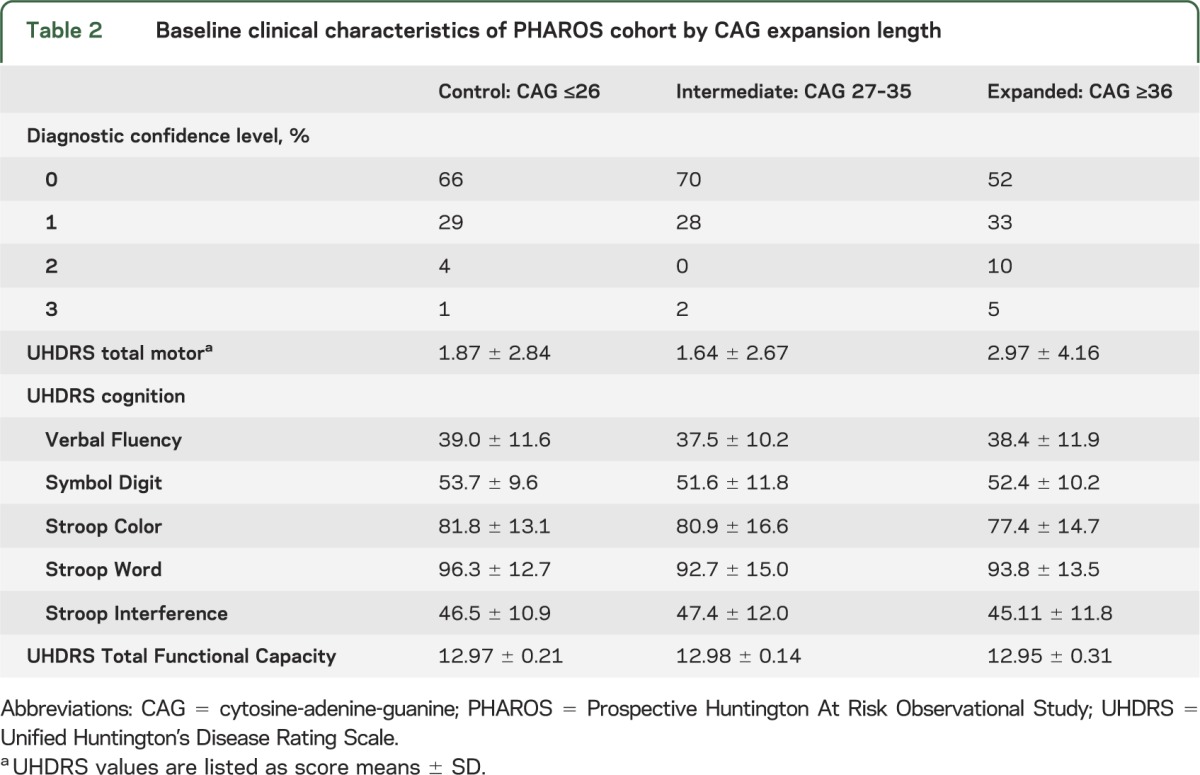
Figure 2. Baseline mean Unified Huntington’s Disease Rating Scale behavioral scores by CAG expansion length.

Figure 3. Mean total behavioral Unified Huntington’s Disease Rating Scale scores over time by CAG expansion length.
DISCUSSION
In a population at risk for HD, we sought to characterize the phenotype associated with the IA. The PHAROS IA group is one of the largest ever studied in a premanifest population. This group was not significantly different from the nonexpanded control group on UHDRS measures of motor, cognition, and function. However, compared to controls at baseline, the IA group had significantly worse apathy and suicidal ideation, and it had higher (worse) scores on 5 of the 9 other behavioral items, as well as the total behavioral score. Such findings were unlikely affected by information bias as both subjects and investigators were blinded to participants' gene status. Our findings suggest that the IA, despite traditionally being considered nonpathologic, may confer an abnormal behavioral phenotype. This is supported by data from the Cooperative Huntington's Observational Research Trial (COHORT), which studied HD-affected individuals and their close relatives.9 Compared to controls, COHORT's IA group had worse depressed mood, suicidal thoughts, and suicidal attempts per history.
The significance of the IA behavioral phenotype is unclear, but it may represent a prodromal stage of HD, with the potential for later development of the motor and cognitive features of the disease. A behavioral prodrome is often seen in expanded individuals10 up to 20 years prior to the onset of the motor features that are traditionally used to define diagnosis.11 The IA group's behavioral features representing a prodrome would support the recent case reports of late-onset HD in IA carriers.5 The notion that motor abnormalities would be of late onset (if at all) in individuals with the relatively short IA is supported by the known inverse correlation between age at onset of motor features and CAG expansion length.12 However, only approximately 60% of the variation in age at onset is determined by the allele expansion length, with genetic and environmental factors accounting for the remainder.13 In late-onset disease, these modifiers can be the determining factor as to whether or not the disease will occur in a lifetime.
Despite the fact that individuals with severe depression were excluded from PHAROS, our IA group was significant for concerning features common to prodromal HD: apathy14 and suicidal ideation.15 Apathy leads to decreased goal-directed behaviors and has a considerable adverse impact on quality of life in manifest HD.16 Suicidal ideation is a strong risk factor for suicide, which itself is known to occur 4 to 8 times more often in HD than in the general population.15 In PREDICT, a prospective study of premanifest subjects aware of their HD gene status, even psychiatric features that were considered subtle and subclinical were negatively associated with daily functioning scores.17 In our study, the IA group's relatively high behavioral scores suggest that they have mental health care needs that are not being met. Currently individuals found to have the IA through predictive genetic testing are believed to be unaffected with no need for follow-up care. However, in light of our findings, perhaps better screening and monitoring for psychiatric symptoms is warranted in this population.
If behavioral features are indeed representative of a prodromal stage for late-onset HD, then the IA is analogous to the “reduced penetrance” CAG repeat range (36–39), and may or may not lead to late-onset disease. As such alleles have a low incidence and often go untested, their penetrance is believed to be grossly underascertained.18 However, one report estimates a 60% penetrance with 39 CAG repeats (in a normal lifespan), progressively decreasing to 14% with 36 repeats.19 It would seem possible that there might be further decrementing probabilities of disease occurrence with each decrease in CAG repeat number, at least into the upper IA range. The values used for defining the intermediate and reduced penetrance alleles are not based on underlying pathology, but on documented cases of HD. However, these cutoffs have had to be adjusted accordingly with recognition of new cases in the past.20 Given the evidence from the recent case reports of IA-associated late-onset HD, the alleles' ranges may warrant further adjustment, so that the category of reduced penetrance extends to include shorter expansion lengths stretching into the IA range. Ultimately, given the wide range of estimated risk of disease associated with these partially penetrant alleles, it might be more informative to replace the use of diagnostic cutoff values in favor of a diagnostic probability spectrum based on expansion length and possibly other relevant genetic or environmental factors as they become known.
An alternative consideration is that the IA group's behavioral features represent, or are a component of, a distinct phenotype. The total behavioral scores did not correlate with the length of the IA expansion (data not shown). In manifest HD, behavioral abnormalities are independent of the CAG expansion length21 as well as the cognitive and motor aspects of the disease.22 Instead, behavioral features are thought to be the result of unknown heritable genetic modifiers,21,23 which IA carriers may also be susceptible to given their common genetic background with expanded individuals. In fragile X syndrome (FXS), another trinucleotide repeat disorder, there is a comparable premutation that is pathogenically independent from the primary mutation.24 Specifically, a loss of function underlies the mental retardation syndrome of FXS, whereas mRNA toxicity is believed to be the cause of the distinct FXS premutation phenotypes, namely fragile X–associated tremor/ataxia syndrome25 and primary ovarian insufficiency.26 The FXS premutation was previously considered to be nonpathologic, like the Huntington IA is. Perhaps the behavioral abnormalities seen in the IA group (as in HD itself) are secondary to a mechanism that is independent from the expansion length that leads to HD's motor and cognitive dysfunction.
Our study had limitations. For one, it was not of sufficient duration to ascertain whether or not any IA carriers would ultimately progress to exhibit other manifestations and warrant a clinical diagnosis of HD. However, this is not surprising given the inverse correlation between the CAG repeat expansion length and the age at motor diagnosis. With the relatively short IA expansions, one would not expect to see any motor features until late in life, and the mean age of the IA group was only 41.5 years. The eventual development of motor and cognitive abnormalities in this population would be consistent with our IA group's behavioral features representing an early prodrome of HD, support the recent case reports of IA carriers manifesting late-onset HD, and reinforce the need to consider adjusting the current genetic criteria for HD's reduced penetrance alleles.
PHAROS's sample size of IA carriers was relatively small, particularly in comparison to the much larger control and expanded groups. At 4 years, less than half (46%) of the IA group remained in the study, somewhat less than the retention rates seen in the expanded (58%) and control (60%) groups. This is not unexpected, given that subjects with psychiatric symptoms such as apathy or depression are more inclined to discontinue study participation.27 The seemingly selective retention of behaviorally healthy participants may have contributed to the slight trend in improved behavioral scores. However, this trend may also be related to the potential benefit of study participation or relatively increased access to psychiatric treatment.
Individuals with behavioral abnormalities may have been underrepresented in PHAROS. People with depressive features may have been relatively less inclined to enroll, and PHAROS excluded potential subjects with psychosis or severe depression. These factors may have selected against participants with an IA, not only resulting in underrepresentation of sample size but also undermining the severity of the phenotype's behavioral abnormalities. In addition, we did not systematically evaluate the impact of medication usage on our findings, thereby not accounting for subjects with effectively treated behavioral symptoms.
Our findings suggest that the IA confers an abnormal behavioral phenotype, despite traditionally being considered nonpathologic. Further study is warranted to better characterize the IA phenotype, its potential development into late-onset HD, and its underlying pathology. Findings from prospective studies may help to inform decisions regarding genetic counseling for individuals with the intermediate CAG repeat expansion, and perhaps help to refine the diagnostic criteria for HD alleles.
Supplementary Material
GLOSSARY
- CAG
cytosine-adenine-guanine
- COHORT
Cooperative Huntington's Observational Research Trial
- FXS
fragile X syndrome
- HD
Huntington disease
- IA
intermediate allele
- PHAROS
Prospective Huntington At Risk Observational Study
- UHDRS
Unified Huntington’s Disease Rating Scale
Footnotes
Editorial, page 2004
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Killoran: drafting/revising the manuscript for content, including medical writing for content; analysis or interpretation of data. Dr. Biglan: drafting/revising the manuscript for content, including medical writing for content; analysis or interpretation of data. Dr. Jankovic: design or conceptualization of the study; drafting/revising the manuscript for content, including medical writing for content. S. Eberly: analysis or interpretation of the data; drafting/revising the manuscript for content, including medical writing for content. E. Kayson: design or conceptualization of the study. Dr. Oakes: design or conceptualization of the study; analysis or interpretation of the data. Dr. Young: design or conceptualization of the study. Dr. Shoulson: design or conceptualization of the study; drafting/revising the manuscript for content, including medical writing for content.
STUDY FUNDING
Supported by NIH/NHGRI/National Institute of Neurological Disorders and Stroke R01HG02449.
DISCLOSURE
A. Killoran reports no disclosures. K. Biglan has received personal compensation for activities with Lundbeck and Theravance Inc as a consultant. Dr. Biglan received research support from Presbyterian Home of Central NY, Susquehanna Nursing and Rehabilitation Center, Marvell Inc, and Google. J. Jankovic has received personal compensation for activities with Allergan, Inc., Chelsea Therapeutics, Serono Inc., Merz Pharma, Lundbeck Research USA, Inc, Teva Neuroscience as a consultant. Dr. Jankovic has received personal compensation in an editorial capacity for Medlink: Neurology in Clinical Practice. Dr. Jankovic has received research support from Allergan, Inc, Allon Therapeutics, Ceregene, Inc., Chelsea Therapeutics; Diana Helis Henry Medical Research Foundation, Serono Inc., Huntington's Disease Society of America, Huntington Study Group, Impax Pharmaceuticals, Ipsen Limited, Lundbeck Research USA, Inc. S. Eberly reports no disclosures. E. Kayson reports no disclosures. D. Oakes has received personal compensation in an editorial capacity for the journal "Lifetime Data Analysis." A. Young has received royalty payments from Novartis. Dr. Young has received research support from Novartis. I. Shoulson has received personal compensation for activities with AstraZeneca, Keryx Pharmaceuticals, Jazz Pharmaceuticals, Neurogen Corp, Alexa Molecular Delivery Corp, Nouvel Pharma, Novartis, and Westat. Dr. Shoulson has received personal compensation in an editorial capacity for Archives of Neurology. Dr. Shoulson has received research support from NIH, NHGRI, National Institute of Neurological Disorders and Stroke, Cephalon, Inc., Pharmacia and Upjohn, Inc. (Pfizer), Parallel Group, and Prestwick Pharmaceuticals, Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72:971–983 [DOI] [PubMed] [Google Scholar]
- 2.Rubinsztein DC, Leggo J, Coles R, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet 1996;59:16–22 [PMC free article] [PubMed] [Google Scholar]
- 3.Potter NT, Spector EB, Prior TW. Technical standards and guidelines for Huntington disease testing. Genet Med 2004;6:61–65 [DOI] [PubMed] [Google Scholar]
- 4.Maat-Kievit A, Losekoot M, Van Den Boer-Van Den Berg H, et al. New problems in testing for Huntington’s disease: the issue of intermediate and reduced penetrance alleles. J Med Genet 2001;38:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ha AD, Jankovic J. Exploring the correlates of intermediate CAG repeats in Huntington disease. Postgrad Med 2011;23:116–121 [DOI] [PubMed] [Google Scholar]
- 6.The Huntington Study Group PHAROS Investigators At risk for Huntington disease. The PHAROS (Prospective Huntington at risk observational study) cohort enrolled. Arch Neurol 2006;63:991–998 [DOI] [PubMed] [Google Scholar]
- 7.Huntington Study Group Unified Huntington’s Disease Rating Scale: reliability and-consistency. Mov Disord 1996;11:136–142 [DOI] [PubMed] [Google Scholar]
- 8.Warner JP, Barron LH, Brock DJ. A new polymerase chain reaction (PCR) assay for the trinucleotide repeat that is unstable and expanded on Huntington’s disease chromosomes. Mol Cell Probes 1993;7:235–239 [DOI] [PubMed] [Google Scholar]
- 9.Ha AD, Beck CA, Jankovic J. Intermediate CAG repeats in Huntington's disease: Analysis of cohort. Tremor Other Hyperkinet Mov [serial online] 2012;2 Available at: http://tremorjournal.org/article/view/64. Accessed May 22, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall J, White K, Weaver M, et al. Specific behavioral manifestations among preclinical Huntington disease mutation carriers. Arch Neurol 2007;64:116–121 [DOI] [PubMed] [Google Scholar]
- 11.Folstein S, Abbott MH, Chase GA, Jensen BA, Folstein MF. The association of affective disorder with Huntington's disease in a case series and in families. Psychol Med 1983;13:537–542 [DOI] [PubMed] [Google Scholar]
- 12.Duyao M, Ambrose C, Myers R, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet 1993;4:387–392 [DOI] [PubMed] [Google Scholar]
- 13.Wexler NS, Lorimer J, Porter J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA 2004;101:3498–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Duijn E, Reedeker N, Giltay EJ, Roos RA, van der Mast RC. Correlates of Apathy in Huntington’s Disease. J Neuropsychiatry Clin Neurosci 2010;22:287–294 [DOI] [PubMed] [Google Scholar]
- 15.Hubers AA, Reedeker N, Giltay EJ, Roos RA, van Duijn E, van der Mast RC. Suicidality in Huntington's disease. J Affect Disord 2012;136:550–557 [DOI] [PubMed] [Google Scholar]
- 16.Krishnamoorthy A, Craufurd D. Treatment of Apathy in Huntington’s Disease and Other Movement Disorders. Curr Treat Options Neurol 2011;13:508–519 [DOI] [PubMed] [Google Scholar]
- 17.Duff K, Paulsen JS, Beglinger LJ, et al. Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol Psychiatry 2007;62:1341–1346 [DOI] [PubMed] [Google Scholar]
- 18.Falush D, Almqvist E, Brinkman R, et al. Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial underascertainment of late-onset cases. Am J Hum Genet 2001;68:373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quarrell OW, Rigby AS, Barron L, et al. Reduced penetrance alleles for Huntington’s disease: a multicentre direct observational study. J Med Genet 2007;44:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semaka A, Creighton S, Warby S, Hayden MR. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. Clin Genet 2006;70:283–294 [DOI] [PubMed] [Google Scholar]
- 21.Vassos E, Panas M, Kladi A, Vassilopoulos D. Effect of CAG repeat length on behavioral disorders in Huntington's disease. J Psychiatr Res 2008;42:544–549 [DOI] [PubMed] [Google Scholar]
- 22.Paulsen J, Ready R, Hamilton J, Mega M, Cummings J. Neuropsychiatric aspects of Huntington's disease. J Neurol Neurosurg Psychiatry 2001;71:310–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groen JL, de Bie RM, Foncke EM, Roos RA, Leenders KL, Tijssen MA. Late-onset Huntington disease with intermediate CAG repeats: true or false? J Neurol Neurosurg Psychiatry 2010;81:228–230 [DOI] [PubMed] [Google Scholar]
- 24.Willemsen R, Levenga J, Oostra BA. CGG repeat in the FMR1 gene: size matters. Clin Genet 2011;80:214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet 2004;74:805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman SL. Premature ovarian failure among fragile X premutation carriers: parent-of-origin effect? Am J Hum Genet 2000;67:11–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moser DK, Dracup K, Doering LV. Factors differentiating dropouts from completers in a longitudinal, multicenter clinical trial. Nurs Res 2000;49:109–116 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.