

Abstract
Adoptive cell transfer (ACT) of genetically engineered T cells expressing cancer-specific T-cell receptors (TCR) is a promising cancer treatment. Here, we investigate the in vivo functional activity and dynamics of the transferred cells by analyzing samples from 3 representative patients with melanoma enrolled in a clinical trial of ACT with TCR transgenic T cells targeted against the melanosomal antigen MART-1. The analyses included evaluating 19 secreted proteins from individual cells from phenotypically defined T-cell subpopulations, as well as the enumeration of T cells with TCR antigen specificity for 36 melanoma antigens. These analyses revealed the coordinated functional dynamics of the adoptively transferred, as well as endogenous, T cells, and the importance of highly functional T cells in dominating the antitumor immune response. This study highlights the need to develop approaches to maintaining antitumor T-cell functionality with the aim of increasing the long-term efficacy of TCR-engineered ACT immunotherapy.
SIGNIFICANCE
A longitudinal functional study of adoptively transferred TCR–engineered lymphocytes yielded revealing snapshots for understanding the changes of antitumor responses over time in ACT immunotherapy of patients with advanced melanoma.
INTRODUCTION
A small percentage of patients with widely metastatic cancers can be cured with a variety of immune-activating approaches. These dramatic, but infrequent, clinical responses are generally mediated by cytotoxic T lymphocytes (CTL) that recognize tumor antigens through their T-cell receptor (TCR). Adoptive cell transfer (ACT)–based therapies bypass many limitations of other cancer immunotherapies by generating ex vivo and then administering to patients large numbers of activated, tumor antigen–specific effector cells. These cellular immune responses to cancer are mediated by CTLs specifically recognizing tumor antigens through their TCR. Tumor antigens are of several classes, including tumor-specific mutations, reexpressed cancer–testis antigens, and lineage-specific antigens. Melanoma frequently expresses proteins of the pigmented pathway, reminiscent of its normal counterpart, the melanocytes, representing lineage-specific antigens such as tyrosinase, MART-1/Melan-A, or gp100, which have been validated as targets for T-cell responses to melanoma (1).
Several groups have shown that the treatment of patients with ACT therapy results in a high frequency of initial tumor responses (2–7). When using T cells with multiple antigen specificities, such as when tumor-infiltrating lymphocytes (TIL) are used for ACT transfer, tumor responses tend to be durable, sometimes lasting years (8). TIL therapy, however, is feasible in only a minority of patients who can undergo surgical resection of a metastatic lesion and who have T cells in the biopsy specimen that can be expanded in the laboratory. A potentially more widely applicable approach is the genetic modification of T cells obtained from peripheral blood. These blood cells can be modified to express natural TCRs or chimeric antigen receptors (CAR) that allow the specific recognition of tumor antigens. Early clinical experiences show that ACT using TCR-engineered T cells has antitumor activity in patients with metastatic melanoma and sarcoma (9–11). However, most of those responses have been transient, despite the persistence of circulating TCR transgenic cells in many cases (9, 10). This observation raises the question of whether these cells lose their antitumor functions or whether other components of the immune system are detrimentally influencing the therapy. As described in patients with HIV infection, the quality of a T-cell response is related to the functional performance of the T cells (12–14), which can be informatively analyzed at a single-cell level with multiplexed technologies (15). Therefore, we conducted a detailed time-course analysis of patient-derived samples, using newly developed multidimensional and multiplexed immune monitoring assays in selected patients receiving TCR-engineered ACT therapy (15, 16). Our analyses revealed that coordinated, time-dependent functional changes of the adoptively transferred TCR transgenic cells and T cells with other antigen specificities exhibited changes that paralleled the clinical outcomes of the patients. This study highlights the need to develop therapeutic approaches to maintaining and fostering antitumor T-cell functionality with the aim of increasing long-term efficacy of ACT immunotherapy.
RESULTS
Clinical Protocol and Characteristics of Patients
To conduct a detailed multidimensional analysis of immune function changes over time, and to study the response and resistance to ACT immunotherapy, we selected 3 of 14 patients enrolled in a phase II clinical trial of MART-1 TCR transgenic ACT therapy. These 3 patients were selected on the basis of their clinical course as representative of the whole group, that is, an initial transient tumor response followed by progression, and also on the basis of the adequacy of samples to be analyzed in different assay platforms. All patients underwent a baseline leukapheresis to collect peripheral blood mononuclear cells (PBMC), which were stimulated with anti-CD3 antibodies and interleukin (IL)-2 ex vivo for 2 days before undergoing 2 rounds of transduction with a retroviral vector carrying the high-affinity MART-1–specific TCR termed F5 (10). With this approach, both CD4+ and CD8+ T cells were genetically engineered to express the MART-1–specific TCR. Cells were then cultured in IL-2 for 3 more days and were cryopreserved after lot release testing following the approved investigator new drug (IND) application number 13859. Once the TCR transgenic cells were generated, patients were admitted to the hospital to receive conditioning chemotherapy with cyclophosphamide and fludarabine (10). This lymphodepletion procedure is designed to provide “space” within the immune system for the TCR transgenic cells to expand, and is followed on day 0 with the reinfusion of up to 1 × 109 MART-1 TCR transgenic cells. On the next day, and on days 14 and 30 after ACT, patients received 3 subcutaneous injections of 1 × 107 MART-126–35 peptide-pulsed dendritic cell (DC) vaccine generated from the same baseline leukapheresis product. They also received up to 14 doses of high-dose IL-2 at 600,000 IU/kg every 8 hours within the first 5 days after ACT (Fig. 1A; Table 1). The lymphodepleting chemotherapy and IL-2 administration were designed to maximize the ability of the infused TCR transgenic lymphocytes to homeostatically expand in vivo. The MART-126–35 peptide-pulsed DC vaccine was designed to provide antigen-specific stimulation based on our prior protocol optimization studies in mouse models (17). The above 4 components form a combined therapy approach that provides a unified effector mechanism—the activated tumor-specific CTLs. Details on the patients’ characteristics, treatment delivery, and outcomes for the 3 patients (F5-1, F5-2, and F5-8) studied herein are provided in Table 1.
Figure 1.
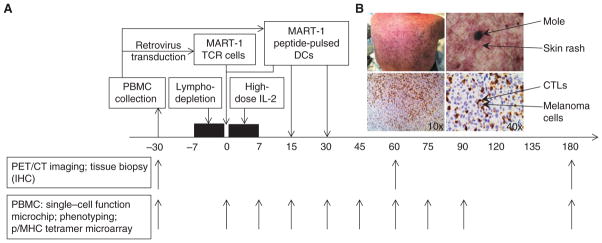
MART-1TCR transgenic T-cell ACT therapy. A, the boxes above the timeline show different modules of the therapy, with arrows pointing to the time they are administered relative to day 0 (the day of the infusion of the TCR-engineered T cells). Arrows below the timeline show the dates when blood samples are collected for different assays. B, photo of one representative patient’s back with skin rash surrounding moles (top). In the immunohistochemical (IHC) staining of a tumor biopsy (bottom), CD8+ CTLs are stained as dark brown and melanoma cells are blue. PET/CT, positron emission tomography/computed tomography.
Table 1.
Patient demographics and treatment
| Patient Study no. | Sex | Age | Prior treatments | Active metastatic sites | Stage | % MART-1+ cells | IFN-γ with K562-A2–MART-1 (pg/mL/106) | Peak of MART-1+ T cells, % | Persistence of MART-1+ T cells, % | IL-2 doses administered, n | Best decrease of tumor sizes from baseline, % | Toxicities | Response at EOS (day 90) | Skin and/or hair depigmentation | Increased TIL infiltrates |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F5-1 | M | 60 | No | Lung, stomach, liver, pancreas, peritoneum, soft tissues | M1c | 84.3 | 2.2 × 105 | 56 (at day 9) | 18.5 (at 3 mo) | 12 | −33 | Grade 3 rash, NPF | PD | Yes | Yes |
| F5-2 | F | 46 | Prime-boost vaccine, HD IL-2 | Skin, LN, bone | M1c | 74.1 | 1.5 × 105 | 59 (at day 9) | 11 (at 6 mo) | 6 | −22 | Grade 3 rash, NPF | SD | Yes | Yes |
| F5-8 | M | 44 | No | LN, liver | M1c | 66.7 | 7.8 × 105 | 51 (at day 7) | 15.5 (at 5 mo) | 11 | −13 | Grade 3 rash, NPF | SD | Yes | NA |
NOTE: EOS, end of study; F, female; HD, high-dose; LN, lymph nodes; M, male; NA, not available; NPF, neutropenic fevers; PD, progressive disease; SD, stable disease.
The adoptively transferred TCR transgenic lymphocytes undergo a rapid in vivo expansion and repopulate the peripheral immune system (peak frequency >50%, 90-day persistence >10%; Table 1), with evidence of target specificity and antitumor activity (tumor reduction of 13%–33%, Table 1 ). Repopulation of the immune system with the MART-1 TCR transgenic T cells resulted in an initial skin rash frequently centered on moles, systemic vitiligo (skin depigmentation) owing to attack on MART-1–specific melanocytes in the skin and hair follicles, and diffuse infiltration of CTLs into melanoma metastases (Fig. 1B; Table 1).
Integrated Single-Cell Functional Analyses and Antigen-Specific CTL Population Enumerations
To capture the time-dependent functional changes of the MART-1 TCR transgenic T cells and certain other T-cell populations that could influence the therapy, we coupled our newly developed single-cell barcode chip (SCBC; ref. 15) with multiparametric fluorescence-activated cell sorting (FACS; Fig. 2A and Supplementary Fig. S1). This approach allowed us to interrogate the functional performance of phenotypically defined, antigen-specific T cells at the single-cell level. We segregated CD4+ and CD8+ T cells with MART-1 specificity, as well as MART-1− T cells with a nonnaïve phenotype, based on 10 cell-surface parameters (Fig. 2A). We then quantitated 19 cytokines and chemokines produced from single cells under stimulation, using the SCBC microchip platform (Supplementary Fig. S1), expanding on the 12-cytokine panel that we had used as a pilot panel for a previous article (15). As an initial example of the potential of our SCBC chip, in that paper, we reported the functionality of MART-1+ T cells from patient F5-2 from study day 30 only, compared with blood lymphocytes from 3 healthy donors. The expanded and modified new panel used herein includes cytokines specific to CTL function (e.g., granzyme B), as well as cytokines characterizing Th1 (IFN-γ, IL-2), Th2 (IL-4, IL-5), Th17 (IL-17), and regulatory T (IL-10, TGF-β) cells (Supplementary Tables S1 and S2).
Figure 2.

FACS scheme for purifying phenotypically defined T cells and general properties of T-cell functions. A, multiparametric FACS purification for selecting phenotypically defined T-cell populations. A representative set of scatterplots is shown with the surface markers used and the cell frequency for each gating. B, hierarchical clustering of the 19 functional cytokines studied on the basis of the single-cell cytokine secretion measurement of CD8+ MART-1+ T cells from all 3 patients, and across all time points. Each functional group is identified and labeled (red). Protein–protein correlations for proteins within the same group and across groups are given below the clustering map. C, one-dimensional scatterplots of 3 representative cytokines produced by single cells, separated by time points. The dotted line represents the gate that separates cytokine-producing and nonproducing cells. The percentages given above the plots denote the frequency of positive cells and the relative mean fluorescence intensity (MFI) of those cells relative to day 7. Each point represents a single-cell assay. The points are color encoded (from purple to red) to represent the number of different proteins produced by each cell. The black trend line shows the total functional intensity of the positive cells for the specific cytokine plotted, computed as the frequency of positive cells, multiplied by their MFI. D, ratio between the MFI (red line) and the molecular number of cytokines (blue line) for polyfunctional T cells (cells with 5 or more functions) and all other cells, for each cytokine. At far right are the mean and median values, averaged over all cytokines. 7-AAD, 7-aminoactinomycin D; CCL, CC chemokine ligand; FSC, forward scatter; GM-CSF, granulocyte macrophage colony-stimulating factor; MIP, macrophage-inflammatory protein; SSC, side scatter.
We first compared the approach of FACS/SCBC with our previously described approach that coupled single-parameter nucleic acid cell sorting (NACS) (16) with SCBC functional analysis (15). The prior approach allowed for analysis of the MART-1 antigen–specific T cells, but did not permit the separation of the CD8+ and CD4+ components from the mixed population. The FACS/SCBC analysis permits such separation, but the FACS step requires a longer time before SCBC loading, which could lead to the loss of protein signal. Therefore, we tested the use of both antigen-specific and mitogen-based T-cell reactivation in FACS/SCBC assays relative to antigen-specific reactivation in NACS/SCBC analyses (Supplementary Fig. S2, Methods) using MART-1+ T cells from a patient (F5-8) who had few CD4+ MART-1+ T cells. Both protocols revealed similar cytokine secretion profiles and time-dependent functional changes, within the range of error bars. Thus, although some signal loss may be associated with the FACS/SCBC assays, much of the critical information content is retained, and the significantly improved phenotype selection is a powerful advantage.
The CD8+ MART-1− T-cell population may contain populations that are specific to melanoma antigens other than MART-1. The detection of such populations has been attributed to epitope spreading, where T cells with antigen specificity other than the one induced by vaccination or ACT appear after tissue damage by the driver clone (18). Thus, we also monitored the frequency of 35 antigen-specific subpopulations against the previously described putative melanoma tumor antigens (Supplementary Table S3) using the NACS peptide/MHC tetramer assays (16). We also conducted multiplexed blood molecular marker assays based on samples from this clinical trial (Supplementary Fig. S1). The results will be reported separately.
General Properties of T-cell Functional Changes Observed in ACT
The 19 measured cytokines and chemokines studied represent a broad range of T-cell functions (Fig. 2B). To capture general trends, we conducted hierarchical clustering of each T-cell type studied on the basis of SCBC data from the 3 patients and all time points (Fig. 2B; Supplementary Table S2). For the CD8+ MART-1+ T cells, we observed a primary clustering into antitumor effector functions (granzyme B, TNF-α, IFN-γ, etc.). Additional functions, such as regulatory (IL-10, TGF-β2) and proinflammatory (IL-6, IL-1β), are resolved with additional clustering ( Fig. 2B ). Further analysis revealed that proteins within the same functional group exhibited a higher correlation than with proteins outside that group (Fig. 2B bottom). These functional groupings are largely conserved across the 4 cell types studied (Supplementary Table S2). Thus, the T cells that we studied tend to exhibit coordinated behaviors.
The cytokine readouts that we measured reflect the kinetics of the immune response process. As an example, Fig. 2C and Supplementary Fig. S3 present one-dimensional scatterplots of the cytokine production intensity of single CD8+ MART-1+ T cells from patient F5-1 over time. Supplementary Figure S4 shows such changes for patients F5-2 and F5-8. Gates that separate protein-secreting and nonsecreting cells are identified by comparing signals from empty chambers and one-cell chambers. The percentage and mean fluorescence intensity (MFI) of granzyme B–producing cells decreased sharply within the first 30 days after ACT, whereas IFN-γ (Fig. 2C ) and TNF-α (Supplementary Fig. S3) were most abundant at day 60. The chemokines MIP-1a (Fig. 2C) and MIP-1b (Supplementary Fig. S3) reflected time-course features of both granzyme B and IFN-γ. Of note, IL-2 secretion remained low (<3% positive cells) across all time points (Supplementary Fig. S3). The total functional intensity for a given cytokine (defined as % positive cells × MFI, and plotted as the black line in Fig. 2C) reveals again these 2 waves of the immune response after ACT, characterized by the production of granzyme B, IFN-γ, and TNF-α. In addition, unique functional properties of the cell types studied are reflected in the cytokine measurements: 3 of the 4 cell types studied could be easily distinguished on the basis of their cytokine profiles (Supplementary Fig. S5).
We noted that higher protein production levels were associated with cells that exhibit a higher polyfunctionality (Fig. 2C). This feature is also quantitatively summarized in Fig. 2D and Supplementary Table S4 for all measured cytokines, where we defined a polyfunctional cell as one producing 5 or more cytokines upon stimulation. Such cells typically make up only 10% of the population of a given cell type, but on average, they secrete 100 times more copies of a given protein than do the remaining 90% of the population (Fig. 2D). The inference is that the highly functional T cells dominate (by about 10-fold) the antitumor immune response.
On the basis of the importance of the polyfunctional T cells, we defined a polyfunctional strength index (pSI) for summarizing the observed T-cell functional changes. For a given cell type, the pSI is calculated as the percentage of polyfunctional T cells relative to all CD3+ T cells, multiplied by the sum of the MFI of each of the 19 assayed cytokines from the polyfunctional subset. This index represents the total functional intensity contributed by all polyfunctional T cells of a given cell type, at a specific time point. We further characterized pSI according to the types of coordinated functions that are revealed through cytokine clustering.
Time-Dependent Functional Changes
We analyzed the samples from 3 patients to compare the functional dynamics of the CD8+ MART-1+ T cells (using the pSI) against the frequency and phenotypic changes of these cells, as well as changes in tumor burden (Fig. 3). All 3 patients exhibited an initial reduction in tumor volume, followed by tumor regrowth, but with different times to tumor relapse (Fig. 3A). In contrast, the patients had evidence of engraftment of the TCR-engineered cells (Fig. 3B). The cells proliferated briskly following ACT by at least one log expansion in total cell numbers, and then diminished gradually. At least 109 MART-1+ T cells, which accounted for more than 10% of blood T lymphocytes, remained in circulation out to day 90 after ACT. The cellular phenotyping, based upon cell-surface marker expression, revealed relatively uniform changes among the 3 patients, from an early differentiation phenotype (naïve, central memory, and effector memory) to a later differentiation phenotype (effector memory RA and effector; Fig. 3C, Methods, and Supplementary Table S5).
Figure 3.

Time-dependent changes in tumor burden, and in number, phenotype, and function of CD8+ MART-1+ T cells. Each row provides results for a different patient (F5-1, F5-2, and F5-8). A, changes in tumor burden, as measured by a modified RECIST method (see Methods). B, changes of frequency (blue) and total number (green insert) of CD8+ MART-1+ T cells. C, phenotypic changes of CD8+ MART-1+ T cells. The percentage of each T-cell phenotype (naïve, central memory, effector memory, effector memory RA, effector) is represented by a different color. D, functional changes (in pSI) of CD8+ MART-1+ T cells. Each cytokine function group is represented by a different color. For F5-2 and F5-8, the y-axis is discontinuous to allow for representation of large functional differences on the same graph.
These observations contrast with the functional changes recorded for polyfunctional T cells, as represented by the pSI plots of Fig. 3D. These plots reveal large-amplitude functional changes as well as clear differences between the patients. For F5-1, at day 7 after ACT, the CD8+ MART-1+ T cells predominantly produced the cytotoxic molecule granzyme B, which accounted for more than 70% of the total functional intensity. However, by day 30, the pSI of these cells declined by about 100-fold. The functional strength of these cells begins to recover by day 45, but that recovery is accompanied by a different set of functions, including TNF-α, IFN-γ, and inflammatory cytokines (such as IL-6), with no cytotoxic granule production. For F5-2, the CD8+ MART-1+ T cells exhibited high antitumor effector functions (IFN-γ and TNF-α) only after day 30. Patient F5-8 exhibited strong antitumor functions (granzyme B, IFN-γ, and TNF-α) initially that rapidly declined by day 30 after ACT.
The functional behavior of the other profiled T-cell types also exhibited sharp dynamic changes. We present such data for patient F5-1 in Fig. 4A–F. Functional changes for patients F5-2 and F5-8 are shown in Supplementary Figs. S6 and S7. A table that summarizes changes between day 60 and day 07 for the 3 patients is provided in Fig. 4G. The pSI of CD4+ MART-1+ T cells drops by approximately 100-fold at day 7 after ACT and shows an unstable functional profile, switching from proliferative-dominant (IL-2) at day 0; to antitumor effector-dominant (IFN-γ, TNF-α) on day 7; to inflammatory, regulatory, and nonspecific-dominant on day 30 ( Fig. 4A). We also recorded an accelerated disappearance of these cells when compared with the CD8+ MART-1+ T cells (Fig. 4B). A similar decrease in the pSI with functional shifting was observed for patient F5-8 (Supplementary Fig. S7 and Fig. 4G ). For F5-2, no cytokine production from this T-cell population was detected. Thus, this group of unnatural T cells—CD4+ T cells expressing TCRs recognizing a MHC class I–restricted antigen—lacked the ability to proliferate and conduct stable functions in vivo.
Figure 4.

Functional changes of other T-cell types for patient F5-1 over time and a summary of functional changes for all 3 patients. A, functional changes of the CD4+ MART-1+ T cells, plotted as a bar graph in pSI. The total frequency of this phenotype is plotted as the orange background. Each cytokine function group is represented by a different color. The percent composition of the functions is provided in the inset. B, the frequency ratio of CD4+ MART-1+ to CD8+ MART-1+ T cells. C, functional changes of the CD8+ MART-1− T cells, with cell frequency presented as the orange background. D, frequency of antigen-specific T cells recognizing melanoma antigen other than MART-1 over the course of the therapy. The total frequency is plotted as the black line to provide an overall view of epitope spreading. The frequency of each antigen specificity detected is provided in the inset, denoted by different colors. E, functional changes of the CD4+ MART-1− T cells, with cell frequency presented as the orange background. F, relative functional changes of the CD4+ MART-1− T cells normalized to those observed at day 7 by each cytokine group, along with cell frequency. G, a summary of functional changes for each T-cell type analyzed across 3 patients. The ratio is calculated by the pSI for a cytokine functional group at day 60 relative to that at day 7. The patients are ordered according to increasing tumor relapse rate. The differences between patients in cell function (solid blue shapes) and in tumor burden (red shapes) are shown graphically in the table.
The lymphodepletion and subsequent recovery process is reflected in the time-dependent functional changes of the MART-1− T cells (Fig. 4C–F). For CD8+ MART-1− T cells from patient F5-1, the composition of the pSI is relatively unchanged across the 90-day period after ACT, and exhibits cytotoxic and other antitumor functions throughout (Fig. 4C). At about day 30 after ACT, 4 populations attributable to epitope spreading were detected (Fig. 4D). The presence of these populations was transient, diminishing again by day 72 after ACT. Patient F5-2 presented similarities to F5-1. For example, similar epitope-spreading dynamics and a durable, antitumor effector function–dominant pSI were both recorded for the CD8+ MART-1− T-cell population for F5-2 (Fig. 4G and Supplementary Fig. S6). In contrast, patient F5-8, who had the lowest level of antitumor response, as well as a rapid tumor relapse (Fig. 4G; Table 1), presented no evidence of epitope spreading and only a transient antitumor pSI that had diminished by day 30 after ACT (Fig. 4A and Supplementary Fig. S7).
In patient F5-1, the pSI of the CD4+ MART-1− T cells diminished during lymphodepletion preconditioning and recovered by day 45. In absolute amplitude, the functional composition of these cells was relatively constant and was dominated by proliferative functions (such as IL-2 and GM-CSF Fig. 4E). However, starting about days 30 to 45, regulatory functions (TGF-β and IL-10) increased most significantly (by 27-fold relative to day 7; Fig. 4F and G). This strong increase of regulatory functions in these non–TCR transgenic T cells was also noted in samples from patient F5-8, as well, at approximately day 60 (Fig. 4G and Supplementary Fig. S7).
DISCUSSION
Little is known about the mechanisms that lead to tumor progression after a response in patients receiving ACT immunotherapy. For many years, the prevailing concepts were based on the outgrowth of tumor escape variants that down-regulated the expression of tumor antigens or molecules of the antigen-expressing machinery, or the preexistence of mutant clones that were insensitive to immune cell attack (19). More recently, several experiences are pointing to direct adaptive responses of tumors resulting from modulation of the tumor target by the T cells themselves, with the tumors changing through a variety of mechanisms to become insensitive to the CTL attack. Examples include the upregulation of PD-L1 (B7-H1) in response to IFNs produced by tumor-infiltrating T cells (20), and the antigen-specific T cells producing cytokines (i.e., TNF-α) that result in melanoma cells that express melanosomal antigens to dedifferentiate so that they may escape (21). These are cancer cell intrinsic mechanisms of resistance in response to adequately activated tumor antigen–specific T cells with full effector functions. A series of cancer cell extrinsic mechanisms of resistance to immunotherapy have also been postulated, with a major focus on the presence of immune-suppressive cells in tumors. The role of regulatory T cells, indoleamine 2,3-dioxygenase–positive DCs, and myeloid-derived suppressor cells in dampening immune responses in tumors has been well established in animal model systems and in patient-derived samples (22). Some of these immune-suppressive cells are attracted or activated by the tumor-mediated secretion of immune-suppressive factors, such as the TGF-β, VEGF, and certain other chemokines.
The study of immune cell intrinsic mechanisms of resistance has been limited by the complexity of repeatedly assaying immune functions over time, and has typically relied on assays quantifying the frequency of tumor-specific T cells, analysis of a single effector molecule, or analyses of surface proteins for identifying different T-cell phenotypes. However, T cells produce a large amount of coordinated proteins in response to antigen recognition, and that immune response, in terms of breadth and depth, is guided by the spectrum and level of the proteins being produced (12). For example, it has been previously shown in mouse models that the functional behavior of polyfunctional T cells best reflects the overall quality of an immune response (23), presumably because multiple T-cell functions are needed to orchestrate a successful immune response.
In the current studies, we interrogated T-cell responses to cancer by applying new-generation, multiplexed immune monitoring assays. These assays yield an unprecedented high-resolution view of T-cell functional dynamics in patient-derived samples. These are highly involved studies, and so we focused on extensively analyzing samples from 3 patients who exhibited different degrees of tumor response after ACT immunotherapy. Our studies suggest that occurring initially is a wave of cytotoxicity-dominated antitumor functions from adoptively transferred CD8+ MART-1–specific T cells. This wave leads to initial melanoma tissue destruction, as detected by a decrease in the size of the metastatic lesions. As has been noted in autoimmune diseases, this initial tissue destruction can lead to the expansion of T cells, with specificity for other melanoma antigens or epitope spreading (18). Epitope spreading within the population of the CD8+ MART-1− T cells was detected about day 30 in the 2 patients who had the best tumor responses, and that population of T cells retained robust antitumor functions out to at least day 90. However, the frequency of tumor-specific CTLs induced by epitope spreading was not maintained. One possibility, consistent with the data reported here, is that the CD4+ T cells, as they recover from the lymphodepletion regimen, may regulate and inhibit the antitumor immune response. This idea is also consistent with a report indicating that a deeper lymphodepletion regimen can improve the outcome for melanoma patients participating in TIL-based ACT trials (24).
A number of factors based on properties of cell phenotype and persistence that are associated with ACT therapy efficacy have been identified (8, 25). Our study supports the notion that T-cell function is another important aspect that requires close scrutiny; even when T cells show similar persistence and phenotypic changes in vivo, they can exhibit dramatically different functional profiles. Although the number of patients in this study was limited, each of the patients was very thoroughly investigated, to the extent that we can begin to associate some of the functional changes with differences in tumor relapse between patients. As shown in Fig. 4G, a strong gain (day 60 compared with day 7) of IFN-γ and TNF-α for CD8+ MART-1+ T cells, granzyme B and IFN-γ for CD8+ MART-1− T cells, antitumor effector and proliferative functions for CD4+ MART-1− T cells, and a wider epitope spreading are, at least for this study, associated with a slower tumor relapse. In contrast, a strong gain of regulatory functions in CD4+ MART-1− T cells following their recovery from the lymphodepletion regimen seems to be associated with a corresponding faster tumor relapse (Fig. 4G).
These results suggest the need to incorporate strategies to maintain the functional properties of the TCR transgenic cells used for ACT therapies. These may include modifications of the culturing system to foster the generation of TCR transgenic cells with improved ability to persist functionally over time ( 26 ), pharmacologic manipulations to provide more prolonged γ-chain cytokine support (such as protracted low-dose IL-2 administration), blockage of negative costimulatory signaling like CTLA4 or PD-1, or additional genetic engineering of the TCR transgenic cells to include cytokines or transcription factors that could maintain their function in vivo. It is also clear that the endogenous T cells can expand and boost the antitumor immune response and can regulate or otherwise influence the function of the infused T cells. Understanding this biology, and learning to control it, seems to be an important research direction for ACT therapy. A broader application of these single-cell functional analyses may prove valuable for probing the successes and failures across the spectrum of cellular immunotherapies. In particular, recent ACT trials that have used either engineered TCRs or CARs directed against antigens with better tumor selectivity than MART-1 have resulted in cases showing complete regression ( 2, 11 ). Directing the tools described herein toward analyzing those successes may provide powerful insights into how best to design such therapies.
METHODS
ClinicalTrial Conduct
Patients were enrolled in the clinical trial after signing a written informed consent approved by the University of California, Los Angeles [UCLA, Los Angeles, CA; IRB (08-02-020 and 10-001212) under an IND filed with the U.S. Food and Drug Administration (IND 13859)]. The study had the clinical trial registration number NCT00910650. Eligible patients had MART-1+ metastatic melanoma by IHC and were HLA-A*0201 positive by intermediate-resolution molecular HLA testing. Objective clinical responses were recorded following a modified Response Evaluation Criteria in Solid Tumors (RECIST; ref. 27), in which tumor burden is quantified by the sum of the largest diameter of each tumor lesion measured either by PET/ CT scan or physical examination. Skin and subcutaneous lesions evaluable only by physical examination were considered measurable if adequately recorded using a photographic camera with a measuring tape or ruler; no minimum size restriction was established for these lesions.
Immunophenotyping and Immunohistochemistry
Calculation of the absolute number of blood circulating MART-1 tetramer-specific T cells was conducted by a dual-platform method, combining the readout from flow cytometry and automated hematology analyzer (28). The flow cytometry–based MHC tetramer assay studies and the adaptation of a multicolor flow cytometry have also been previously described (29–31). T cells were classified using a panel of antibodies as described in Supplementary Table S5. Naïve cells were classified as CD45+/CCR7+/CCR5−/PD1−, CD27+/ CD28−/CD62L+, and CD45+/CCR7+/CCR5−/PD1+; central memory as CD45RO−/CD25−/HLA-DR−/CD127+; effector memory as CD45−/ CCR7−/CCR5+/PD1+, CD45−/CCR7−/CCR5−/PD1+, CD45−/CCR7−/ CCR5−/PD1−, CD45−/CCR7−/CCR5−/PD1−, and CD27−/CD28−/ CD62L−; effector memory RA (EMRA) in CD8+ as CD45RA+/CCR7−/ CCR5−/PD1+, and CD45RA+/CCR7−/CCR5+/PD1+; and effector as CD45RO+/CD25+/HLA-DR+/CD127−, CD45RO+/CD25+/HLA-DR−/CD127−, and CD45RO+/CD25−/HLA-DR−/CD127− (32, 33). Immunohistochemical staining was conducted following standard methods as previously described (34, 35).
Enumeration of Antigen-Specific T Cells
Our previously described peptide/MHC tetramer cell sorting approach (NACS) was used to enumerate antigen-specific T cells from cryopreserved PBMCs obtained from patients through peripheral blood draws and leukaperesis, as we have previously described (16). Briefly, NACS chips were generated by incubating p/MHC tetramer–ssDNA conjugate cocktail for 1 hour. Then 1 million PBMCs were added on the chip in RPMI medium for 20 minutes. Later, the slide was washed with RPMI medium and stained with CD3, CD4, and CD8 antibody cocktail for 30 minutes. Then, the sample was fixed with paraformaldehyde /PBS buffer for imaging with a Nikon TI fluorescence microscope. The p/MHC tetramer was manufactured in-house. The method was shown to have good assay performance in comparison with flow cytometry analysis (ref. 16; Supplementary Fig. S8). The sensitivity of these NACS arrays in detecting a particular tumor antigen–specific T-cell population from all CD3+ T cells is approximately 0.1% (16).
Purification of Phenotypically Defined T Cells
We used 2 methods to purify T cells for SCBC functional assays. For the NACS/SCBC protocol, the MART-1+ or CD3+ cells were purified using p/MHC tetramer or CD3 antibody, as described above. Then cells were released and loaded on the SCBC chip within 30 minutes with MART-1 tetramer/CD28 antibody (10−7 mol/L and 2 μg/mL, respectively; ref. 36) or CD3/CD28 antibody (10 and 2 μg/mL, respectively) stimulation.
For the FACS/SCBC protocol, the T cells were sorted using a BD Aria II machine. Cells were gated on FSC-A, FSC-H, SSC-A (for singlet, lymphocyte identification), 7-AAD (viability), CD3 (clone: UCHT-1), CD4 (OKT-4), CD8 (HIT8a), MART-1 tetramer, CD45RA (HI100), and CCR7 (G043H7). The machine was calibrated with BD -positive and -negative CompBeads before use. Before sorting, PBMCs had a viability of more than 80%, and the 4 collected T-cell populations were gated to achieve more than 95% viability and purity. The following cell populations were sorted and analyzed: CD3+CD4+MART-1+, CD3+CD8+MART-1+, CD3+CD4+ nonnaïve MART-1−, and CD3+CD8+ nonnaïve MART-1−. Naïve T cells were classified as CD45RA+ and CCR7+. Then, the cells were washed before stimulation with MART-1 tetramer/CD28 antibody, MART-1 tetramer, CD3/CD28 antibodies, and CD3 antibody, respectively. The concentrations used were as follows: MART-1 tetramer (10−7 mol/L; ref. 36), CD3 antibody (10 μg/mL), and CD28 antibody (2 μg/mL). All cell samples were also nonspecifically stimulated with phorbol 12-myristate 13-acetate (PMA; 5 ng/mL) and ionomycin (500 ng/mL), given the long time and low-concentration staining required for FACS previously on chip functional assays that may lead to a low level of spontaneous cytokine production. However, these could be recovered by mitogen stimulation, as shown in Supplementary Fig. S2.
Integrated Functional Assays of Single T Cells
We integrated upstream cell purification techniques with the SCBC (15) to enable the study of the functional proteomics from phenotypically defined single cells. The chips used here had a capacity of 1,360 microchambers, and permitted the simultaneous measurement of 19 cytokines from single cells. The chip was first blocked with 3% bovine serum albumin (BSA)/PBS buffer before hybridizing with antibody–DNA cocktails (15). Each step takes an hour. After PMBCs were stimulated and loaded onto an SCBC chip, the chip was imaged using high-resolution bright field microscopy. Cells were incubated on the chip for 12 hours at 37°C in a 5% CO2 cell incubator. Then they were rapidly washed out. The assay was completed by applying secondary biotinylated antibodies and streptavidin-Cy3 and then a final wash with 3% BSA/PBS buffer in sequence. Every step in this part of the process also takes an hour. Finally, the slide was washed with PBS and 50/50 PBS/deionized water in sequence before spin drying and scanning by a GenePix 4400A scanner (Molecular Devices). Detailed calibration and validation have been provided previously, where the measurement accuracy (coefficient of variation) of any given protein within a single-cell assay is approximately 10% (15) and the assay sensitivity is several hundred molecules (15).
Computational Algorithm and Statistical Analysis
Custom routines written in the R software package were used to process, analyze, and visualize the single-cell functional assay results. The algorithm converts original scanned fluorescence images into data files containing the fluorescence intensities for each assayed protein within a given microchamber, and then matches them with the number of cells counted from videos of the chip collected. Data from empty chambers are used to measure the background level for each protein. These data were used to generate protein abundance histograms, which are fitted by normal distributions and nonparametric methods, judged by goodness of fit (37). The mean of the histogram, identified by the best fit, is used as the background level. Single-cell data were then normalized by subtracting this background, so that different samples can be compared. The single-cell data were then fitted by finite mixture models, and the gate that separates the cytokine-producing and non–cytokine-producing cells was identified (37, 38). To ensure robustness, the results were individually checked. These data were then used for subsequent hierarchical clustering and principal component analysis (38), as well as the analysis of the pSI of the cells. Statistics and visual presentation were automatically generated by the algorithm.
The pSI is defined as the total functional intensity contributed by all polyfunctional T cells of a given cell type, at a specific time point. Therefore, for a given cell type, the pSI is calculated as the percentage (%) of polyfunctional T cells relative to all CD3+ T cells, multiplied by the sum of the MFI of each of the 19 assayed cytokines from the polyfunctional subset, that is,
Then, we further plot pSI as a segmented bar showing the contribution of each group of cytokines identified by hierarchical clustering. When comparing polyfunctional T cells with other T cells, fluorescence intensity is converted into molecular number, using the calibration curves provided in ref. 15.
Supplementary Material
Acknowledgments
The authors thank Steven A. Rosenberg, Richard Morgan, Laura Johnson, and Mark Dudley (all from the National Cancer Institute Surgery Branch) for access to the clinical grade retroviral vector master cell bank and their guidance in establishing the TCR-engineered ACT protocol; Carl June and Michael Kalos at University of Pennsylvania; and Jonathan Braun at UCLA for valuable discussions. The authors also thank Erika von Euw, Joanne Cox, and Narsis Attar for the manufacture of cell therapies; Elizabeth Seja and Arturo Villanueva for study coordination and data management at UCLA; Li Cheung and Rochelle Diamond at the Caltech flow cytometry facility; Bruz Marzolf at the Institute for Systems Biology (Seattle, WA); the UCLA Institute of Molecular Medicine; and the UCLA flow cytometry core. Finally, the authors thank the Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research at UCLA for clinical trial materials.
Grant Support
This work was funded by National Cancer Institute grants 5U54 CA119347 (to J.R. Heath), P50 CA086306 (to A. Ribas), P01 CA132681 (to D. Baltimore, A. Ribas, and J.R. Heath), and R01 CA170689-01 (to J.R. Heath and A. Ribas); the Jean Perkins Foundation (to J.R. Heath); the California Institute for Regenerative Medicine New Faculty Award RN2-00902-1 (to A. Ribas); the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA (to O.N. Witte and A. Ribas); The Seaver Institute (to A. Ribas); the PhaseOne Foundation (to A. Ribas); the Louise Belley and Richard Schnarr Fund (to A. Ribas); the Wesley Coyle Memorial Fund (to A. Ribas); the Garcia-Corsini Family Fund (to A. Ribas); the Caltech/UCLA Joint Center for Translational Medicine (to A. Ribas and J.R. Heath); the Melanoma Research Alliance (to A. Ribas, D. Baltimore, and J.R. Heath); and a Rosen Fellowship (to C. Ma). The UCLA Jonsson Comprehensive Cancer Center Flow Cytometry Core Facility is supported by NIH awards CA-16042 and AI-28697.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: C. Ma, A. Ribas, J.R. Heath
Development of methodology: C. Ma, A.F. Cheung
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C. Ma, A.F. Cheung, T. Chodon, R.C. Koya, B. Comin-Anduix, A. Ribas
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Ma, A. Ribas
Writing, review, and/or revision of the manuscript: C. Ma, A. Ribas, J.R. Heath
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Z. Wu, C. Ng, E. Avramis, A.J. Cochran
Study supervision: C. Ma, O.N. Witte, D. Baltimore, B. Chmielowski, J.S. Economou, A. Ribas, J.R. Heath
References
- 1.Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte–associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol. 2005;23:8968–77. doi: 10.1200/JCO.2005.01.109. [DOI] [PubMed] [Google Scholar]
- 2.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chornic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–81. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotech. 2012;30:611–4. doi: 10.1038/nbt.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle CJ, Riddell SR. Genetically retargeting CD8+ lymphocyte subsets for cancer immunotherapy. Curr Opin Immunol. 2011;23:299–305. doi: 10.1016/j.coi.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–58. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 13.Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat Med. 2008;14:623–8. doi: 10.1038/nm.f.1774. [DOI] [PubMed] [Google Scholar]
- 14.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–9. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–43. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwong GA, Radu CG, Hwang K, Shu CJ, Ma C, Koya RC, et al. Modular nucleic acid assembled p/MHC microarrays for multiplexed sorting of antigen-specific T cells. J Am Chem Soc. 2009;131:9695–703. doi: 10.1021/ja9006707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koya RC, Mok S, Comin-Anduix B, Chodon T, Radu CG, Nishimura MI, et al. Kinetic phases of distribution and tumor targeting by T cell receptor engineered lymphocytes inducing robust antitumor responses. Proc Natl Acad Sci U S A. 2010;107:14286–91. doi: 10.1073/pnas.1008300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribas A, Timmerman JM, Butterfield LH, Economou JS. Determinant spreading and tumor responses after peptide-based cancer immunotherapy. Trends Immunol. 2003;24:58–61. doi: 10.1016/s1471-4906(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 19.Ferrone S, Marincola FM. Loss of HLA class I antigens by melanoma cells: molecular mechanisms, functional significance and clinical relevance. Immunol Today. 1995;16:487–94. doi: 10.1016/0167-5699(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 20.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–6. doi: 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 22.Chow MT, Möller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22:23–32. doi: 10.1016/j.semcancer.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Seder RA, Darrah PA, Patel DT, De Luca PM, Lindsay RWB, Davey DF, et al. Multifunctional T(H)1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–50. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 24.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res. 2011;17:5343–52. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic induction of CD8+ T cell memory: better living through chemistry. Sci Transl Med. 2009;1:11ps2. doi: 10.1126/scitranslmed.3000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 28.Hultin LE, Chow M, Jamieson BD, O’Gorman MRG, Menendez FA, Borowski L, et al. Comparison of interlaboratory variation in absolute T-cell counts by single-platform and optimized dual-platform methods. Cytometry B Clin Cytom. 2010;78B:194–200. doi: 10.1002/cyto.b.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comin-Anduix B, Gualberto A, Glaspy JA, Seja E, Ontiveros M, Reardon DL, et al. Definition of an immunologic response using the major histocompatibility complex tetramer and enzyme-linked immunospot assays. Clin Cancer Res. 2006;12:107–16. doi: 10.1158/1078-0432.CCR-05-0136. [DOI] [PubMed] [Google Scholar]
- 30.Comin-Anduix B, Lee Y, Jalil J, Algazi A, de la Rocha P, Camacho L, et al. Detailed analysis of immunologic effects of the cytotoxic T lymphocyte-associated antigen 4-blocking monoclonal antibody tremelimumab in peripheral blood of patients with melanoma. J Transl Med. 2008;6:22. doi: 10.1186/1479-5876-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tumeh PC, Koya RC, Chodon T, Graham NA, Graeber TG, Comin-Anduix B, et al. The impact of ex vivo clinical grade activation protocols on human T-cell phenotype and function for the generation of genetically modified cells for adoptive cell transfer therapy. J Immunother. 2010;33:759–68. doi: 10.1097/CJI.0b013e3181f1d644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Appay V, van Lier RAW, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73A:975–83. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 33.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 34.Ribas A, Comin-Anduix B, Chmielowski B, Jalil J, de la Rocha P, McCannel TA, et al. Dendritic cell vaccination combined with CTLA4 blockade in patients with metastatic melanoma. Clin Cancer Res. 2009;15:6267–76. doi: 10.1158/1078-0432.CCR-09-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, et al. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res. 2011;17:4101–9. doi: 10.1158/1078-0432.CCR-11-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cochran JR, Cameron TO, Stern LJ. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity. 2000;12:241–50. doi: 10.1016/s1074-7613(00)80177-6. [DOI] [PubMed] [Google Scholar]
- 37.Reynolds DA, Rose RC. Robust text-independent speaker identification using Gaussian mixture speaker models. IEEE Trans Audio Speech Lang Processing. 1995;3:72–83. [Google Scholar]
- 38.Johnson R, Wichern DW. Applied multivariate statistical analysis. 6. Upper Saddle River (NJ): Pearson; 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.