

Abstract
The development of a miniaturized sensing platform for the selective detection of chemical odorants could stimulate exciting scientific and technological opportunities. Oligopeptides are robust substrates for the selective recognition of a variety of chemical and biological species. Likewise, semiconducting nanowires are extremely sensitive gas sensors. Here we explore the possibilities and chemistries of linking peptides to silicon nanowire sensors for the selective detection of small molecules. The silica surface of the nanowires is passivated with peptides using amide coupling chemistry. The peptide/nanowire sensors can be designed, through the peptide sequence, to exhibit orthogonal responses to acetic acid and ammonia vapors, and can detect traces of these gases from “chemically camouflaged” mixtures. Through both theory and experiment, we find that this sensing selectivity arises from both acid/base reactivity and from molecular structure. These results provide a model platform for what can be achieved in terms of selective and sensitive “electronic noses.”
Introduction
Progress in the development of highly selective and sensitive sensors has recently accelerated due to increased interest in potential chemical and biological threats.1–12 While DNA and protein biomolecular sensors can exploit well-established “lock-and-key” interactions to achieve selectivity, achieving high selectivity and sensitivity in gas phase sensors has until recently had to rely on physical (i.e., chromatographic) separation methods or spectroscopic fingerprinting techniques.13 However, the associated instrumentation is limited in portability, precludes the possibility of implantable or wearable sensors, and usually requires skilled human operators. “Electronic noses” offer a promising alternative, with the potential for continuous real-time monitoring and discrimination of large families of gases. These vapor analyzers are designed to mimic the olfactory system via the integration of sensor arrays (typically conducting polymer14,15 or metal oxide16 thin films) and pattern recognition algorithms.15–18 The sensors are designed in a combinatorial fashion to yield varying responses to different analytes. An alternative is to attempt highly specific sensing for the deconvolution of molecular signatures from interfering gas mixtures, without requiring an external analytical filter, pattern recognition algorithms, or advance calibration. For the case of traditional e-noses, selectivity is increased by employing larger sensor libraries. In this paper, we achieve selectivity by focusing on the specific molecular interactions between the sensor elements and the target gas molecules. Mammalian olfactory systems apparently use a combination of both approaches.
Odorant perception in the mammalian olfactory system results from an aggregate response of many intricate biochemical and electrophysiological signaling events. Mammals contain ca. 1000 genes expressing for odor reception, though not all of these may encode functional odor receptor binding proteins. Research suggests that odor discrimination begins with molecular feature detection upon binding to receptor proteins.19 This arrangement, in conjunction with the complex processing capabilities of the brain, renders the mammalian olfactory system one of the most effective sensing structures. Indeed, most mammals can discriminate 10 000 or more distinct odors at detection levels of only a few parts per billion (ppb).20 Successful attempts have been made to mimic these binding domains via the use of peptide aptamers,21–24 which are biorecognition molecules that can be chemically engineered to bind specific targets. In this respect, peptides are particularly interesting because of their broad chemical diversity (acidity, hydrophobicity, etc.) that can be achieved within a relatively compact size.25,26 Oligopeptides have been coated onto piezoelectric crystal mass sensors to achieve selectivity to various saturated vapors.21,22 The peptide sequences were rationally designed to mimic the putative binding sites of a human olfactory protein modeled by molecular simulation methods.
Recently, electronic noses based on arrays of semiconducting nanowires5,18 and nanotubes8,27 have been implemented. These “nano-noses” boast ppb sensitivities, a consequence of the nanostructure diameters being comparable to the width of the surface space charge region. In some cases, these nanosensors have shown some selectivity to certain molecules, via the use of chemoselective polymer coatings,28,29 surface chemistry functionalizations,5 and varying metal oxide material compositions.18 However, a general scheme for achieving a high degree of specificity to any given target molecule would be highly desirable. Here we demonstrate that covalently coupling oligopeptides to the surfaces of silicon nanowires (SiNWs) provides an interesting model system for selective sensors, by displaying high degrees of selectivity to acetic acid (AcOH) and ammonia (NH3) small molecules. We combine theoretical modeling with experiment in an effort to understand the selectivity mechanism of our peptides to their target analytes and also to separate the contributions to the sensor responses that arise from analyte physisorption, analyte/peptide acid–base interactions, and analyte/peptide structural interactions. We find that three-element NW/peptide sensor arrays can separately detect AcOH and NH3 from gas mixtures, even in the presence of background gases.
Methods
Nanowire Fabrication
The SiNW arrays were fabricated as previously described,5,30,31 and all fabrication was done within a class 1000 or class 100 clean room environment. A typical array of nanowires fabricated by this technique is shown in Figure 1. The starting material for this process was an intrinsic, 320 Å thick silicon-on-insulator (SOI) film (<100> orientation) (Simgui, Shanghai, China) with a 2500 Å buried oxide. After thorough cleaning and rinsing with deionized water, the substrate was coated with p-type spin-on dopants (SODs) (Boron A, Filmtronics, Inc., Butler, PA). Dopants were diffused into the SOI film using rapid thermal processing (RTP) at 800 ° C for 3 min. Four-point resistivity measurements, correlated with tabulated values, yielded a doping level of ~1018/cm3. Separately, a superlattice consisting of 800 layers of alternating GaAs and AlxGa(1−x)As thin films was prepared (IQE, Ltd., Bethlehem, PA). The superlattice was cleaved along a single crystallographic plane and thoroughly cleaned by sonication in methanol and gentle swabbing. The exposed edge was immersed in NH3/H2O2/H2O (1:20:750 v/v) for 10 s to selectively etch the GaAs regions (etch depth ~30 nm). The resulting edge of the superlattice thus consisted of AlxGa(1−x)As plateaus separated by GaAs valleys. Pt metal was deposited using electron beam evaporation onto the edge of the AlxGa(1−x)As ridges, with the edge of the superlattice held at a 45° angle to the incident flux of Pt atoms. The Pt-coated superlattice edge was then brought into contact with the doped SOI substrate spin-coated (6000 rpm, 30 s) with a thin-film PMMA/epoxy (1:50 w/w). The superlattice/epoxy/SOI sandwich was dried on a hot plate (150 ° C, 40 min), and the superlattice was released by a selective etch in H3PO4/H2O2/H2O (5:1:50 v/v, 4.5 h) solution, leaving a highly aligned array of 400 Pt NWs on the surface of the SOI substrate. These Pt NWs served as protective masks for a reactive ion etch (RIE) process to produce aligned, single-crystal SiNWs (CF4/He, 20/30 sccm, 5 mTorr, 40 W, 3.5 min). The Pt NWs were dissolved in aqua regia (30 min) to produce an array of 400 SiNWs. Finally, the substrate was cleaned in ALEG solution (Mallinckrodt Baker, Phillipsburg, NJ) and rinsed with DI water to remove residual epoxy.
Figure 1.

(A) Optical image of microfluidic functionalization channels (vertical conduits) intersecting nanowire sensor devices. The nanowire islands (horizontal blue bars) are electrically contacted by metal leads (yellow lines). (Inset) Scanning electron micrograph of the nanowire film. (B) Characterization of the bare silicon-on-insulator (SOI, red), amine-terminated (APTES, green), and peptide-coupled (blue) surfaces by X-ray photoelectron spectroscopy. (Inset) Water contact angle goniometric measurements of the surfaces.
SiNW Sensor Fabrication
A typical set of NW sensors employed for this work is shown in Figure 1. The chip containing the SNAP wire arrays was treated to mild O2 plasma (300 mTorr, 30 W, 60 s), then immersed in buffered oxide etch (BOE) for 3 s to remove oxides and promote the formation of ohmic contacts. Source and drain electrodes were formed by electron-beam evaporating 1000 Å Ti uniformly across the chip, and then patterning the Ti through a photoresist mask (Shipley 1813, MicroChem Corp., Newton, MA) via wet etching (1:1:10 HF/H2O2/DI v/v, 5 s). The resulting device channels were 5 μm in length. A new photoresist mask was applied to expose unwanted regions of the NW array for sectioning into device islands. The Si was removed via RIE (SF6, 20 sccm, 20 mTorr, 30 W, 1 min), and the photoresist was removed in acetone.
Peptide Synthesis
Peptides were synthesized on Fmoc-Rink amide MBHA resin (0.67 mmol/g, Anaspec) using conventional solid phase synthesis strategy with Fmoc protection chemistry.32 TFA-deprotected peptides were purified by HPLC on a C18 reversed phase column (Varian Dynamax semipreparative column, 25 cm × 2.15 cm). The column was eluted with 0.1% trifluoroacetic acid and a three-part linear gradient of acetonitrile, rising from 0–25% over 30 min and 25–100% over 30 min and holding at 100% over 20 min. The pure acetic acid and ammonia recognition peptides Fmoc-RVNEWVID and Fmoc-DLESFLD (where the final D is a linking residue) each eluted at 56 and 61 min, respectively. The reverse peptide sequences eluted at the same times. All purified peptide products were verified to have the correct molecular weight as determined by mass spectrometry.
X-ray Photoelectron Spectroscopy
X-ray photoelectron spectroscopy (XPS) was utilized to evaluate bulk Si(100) surfaces at each step of the functionalization outlined in Scheme 1, using a chlorinated peptide Fmoc-X7D (where X = 4-chlorophenylalanine). All XPS measurements were performed in an ultrahigh vacuum chamber of an M-probe surface spectrometer that has been previously described.33 Monochromatic Al KαX-rays (1486.6 eV) were used to irradiate the sample incident at 35° from the surface. ESCA-2000 software was used to collect and analyze the data. To gain an overview of the species present in the sample, survey scans were run from 0 to 1000 binding eV (BeV). The C 1s (282–292 BeV), Cl 2p (196–206 BeV), and N 1s (393–407 BeV) regions were investigated in detail.
Scheme 1.
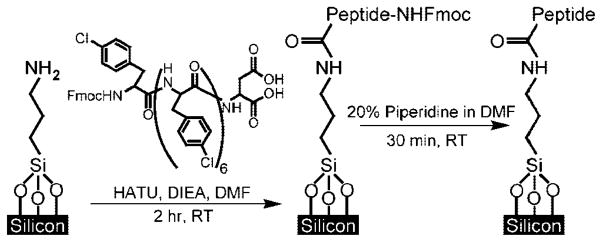
Covalent Attachment of Peptides to SiNWs
Contact Angle Measurements
The sessile water contact angle on the functionalized Si(100) surfaces was used to check the fidelity of surface chemistry as described in Scheme 1. Contact angle measurements were obtained with a Naval Research Laboratory contact angle Goniometer Model #100-00 (Rame-Hart, Inc., Net-cong, NJ) at room temperature. All measurements were repeated 10 times and averaged to obtain the surface contact angle.
Microfluidics Fabrication
Soft lithography microfluidics chips were fabricated as described by others.34 A silicon wafer (Virginia Semiconductor, Inc., Fredericksburg, VA) was thoroughly cleaned in acetone and isopropanol and spin-coated with SU-8 2015 (1750 rpm, 30 s) (MicroChem Corp., Newton, MA). The photoresist was baked at 65 ° C for 2 min and 95 ° C for 4 min and then exposed to a microfluidic channel pattern by conventional photolithography. The resist was post-baked at 65 ° C for 1 min and 95 ° C for 8 min, developed for 4 min in SU-8 Developer (MicroChem), rinsed with isopropanol, and hard baked at 180 ° C for 15 min. The SU-8-patterned wafer was then coated with PDMS prepolymer and cured.
Surface Functionalization
The functionalization of NWs with peptides is outlined in Scheme 1. The SiNW sensor chip was treated to an O2 plasma oxidation step (300 mTorr, 30 W, 60 s), then immersed in a 1% toluene solution of the surface modifying reagent 3-aminopropyltrimethoxysilane (United Chemical Technologies, Bristol, PA) to generate amine-terminated SiNW surfaces. The chip was allowed to react for 50 min, then rinsed thoroughly in toluene and isopropanol, and finally heated at 110 ° C for 10 min. Separately, a silicon wafer (Virginia Semiconductor, Inc., Fredericksburg, VA) was thoroughly cleaned in acetone and isopropanol and spin-coated with SU-8 2015 (1750 rpm, 30 s) (MicroChem Corp., Newton, MA). The photoresist was baked at 65 ° C for 2 min and 95 ° C for 4 min, and then exposed to a microfluidic channel pattern by conventional photolithography. The resist was post-baked at 65 ° C for 1 min and 95 ° C for 8 min, developed for 4 min in SU-8 Developer (MicroChem), rinsed with isopropanol, and hard baked at 180 ° C for 15 min. The SU-8-patterned wafer was then coated with PDMS prepolymer and cured. Subsequently, a PDMS stamp containing microfluidic channels was aligned to the sensor device chip such that the channels intersected with the sensors. A coupling solution of 20 mM Fmoc-peptide in DMF was prepared to contain 2 equiv of 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU, relative to peptide). Fresh oligopeptide coupling solutions and 6 equiv of N,N-diisopropyl-ethylamine (DIEA, relative to peptide) were injected into the channels and allowed to react for 2 h at room temperature. The PDMS channel was removed, the chip rinsed thoroughly with DMF, and then immersed in a 20% piperidine solution in DMF for 30 min to remove the Nα-Fmoc protecting group moiety.
Sensor Characterization
Electrical characterization of sensors was achieved with a preamplifier (Stanford Research Systems, Sunnyvale, CA) interfaced to a DAC card and BNC adapter breakout panel (National Instruments, Austin, TX). Data were collected with a custom-programmed software routine (National Instruments LabVIEW). The sensor chips were wire-bonded to a chip carrier and placed in a home-built gas delivery chamber with electrical feed-through. Prior to all experiments, the chips were placed under vacuum while the chamber was heated to a temperature of ~70 ° C for >1 h to dehydrate the surface. Acetic acid, ammonia, acetone, trimethylamine, and/or carbon dioxide vapors (Matheson Tri-Gas, Newark, CA) were introduced at a flow rate of 300 sccm, following pure N2 flowing at a rate of 1000 sccm.
Theoretical Modeling
Optimized geometries and Mulliken charges were calculated with Becke–Lee–Yang–Parr 3 parameter (B3LYP) hybrid density functional theory (DFT) using the 6-31g** basis set, 1160 and 1450 basis functions for DLESFLD and RVNEWVID, respectively. With the exception of ARG1 and GLU4 in RVNEWVID, all amino acids were modeled in their neutral state. The ARG1(+)–GLU4(−) salt bridge was calculated to be 11.5 kcal/mol stable. Quantum geometries and charges were input into a Boltzmann jump search of the global three-dimensional stable conformation, using the DREIDING35 force field without Coulomb cutoffs. The procedure consisted of two steps: (1) 10 anneal molecular dynamics cycles, from 200 to 500 K in 100 K steps, followed by quench minimization, anneal period of 1000 time steps, each 1 fs, followed by (2) 2000 Boltzmann jump sequences, with an average of 30 perturbations per sequence, an adjustable dihedral window of 10° for all rotatable bonds, and an acceptance maximum temperature of 5000 K relative to the current minimum. Cluster analysis showed that the 100 lowest energy conformations were indistinguishable. Stable peptide structures were then exposed to ammonia and acetic acid target analytes in various orientations using the Molecular Silverware algorithm,36 against 180 recognition moieties on the peptide surface. The binding energetics of each of over 47 000 peptide moiety/analyte orientation pairs were sorted, and the best binding configurations were chosen for quantum calculations using shortened versions of the peptides: ARG1-GLU4 for RVNEWVID, and ASP1-GLU3 for DLESFLD. Reported reaction energies are the difference between the neutral and the acid/base reaction products with fully minimized geometries (B3LYP/6-31g**). Quantum calculations were performed using Jaguar 7.0 (Schrödinger, LLC, Portland, OR), Boltzmann jump conformational searches were conducted with the Cerius2 package (Accelrys Software Inc., San Diego, CA), and binding dockings were done with an in-house coded Molecular Silverware algorithm.
Results and Discussion
Coupling of Peptides to Nanowires
SiNW surfaces terminate in intrinsic silica, which has a well-established chemistry,3,10 that permits NW surface modification without strongly affecting the semiconducting core. The SiNWs were fabricated using the superlattice nanowire pattern transfer (SNAP) method,30 which can be harnessed to produce highly regular arrays of virtually any material that can be obtained as a high quality thin film.31,37 The NW arrays comprised 400 high aspect ratio (>105), 16 nm wide SiNWs at a pitch of 33 nm, with a p-type doping level of ~1018/cm3. These NWs perform as excellent field-effect transistors on both solid and flexible plastic substrates, with mobilities comparable to that of bulk silicon.5,31 The SiNWs were fashioned into sensor devices via conventional microfabrication techniques. A contact metal layer of 1000 Å Ti was uniformly evaporated across the entire chip and subsequently patterned via photolithography and HF etching to form source/drain finger electrodes across the SNAP wire array. The NW film was then sectioned into individual sensor elements using photolithography and etching. SNAP SiNW sensors are capable of detecting ppb levels of NO2, even on plastic substrates.5
Peptides were synthesized by the fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesis method, in which Fmoc-protected amino acid residues are sequentially linked on a solid bead support via repeated cycles of coupling and deprotection.32 The peptides remain immobilized on the beads until cleaved by trifluoroacetic acid. Peptides were subsequently purified to >95% by HPLC using a C18 semipreparative column. Peptides were then immobilized onto the NWs using amide coupling (Scheme 1).38 First, the nanowire surfaces were chemically modified by immersion of the chip in an amino silane (APTES) modifying reagent. Next, oligopeptides were synthesized with the desired recognition sequences, plus an aspartic acid “linking residue” tail at the carboxy terminus. The peptides were dissolved in DMF, mixed with coupling reagents, and immediately injected into PDMS microfluidic chambers aligned to the device islands (Figure 1a). Once this coupling reaction was complete (2 h), the microfluidic channels were removed and the chip was thoroughly rinsed to remove uncoupled peptide. Finally, the chip was treated to a piperidine solution to cleave the Fmoc protecting group.
X-ray photoelectron spectroscopy (XPS) measurements on silicon-on-insulator (SOI) wafer pieces, treated to identical surface reaction protocols as the SiNW sensors, were employed to monitor this coupling chemistry. We used a chlorophenylalanine 7-mer peptide (Scheme 1) to provide a chlorine signature for XPS studies. As shown in Figure 1b, neither the bare SOI coupon nor the APTES-treated chip show significant chlorine presence. By contrast, the chloropeptide-modified substrate displays characteristic Cl 2p peaks in the expected 2:1 2p3/2/2p1/2 ratio, centered at 203 eV.39 Attempts to recover these peaks without the use of coupling reagents failed, indicating that the peptides are covalently coupled rather than physisorbed. Water contact angle goniometric measurements confirm a sharp increase in surface hydrophobicity with peptide presence (Figure 1b), due to the phenyl groups.
Selectivity of Peptide–NW Hybrid Sensors
Confirmation of the peptide–NW linkage permitted us to characterize the performance of these hybrid materials as selective sorption-based vapor sensors. The sensor chips were placed in a custom-built gas delivery chamber equipped with electrical feed-throughs. Humidity and desorption of water affected our readings,21 so all experiments were prepared under dry conditions, in which the chips were placed under vacuum for 1 h with mild heating to establish a dry baseline. The sensing experiments were run under pure flowing N2 at room temperature. The peptide–NW sensors were exposed to target molecules using a flow-through technique. Target binding onto the NW surface can take place via direct physisorption (this should occur for all odorants) or via the potentially selective adsorption at peptide binding sites. To separate these two processes, we included in our sensing experiments a “peptide-free” NW sensor functionalized with just APTES. The adsorption selectivity of a peptide/NW sensor to the target molecules was ascertained after normalization of that sensor response against the APTES-functionalized NW sensor. We chose acetic acid and ammonia target molecules for this initial work because (1) peptide sequences against both have been identified,21 (2) they are sufficiently reactive to elicit electrical response in the sensors, yet subtle enough for exploring the chemical space of peptide recognition sites, and (3) they can serve as exhaled breath40 disease biomarkers for asthma (acetic acid)41 and kidney diseases (ammonia).42
Three-component NW sensor libraries were fabricated to evaluate the selectivity of NW–peptide sensors. The components comprised one sensor modified with an acetic acid recognition peptide sequence (RVNEWVID),21 one with an ammonia recognition sequence (DLESFLD), and a third element with APTES modification (no peptide). Figure 2a shows normalized responses of all three NW sensors to 100 ppm levels of acetic acid and ammonia vapors diluted in N2. Upon exposure to ammonia for 5 min, the APTES module exhibited a strong 90% decrease in current. After exposure to acetic acid, this sensor exhibited an 11.5% increase in conductance. We assign these responses as arising from physisorption of the analyte onto the NW surface.43–45 These responses were reproducible and reversible, although sensor recovery required vacuum pumping and gentle heating. Sensor responses normalized to these APTES signals are shown in Figure 2b. Strikingly, the NH3 recognition peptide displays ca. 75:1 selectivity toward ammonia over acetic acid. This specificity is clearly reversed in the AcOH recognition peptide, with a selectivity ratio of 3.75:1 for the affinity of the acetic acid peptide to AcOH relative to NH3, a value that is in good agreement with previous work.21
Figure 2.
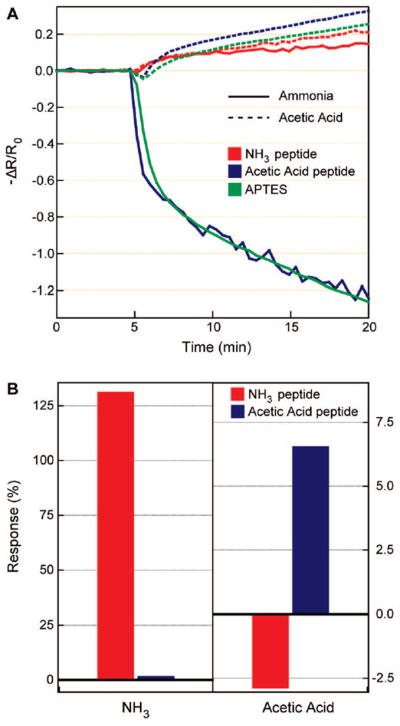
(A) Electrical responses of an ammonia recognition peptide–nanowire sensor (red), an acetic acid recognition peptide–nanowire sensor (blue), and an amine-terminated nanowire sensor (green) to 100 ppm ammonia (solid) and acetic acid (dashed) vapors, introduced to the sensing chamber after 5 min of flowing N2. (B) Conductance responses of the peptide–nanowire hybrid sensors, averaged over a 5 min time window of target vapor exposure (starting 10 min after the analyte gas exposure), and normalized to the amine-terminated sensor. The abscissa is labeled with the analyte vapors.
Mechanistic Investigations of Selective Sensing
Previous work focused primarily on analyte–peptide molecular shape interactions to describe the mechanism of selectivity.21 Our initial calculations suggested that molecular shape did not fully account for the observed data. In order to more deeply understand the mechanism of specificity exhibited by the peptide–NW sensors, a combination of classical molecular dynamics followed by Becke–Lee–Yang–Parr 3 parameter (B3LYP) density functional theory (DFT) calculations was performed.46 A three-step modeling procedure was implemented: first, the unbiased (vacuum) conformations of the relevant peptide sequences were determined using a Boltzmann jump conformational search, absent the target molecules. The targets were then introduced via the previously described “Molecular Silverware” algorithm,36 which allows for an efficient sampling of small molecule binding sites over the entire peptide topography. Finally, once an optimal binding arrangement of the analyte with the peptide was determined, the magnitude of the binding energy was estimated by molecular mechanics and verified on a minimal binding motif using DFT calculations.
Figure 3 shows the results of the molecular modeling studies. Interestingly, our calculations reveal that acid/base binding equilibria among the peptides and odorant compounds are significant factors in achieving selectivity. For example, the acetic acid binding peptide RVNEWVID (Figure 3a) contains a GLU4-ARG1 “salt bridge” or ionic attraction following proton transfer, which is stable (~10 kcal/mol) in vacuum and at room temperature. This suggests that secondary structure is important even in these short-chain sequences. In the presence of acetic acid vapor (Figure 3a, right panel), the salt bridge is in equilibrium with the open free peptide conformation (neutral GLU and positively charged ARG). By contrast, the acid binding peptide remains unaffected following exposure to ammonia, and the salt bridge stays intact. Our binding energy calculations confirm and quantify these results: reaction of the acetic acid analyte with the RVNEWVID peptide is exothermic, −5.5 kcal/mol of energy, while reacting ammonia with the peptide is a highly unfavorable uphill energetic process (+15.1 kcal/mol).
Figure 3.

(A) The left panel shows the lowest conformation of the acetic acid binding peptide, which contains a GLU4-ARG1 salt bridge (dashed yellow lines). The right panel shows preferential binding of acetic acid to the ARG1 N-terminus. (Inset) The reaction products: neutral GLU4 and protonated ARG1, stabilized by acetate. (B) The left panel shows the lowest conformation of the ammonia binding peptide. The polar and nonpolar amino acids align on opposing sides. The right panel shows ammonia binding at the neutral N-terminus ASP1. (Inset) Ammonium stabilized by hydrogen bonds to the deprotonated aspartic acid and the N-terminus.
The DLESFLD ammonia-binding peptide forms an interesting structure in which the nonpolar groups (leucines and phenylalanine) stack on one side of the peptide and the polar groups (aspartic and glutamic acids, serine) line up on the other (Figure 3b). It was found that ammonia reacts favorably (−7.4 kcal/mol) with the N-terminal aspartic acid residue to form a unique hydrogen-bond center at the terminal acid and amine groups (Figure 3b, right panel). By contrast, the reaction of acetic acid with this peptide has a near-zero enthalpy of reaction (+0.4 kcal/mol), and the ratio of protonated to deprotonated peptide is 1:3 at room temperature. Overall, these results correlate very well with the observed experimental response patterns, in that more energetically favorable reactions yield larger resistivity changes in the sensors. They also suggest peptide structures for achieving selectivity to nonreactive and nonpolar compounds, for example, by designing “looped” peptides connected by salt bridges that break in the presence of the target analyte.
Key experimental tests provided further insight into the mechanism of binding. First, to assess whether the selectivity depends on surface peptide orientation, we compared the responses of the peptides to reversed peptide sequences, IVWENVRD and LFSELDD (Figure 4a). These reverse sequences respond nearly identically as the original peptides to acetic acid and ammonia. This suggests that side chain chemistries contribute more to the specificity than the peptide backbones, and modulation of the backbone dipole upon analyte binding is not a factor in eliciting the electronic response. Next, we examined other reactive pairs, by comparing the activity of the ammonia binding peptide (DLESFLD) to equal-length peptides composed entirely of the presumed binding sites,21 aspartic acid (D6D, pKa = 4.0), glutamic acid (E6D, pKa = 4.1), and phenylalanine (F6D), following exposure to 100 ppm ammonia (pKb = 4.75) and trimethylamine (TMA, pKb = 4.13). As shown in Figure 4b, despite its higher basicity, TMA generates a 3× smaller conductivity response in the ammonia–peptide device relative to equal concentrations of ammonia gas, in good agreement with previous reports.21 Here, our experiments contrast with theory, as we calculated that TMA should react with a large exothermic energy (−12.7 kcal/mol) with the NH3 peptide. This suggests that molecular shape does play a strong role in determining selectivity, as TMA is significantly bulkier than ammonia, and the methyl groups of TMA reduce the number of available hydrogen bonds in the product.
Figure 4.

(A) Conductance responses of the acetic acid recognition (blue) and reversed acetic acid recognition (dotted blue) peptide–nanowire sensors to 100 ppm acetic acid, and the ammonia recognition (red) and reversed ammonia recognition (dotted red) sensors to 100 ppm ammonia. The abscissa is labeled with the target analyte vapors. (B) Responses of ammonia recognition, all-aspartic acid (D6), all-glutamic acid (E6), and all-phenylalanine (F6) peptide-coupled sensors to 100 ppm NH3 (red) and trimethylamine (TMA, purple).
This conclusion was confirmed by noting that sensors containing pure aspartic and glutamic acids respond 1/2 and 1/3, respectively, as strongly to ammonia vapor as the ammonia-binding peptide sequence. The response of these acids to TMA is similarly diminished. This significant decrease in response highlights the importance not just of acid/base reactivity in determining selectivity but also molecular secondary structures, as the ability of the peptide to generate a hydrogen-bonded reception center may be compromised in the pure acid sequences. Finally, we note that, while the hydrophobic F6D peptide responds half as strongly to ammonia as D6D, it responds equally as strongly to TMA as D6D. This is interesting as it suggests that phenyl groups are better able to “solvate” the bulky methyl groups of TMA and thus provides inspiration for designing peptides to nonreactive, nonpolar molecular species.
Simultaneous and Selective Detection from Molecular Mixtures
We also performed target analyte detection experiments in compound chemical backgrounds. Figure 5a plots the response of the sensors to AcOH diluted in a 10-fold excess acetone background. Specifically, an acetic acid peptide-coated sensor and an APTES-only sensor were exposed to continuously flowing 1000 ppm acetone. Both sensors exhibited ~10% conductance increases after 15 min, resulting in a net zero differential response; 100 ppm acetic acid was then injected into the mixture. After 1 min of AcOH influx, the acetic acid peptide-coupled sensor surpassed the APTES response, eventually saturating in a ~7% peptide–APTES conductance gap (consistent with Figure 2b). As acetone and acetic acid are of similar structures and molecular weights (58 vs 60 amu, respectively), we interpret this result as arising from the difference in reactivity between the two molecules.
Figure 5.

(A) Electrical response of an acetic acid recognition peptide–nanowire sensor (blue) and an amine-terminated peptide-free nanowire sensor (green) to 1000 ppm acetone (introduced at time 5 min) and 100 ppm acetic acid (introduced at time 20 min). The black curve is the differential response, obtained by subtracting the green curve from the blue curve. (B) Electrical responses of an acetic acid recognition peptide–nanowire sensor (blue) and an ammonia recognition sensor (red) to sequential influxes of 6% CO2, 100 ppm acetic acid, and 100 ppm ammonia, introduced at the times indicated.
As a closer approximation toward medical applications, we investigated the performance of our sensors in simulated breath backgrounds. Exhaled human breath contains a mixture of ca. 6% CO2 with hundreds of volatile organic compounds (VOCs), mostly in sub-ppm concentrations.40,47 Previous reports have successfully microanalyzed the contents of human breath to identify molecular markers for conditions such as cancers,48 cardiopulmonary19,49 and kidney diseases,42 microbial infections,50 and acute asthma.41 Such demonstrations typically employ GC/MS for molecular separation and identification. Peptide–NW selective sensors on biocompatible plastic substrates provide the potential for implantable, low cost, and continuous monitoring of exhaled breath content at high sensitivities. We tested the responses of peptide–NW hybrid sensors to low levels of NH3 and AcOH molecular markers in a background of 6% CO2. Figure 5b plots the result: exposure of the NH3 and AcOH peptide sensors to CO2 produces no detectable difference in response. By contrast, injection of AcOH in the CO2 background activates the AcOH peptide and subsequent exposure of this mixture to NH3 triggers the ammonia targeting device (consistent with Figure 2b), although at a reduced response due to dilution in the gas mixture. This is a key initial demonstration toward enabling these devices for continuous breath analysis. Nevertheless, the exclusion of humidity in these experiments suggests a motivation for the design of peptides that can be used to normalize against moisture content, in the same way that the APTES/NW sensor was utilized here to normalize against physisorption.
Conclusion
Peptides can be covalently coupled to surfaces of NW sensor arrays using straightforward on-chip modifying reaction chemistry. Upon exposure to target small molecules, the hybrid materials demonstrate the ability to orthogonally sense at low concentrations. Theoretical and experimental investigations into the mechanisms of response show that acid/base reactivity and structural interactions are equally important in realizing analyte selectivity. Molecular detection experiments in chemical mixtures demonstrate the ability to discriminate molecules from “noisy” backgrounds. These results serve as a model platform in the use of sensors for targeted applications such as non-invasive breath monitoring for molecular disease indicators, as well as food spoilage or chemical threat detectors. Finally, through a combination of advanced theoretical modeling and peptide design, we hope to overcome the limitations of our approach, by generating peptides which can be utilized as normalization devices against humidity effects, as well as other peptides that are selective to nonreactive VOCs.
Supplementary Material
Acknowledgments
We thank S. Millward, W. Dichtel, and W. S. Yeo for useful discussions. The XPS measurements were carried out at the Molecular Materials Research Center of the Beckman Institute at Caltech. Peptide purification by HPLC was performed in the Beckman Institute Laser Resource Center at Caltech. M.C.M. thanks the Intelligence Community Postdoctoral Research Fellowship Program for financial support. H.D.A. thanks the National Science Foundation Graduate Research Fellowship Program for financial support. R.D.R. thanks the Gates Millennium Scholars Program for financial support. J.R.H. acknowledges primary support of this work via a contract from the MITRE Corporation, and support from the National Cancer Institute (#5U54 CA119347).
Footnotes
Supporting Information Available: XPS data, molecular modeling coordinates, computed reaction energies, and quantum mechanically determined charges. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Yang W, Auciello O, Butler JE, Cai W, Carlisle JA, Gerbi JE, Gruen DM, Knickerbocker T, Lasseter TL, Russell JN, Smith LM, Hamers RJ. Nat Mater. 2002;1:253–257. doi: 10.1038/nmat779. [DOI] [PubMed] [Google Scholar]
- 2.Bao Z, Weatherspoon MR, Shian S, Cai Y, Graham PD, Allan SM, Ahmad G, Dickerson MB, Church BC, Kang Z, Abernathy HW, III, Summers CJ, Liu M, Sandhage KH. Nature. 2007;446:172–175. doi: 10.1038/nature05570. [DOI] [PubMed] [Google Scholar]
- 3.Cui Y, Wei Q, Park H, Lieber CM. Science. 2001;293:1289–1292. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 4.Kong J, Franklin NR, Zhou C, Chapline MG, Peng S, Cho K, Dai H. Science. 2000;287:622–625. doi: 10.1126/science.287.5453.622. [DOI] [PubMed] [Google Scholar]
- 5.McAlpine MC, Ahmad H, Wang D, Heath JR. Nat Mater. 2007;6:379–384. doi: 10.1038/nmat1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponzoni A, Comini E, Sberveglieri G, Zhou J, Deng SZ, Xu NS, Ding Y, Wang ZL. Appl Phys Lett. 2006;88:203101–203103. [Google Scholar]
- 7.Schedin F, Geim AK, Morozov SV, Hill EW, Blake P, Katsnelson MI, Novoselov KS. Nat Mater. 2007;6:652–655. doi: 10.1038/nmat1967. [DOI] [PubMed] [Google Scholar]
- 8.Staii C, Johnson AT, Chen M, Gelperin A. Nano Lett. 2005;5:1774–1778. doi: 10.1021/nl051261f. [DOI] [PubMed] [Google Scholar]
- 9.Zhang D, Liu Z, Li C, Tang T, Liu X, Han S, Lei B, Zhou C. Nano Lett. 2004;4:1919–1924. [Google Scholar]
- 10.Bunimovich YL, Shin YS, Yeo WS, Amori M, Kwong G, Heath JR. J Am Chem Soc. 2006;128:16323–16331. doi: 10.1021/ja065923u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuzmych O, Allen BL, Star A. Nanotechnology. 2007;18:375502–375508. [Google Scholar]
- 12.Thorpe MJ, Moll KD, Jones RJ, Safdi B, Ye J. Science. 2006;311:1595–1599. doi: 10.1126/science.1123921. [DOI] [PubMed] [Google Scholar]
- 13.Fraden J. Handbook of Modern Sensors: Physics, Designs, and Applications. Springer; New York: 2004. [Google Scholar]
- 14.Stella R, Barisci JN, Serra G, Wallace GG, De Rossi D. Sens Actuators B. 2000;63:1–9. [Google Scholar]
- 15.Freund MS, Lewis NS. Proc Natl Acad Sci USA. 1995;92:2652–2656. doi: 10.1073/pnas.92.7.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martín YG, Oliveros MCC, Pavón JLP, Pinto CG, Cordero BM. Anal Chim Acta. 2001;449:69–80. [Google Scholar]
- 17.Gardner JW, Bartlett PN. Meas Sci Technol. 2000;11:1087. [Google Scholar]
- 18.Sysoev VV, Button BK, Wepsiec K, Dmitriev S, Kolmakov A. Nano Lett. 2006;6:1584–1588. doi: 10.1021/nl060185t. [DOI] [PubMed] [Google Scholar]
- 19.Buck L. Cell. 2000;100:611–618. doi: 10.1016/s0092-8674(00)80698-4. [DOI] [PubMed] [Google Scholar]
- 20.Reed RR. Neuron. 1992;8:205–209. doi: 10.1016/0896-6273(92)90287-n. [DOI] [PubMed] [Google Scholar]
- 21.Wu TZ, Lo YR, Chan EC. Biosens Bioelectron. 2001;16:945–953. doi: 10.1016/s0956-5663(01)00215-9. [DOI] [PubMed] [Google Scholar]
- 22.Mascini M, Macagnano A, Monti D, Del Carlo M, Paolesse R, Chen B, Warner P, D’Amico A, Di Natale C, Compagnone D. Biosens Bioelectron. 2004;20:1203–1210. doi: 10.1016/j.bios.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 23.Buerger C, Nagel-Wolfrum K, Kunz C, Wittig I, Butz K, Hoppe-Seyler F, Groner B. J Biol Chem. 2003;278:37610–37621. doi: 10.1074/jbc.M301629200. [DOI] [PubMed] [Google Scholar]
- 24.Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R. Nature. 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- 25.Banga AK. Therapeutic Peptides and Proteins: Formulation, Processing, and Delivery Systems. CRC Press; Boca Raton, FL: 2006. [Google Scholar]
- 26.Stryer L. Biochemistry. W. H. Freeman and Company; New York: 1995. [Google Scholar]
- 27.Cattanach K, Kulkarni RD, Kozlov M, Manohar SK. Nanotechnology. 2006;17:4123–4128. doi: 10.1088/0957-4484/17/16/022. [DOI] [PubMed] [Google Scholar]
- 28.Qi P, Vermesh O, Grecu M, Javey A, Wang Q, Dai H, Peng S, Cho KJ. Nano Lett. 2003;3:347–351. doi: 10.1021/nl034010k. [DOI] [PubMed] [Google Scholar]
- 29.Snow ES, Perkins FK, Houser EJ, Badescu SC, Reinecke TL. Science. 2005;307:1942–1945. doi: 10.1126/science.1109128. [DOI] [PubMed] [Google Scholar]
- 30.Melosh NA, Boukai A, Diana F, Gerardot B, Badolato A, Petroff PM, Heath JR. Science. 2003;300:112–115. doi: 10.1126/science.1081940. [DOI] [PubMed] [Google Scholar]
- 31.Wang D, Sheriff BA, Heath JR. Nano Lett. 2006;6:1096–1100. doi: 10.1021/nl052558g. [DOI] [PubMed] [Google Scholar]
- 32.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; Oxford: 2000. [Google Scholar]
- 33.Haber JA, Lewis NS. J Phys Chem B. 2002;106:3639–3656. [Google Scholar]
- 34.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 35.Mayo SL, Olafson BD, Goddard WA. J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 36.Blanco M. J Comput Chem. 1991;12:237–247. [Google Scholar]
- 37.Xu K, Heath JR. Nano Lett. 2008;8:136–141. doi: 10.1021/nl072227z. [DOI] [PubMed] [Google Scholar]
- 38.Sakaniwa D, Ohe T, Misumi T, Monjushiro H, Onoda A, Yamamura T. Chem Lett. 2005;34:1634–1635. [Google Scholar]
- 39.Cheng CC, Guinn KV, Herman IP, Donnelly VM. J Vac Sci Technol A. 1995;13:1970–1976. [Google Scholar]
- 40.Phillips M. Anal Biochem. 1997;247:272–278. doi: 10.1006/abio.1997.2069. [DOI] [PubMed] [Google Scholar]
- 41.Effros RM, Casaburi R, Su J, Dunning M, Torday J, Biller J, Shaker R. Am J Respir Crit Care Med. 2006;173:386–392. doi: 10.1164/rccm.200507-1059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spanel P, Davies S, Smith D. Rapid Commun Mass Spectrom. 1998;12:763–766. doi: 10.1002/(SICI)1097-0231(19980630)12:12<763::AID-RCM229>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 43.Hair ML. Infrared Spectroscopy in Surface Chemistry. Dekker; New York: 1967. [Google Scholar]
- 44.Marquis BT, Vetelino JF. Sens Actuators B. 2001;77:100–110. [Google Scholar]
- 45.Gong H, Wang YJ, Teo SC, Huang L. Sens Actuators B. 1999;54:232–235. [Google Scholar]
- 46.Car R, Parrinello M. Phys Rev Lett. 1985;55:2471–2474. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- 47.Turner AP, Magan N. Nat Rev Microbiol. 2004;2:161–166. doi: 10.1038/nrmicro823. [DOI] [PubMed] [Google Scholar]
- 48.Di Natale C, Macagnano A, Martinelli E, Paolesse R, D’Arcangelo G, Roscioni C, Finazzi-Agrò A, D’Amico A. Biosens Bioelectron. 2003;18:1209–1218. doi: 10.1016/s0956-5663(03)00086-1. [DOI] [PubMed] [Google Scholar]
- 49.Skrupski VA. Clin Lab Diagn. 1995;4:35–38. [Google Scholar]
- 50.Klein PD, Malaty HM, Martin RF, Graham KS, Genta RM, Graham DY. Am J Gastroenterol. 1996;91:690–694. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.