

Abstract
BACKGROUND:
Vitronectin (VN) is an abundant acute-phase plasma protein that regulates cell adhesion and migration as well as interactions with components of the plasminogen activator/plasmin system, specifically plasminogen activator inhibitor type 1. This system plays a major role in tissue remodelling regulating wound healing after myocardial infarction.
OBJECTIVES:
To investigate the feasibility of using VN knockout mice (VN−/−) to study the role of VN on ventricular remodelling following myocardial infarction.
METHODS:
Specifically bred VN−/− mice and normal wild-type (VN+/+) mice underwent coronary artery ligation and were assessed 28 days postligation using echocardiography and morphometric histology.
RESULTS:
No difference was observed between VN−/− mice and VN+/+ mice with respect to gross phenotype, weight, coronary anatomy or echocardiographically measured ejection fraction (56%). Following myocardial infarction, VN−/− mice exhibited less ventricular dilation and less impairment in echocardiographic ejection fraction compared with VN+/+ mice (48% versus 41%; P=0.01). VN−/− mice also exhibited smaller infarcts on morphometric analysis.
CONCLUSIONS:
The results of the present study confirmed the feasibility of using coronary artery ligation in VN knockout mice to investigate the role of VN in post-myocardial infarction remodelling. The absence of VN appears to result in favourable effects on wound healing. These data suggest that this model may offer novel insights into the role of VN in the regulation of myocardial remodelling.
Keywords: Myocardial infarction, Remodelling, Vitronectin
Post-myocardial infarction (MI) wound healing is a complex process during which the size, shape and function of the heart are modulated by mechanical, neurohormonal and genetic factors. Vitronectin (VN) is an acute-phase plasma protein with multiple ligands that accumulates at various sites of inflammatory cell-induced damage and necrosis (1,2). VN is commonly present in the plasma exudate at sites of tissue injury and has been implicated in complement activation and wound healing. In addition, VN provides abundant attachment sites for multiple cell types by binding various integrins, thus modulating adhesion and migration of cells in the damaged area (3,4). It has also been suggested that deposition of VN may regulate the fibrinolytic response in the provisional wound matrix by binding and stabilizing PAI-1 (5,6). However, little is currently known regarding its role in post-MI remodelling.
Post-MI remodelling involves modification of the extracellular matrix (ECM), which is achieved, in part, through a tight regulation of proteases and the plasminogen activator (PA)/plasmin system (7,8). The PA/plasmin system, and the matrix metalloproteinase (MMP) system that it regulates, is involved in cell migration and activation of proteases and inflammatory cytokines, as well as ECM synthesis and degradation (9–12). Plasminogen knockout mice demonstrate a lack of immune cell activation and infiltration into the infarct zone post-MI, resulting in reduced removal of necrotic tissue and more fibrous healing (8). Urokinase PA (u-PA) knockout mice exhibit impaired scar formation and infarct revascularization, and increased death from cardiac failure due to depressed contractility, arrhythmias and ischemia (11). Furthermore, an increase in PA inhibitor type 1 (PAI-1), the major inhibitor of PAs, is associated with impaired remodelling and increased cardiac fibrosis post-MI, which contributes to progressive left ventricular (LV) dysfunction (11,13–15). In infarcted hearts, PAI-1 is derived from various cardiac cell types including cardiomyocytes, endothelial cells and fibroblasts (13,16). Secreted PAI-1 plays a major role in post-MI fibrosis by preventing the activation of PAs and, hence, MMPs (13,14). Significant increases in PAI-1 levels have been observed in scars from failing and aging hearts (13,17).
By forming complexes with PAI-1, VN stabilizes the active conformation of PAI-1, extending its half-life and, therefore, reducing plasmin generation (5,18). Moreover, the binding of VN to PAI-1 causes a conformational change in VN, promoting VN multimerization, which stimulates additional PAI-1 binding (5). Prolonged accumulation of VN at sites of tissue damage reflects chronic injury associated with fibrosis (19–21). Deposition of plasma VN conceivably regulates the fibrinolytic response in the provisional wound matrix by binding and stabilizing PAI-1 (22,23). Moreover, the absence of VN may lead to a deficiency in PAI-1, which has been shown to reduce myocardial (13), pulmonary (21), nerve (24) and dermal (25) fibrosis, and to accelerate regeneration and wound healing. Thus, VN, through interaction with PAI-1, plays a major role in tissue healing following injury and may also be involved in regulating post-MI remodelling through both plasmin-dependent and plasmin-independent mechanisms. We hypothesized that the absence of VN would improve post-MI remodelling and reduce cardiac fibrosis.
In the present study, we examined the feasibility of using coronary artery ligation in VN knockout (VN−/−) mice as a model to investigate the effects of VN on myocardial healing and function following infarction. Following left anterior descending artery (LAD) ligation, we compared the wound healing response of wild-type (VN+/+) mice with VN−/− mice. Initial findings showed that the absence of VN significantly reduced fibrous scar formation, promoted LV healing and reduced cardiac dysfunction. Our findings demonstrate that this mouse model appears to be valid, and suggest a novel role for VN in the regulation of ventricular remodelling following MI.
METHODS
Evaluation of VN−/− mouse model
The animal experiments were performed in an accredited facility with approval from the University of British Columbia Committee on Animal Care (Vancouver, British Columbia). VN−/− mice were purchased from a commercial source (Jackson Laboratories, USA). Animals were cross-bred with VN+/+ C57B6/6J mice and the heterozygotic offspring were cross-bred again. Offspring were then genotyped using tail clippings and appropriately segregated to create sustainable breeding colonies of homozygotic VN+/+ and VN−/− mice.
Mouse model of LAD ligation
The mouse model of wound healing following acute MI has been described previously (26). Twelve-week-old C57B6/6J VN+/+ and VN−/− mice (body weight 20 g to 25 g) underwent surgical ligation of the LAD under general anesthesia. Coronary artery anatomy was defined as branching (into the LAD and large diagonal branch) or nonbranching according to previously described patterns (27). The LAD was then tied either above or below the first diagonal branch. A qualitative estimate of infarct size was made by the operator through a visual estimation of pale demarcation. Mice were allowed to recover and were observed for varying periods before they were euthanized. Echocardiography was performed 28 days post-MI, after which the mice were euthanized.
Echocardiographic analysis
Echocardiographic evaluation was performed in noninfarcted control mice and infarcted mice 28 days after infarction using a 700 VEVO system (VisualSonics, Canada) with a 30 MHz probe, probe holder and data analysis unit. Animals were lightly sedated with a 0.1 mL/30 g solution of a mixture of ketamine (300 μL) and xylazine (40 μL) (one-third of the dose used for ligation purposes). The lightest sedation compatible with keeping animals sedated while in the supine position was used. After chest hair was removed, the animal was secured in the left lateral position on the examination table. Liberal quantities of ultrasound jelly were applied to the chest wall. Multiple echocardiographic images were obtained using different probe and table positions including a parasternal long axis and short axis views at the levels of the submitral valve, papillary muscle, subpapillary muscle and apex. Three separate measurements were obtained and the mean was determined for each animal. Ejection fraction was calculated according to Simpson’s method. Offline analysis was performed in a blinded manner using only numeric identifiers.
Histology and morphometry
Following echocardiography, animals were again fully anesthetized, had their chest hair removed and were intubated and immobilized under a dissecting microscope. The chest wall was opened using a long midsternal incision, which was extended caudally into the abdomen. A visual estimation of infarct size was made by the operator and recorded (none, small, medium, large). The beating heart was removed and placed briefly in normal saline to rinse out blood before being placed in KCl solution to arrest the heart in diastole. The aorta was then attached to a cannula and the heart was perfused with normal saline to remove any residual blood before being immersed in formalin for 24 h and embedded in paraffin for histological and morphometric analysis. Paraffin-embedded heart tissue was sectioned into 5 μm thick slices and stained using picrosirius red. Slides were scanned under high resolution (300 dpi) and analyzed using a computerized morphometry system (Image Pro Plus, Media Cybernetics, USA). The area occupied by the infarct was measured and expressed as the ratio of picrosirius red-positive scar area to total LV area. Measurements were performed in serial sections at intervals of 500 μm, beginning at the midventricle and ending at the apex. Final infarct size was expressed as a percentage of the total area of four to six sections from each heart. LV, right ventricular and septum geometries were determined as previously described (28).
Proteinalysis and immunohistochemistry
Protein levels were measured using Western blot analysis as previously described (29). Briefly, Triton X-insoluble heart extracts (20 μg/lane) were treated with lysis buffer (100 mM Tris buffer, pH 8.3, containing 2% sodium dodecylsulfate, 5 mM EDTA, 10% glycerol and 15 mM dithiothreitol) and fractionated using polyacrylamide gel electrophoresis. After fractionation, the gels were electrophoretically transferred onto nitrocellulose membranes. Subsequently, the membranes were blocked using phosphate-buffered saline containing 10% casein and incubated with antibodies to VN, PAI-1 or tissue PA for 1 h. The membranes were then incubated with alkaline phosphatase-conjugated streptavidin and processed for colour development using the Fast Red system. The relative intensity of the bands was compared using ImageJ software version 1.38 (National Institutes of Health, USA) and tabulated using SigmaPlot software version 8.0 (Systat Software, USA).
Paraffin-embedded heart tissue sections were subjected to routine immunohistochemistry using purified antibodies to VN or pre-immune immunoglobulin G obtained from Molecular Innovations (USA). Staining was visualized using the chromogen Vector Red (Vector Laboratories, USA).
Statistical analysis
All data were entered into spreadsheets (Excel, Microsoft Corporation, USA) with a unique numeric subject identifier. Tables and charts were prepared using Microsoft Excel and Word (Microsoft Corporation, USA). All values are expressed as mean ± SD or mean ± SE. GraphPad InStat version 3.00 (GraphPad Software, USA) for Windows 95 (Microsoft Corporation, USA) was used to perform statistical analyses. Statistical significance was assessed using two-tailed Student’s t tests and comparisons among multiple groups were performed using one-way ANOVA; P<0.05 was considered to be statistically significant. The visual estimations of infarct size were compared using χ2 analysis.
RESULTS
Colonies of VN+/+ and VN−/− mice were established after cross-breeding and appropriate selection based on genotyping of tail clippings. Control, noninfarcted animals were assessed using echocardiography and pressure-wave analysis. No differences were noted with regard to gross phenotype, weight, coronary artery anatomy, echocardiographical parameters or pressure-wave parameters (Table 1).
TABLE 1.
Echocardiographic evaluation of control (noninfarcted) mice
| Characteristic | VN+/+ (n=11) | VN−/− (n=10) | P |
|---|---|---|---|
| Weight, g | 22±5 | 23±4 | 0.41 |
| Age, weeks | 13±3 | 14±4 | 0.64 |
| M-mode measurements | |||
| IVS, diastole, mm | 0.91±0.25 | 0.90±0.29 | 0.98 |
| LVID, diastole, mm | 3.36±0.51 | 3.24±0.23 | 0.52 |
| IVS, systole, mm | 1.18±0.27 | 1.17±0.24 | 0.96 |
| LVID, systole, mm | 2.10±0.42 | 2.00±0.15 | 0.50 |
| LVPW, diastole, mm | 0.93±0.20 | 1.09±0.20 | 0.08 |
| LVPW, systole, mm | 1.29±0.21 | 1.42±0.24 | 0.23 |
| Heart rate, beats/min | 219±65 | 231±54 | 0.66 |
| Simpson’s measurements | |||
| End-systolic volume, μL | 28.8±10.7 | 27.5±8.2 | 0.77 |
| End-diastolic volume, μL | 66.1±22.5 | 62.1±15.4 | 0.64 |
| Stroke volume, μL | 37.3±13.2 | 34.6±8.4 | 0.58 |
| Ejection fraction, % | 56.3±7.0 | 55.9±4.9 | 0.89 |
| Cardiac output, μL/min | 9.0±6.5 | 8.1±3.2 | 0.71 |
Data presented as mean ± SD unless otherwise indicated. IVS Interventricular septal thickness; LVID Left ventricular internal diameter; LVPW Left ventricular posterior wall thickness; VN−/− Vitronectin knockout; VN+/+ Wild-type
Coronary artery ligation
Twelve-week-old VN−/− and VN+/+ mice were subjected to LAD ligation. There were no significant differences in age, weight, phenotype or visual estimation of initial infarct size between the groups at the time of ligation (Table 2). A single main artery was present in 29% of VN+/+ mice and 32% of VN−/− mice (difference not statistically significant).
TABLE 2.
Evaluation of infarcted mice one month after coronary artery ligation
| Baseline | VN+/+ (n=27) | VN−/− (n=31) | P |
|
| |||
| Age, weeks | 11.4±0.9 | 10.6±0.7 | ns* |
| Weight, g | 29.2±1.8 | 27.9±2.2 | ns* |
| Perioperative mortality, n | 1 | 1 | ns† |
| Site of ligation, n | ns† | ||
| Mid-LAD | 23 | 26 | |
| Main LAD | 3 | 5 | |
| Infarct size, n | ns† | ||
| Large | 3 | 7 | |
| Medium | 20 | 23 | |
| Small | 3 | ||
| Postoperative mortality, n | 2 | 3 | ns† |
| Total mortality (28 days), n (%) | 3 (11) | 4 (13) | ns† |
| 28 days post-MI | VN+/+(n=24) | VN−/− (n=27) | |
|
| |||
| Days postligation | 28.6±0.9 | 28.4±0.7 | ns* |
| Weight, g | 29.0±6.4 | 28.5±6.0 | ns* |
| Infarct size | |||
| Large | 10 | 5 | 0.07† |
| Medium | 10 | 13 | |
| Small | 3 | 7 | |
| None | 1 | 2 | |
| Simpson’s measurements | |||
| End-systolic volume, μL | 68.7±25.2 | 49.0±14.0 | 0.01 |
| End-diastolic volume, μL | 114.5±30.1 | 94.9±21.7 | 0.03 |
| Stroke volume, μL | 45.9±8.8 | 45.9±11.9 | ns |
| Ejection fraction, % | 41.4±7.6 | 48.4±7.0 | 0.005 |
| Heart rate, beats/min | 219±61 | 215±73 | ns |
| Cardiac output, μL/min | 9.7±1.9 | 10.0±4.5 | ns |
Data presented as mean ± SD unless otherwise indicated.
Student’s t test used for the comparison of means;
χ2 test for proportions. LAD Left anterior descending artery; MI Myocardial infarction; ns Not statistically significant; VN−/− Vitronectin knockout; VN+/+ Wild-type
VN−/− mice exhibit a smaller reduction in cardiac function relative to VN+/+ mice post-MI
There were no differences in mortality or weight 28 days after LAD ligation. Mortality was low (8% early, 12% late); the majority of the early deaths were likely related to arrhythmias or acute heart failure resulting in death either intraoperatively or within 24 h postoperatively. Later deaths in both groups were associated with cardiac rupture and massive hemopericardium. There was a trend toward smaller infarcts by qualitative assessment; however, this did not reach statistical significance. Echocardiographical parameters demonstrated that VN+/+ mice had larger end-systolic and end-diastolic volumes and a significantly reduced ejection fraction (41% versus 48%) compared with VN−/− mice.
VN deposition occurs within infarct zones
Histopathological evaluation at various time points revealed that in VN+/+ mice (n=8), VN localized to the myocardial infarct zone, peaking at seven days, and was predominantly present in the cytoskeletal (sodium dodecylsulfate-soluble) fraction, suggesting that the VN was bound to cytoskeletal components. VN was absent, as expected, in VN−/− mice (n=8) (Figure 1).
Figure 1).
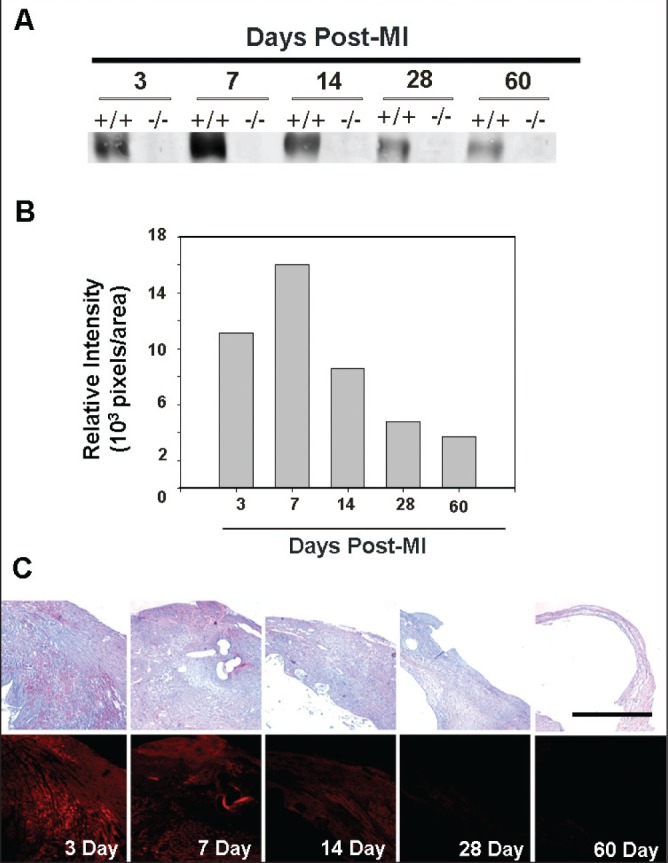
Vitronectin (VN) deposition following myocardial infarction (MI). Influx of VN into the cytoskeletal (sodium dodecylsulfate-soluble) fraction of the myocardial infarct zone, peaking at seven days, in wild type (+/+) mice (n=8) in both serial histological sections and Western blot analysis. VN is absent in the VN knockout (−/−) mice (n=8). The black bar represents 1 mm
VN−/− mice exhibit attenuated ventricular remodelling post-MI
Morphometric analysis of picrosirius red staining of sequential cardiac sections, defining collagen deposition as a percentage of the LV area, showed significantly greater staining (P<0.01) in infarcts in VN+/+ mice compared with VN−/− mice (VN+/+, n=8; VN−/−, n=8) (Figure 2). The thickness of the LV free wall decreased following infarction, while the LV internal diameter increased, as would be expected, with compensatory hypertrophic thickening of the noninfarcted septum and right ventricular wall, although none of these trends reached statistical significance.
Figure 2).
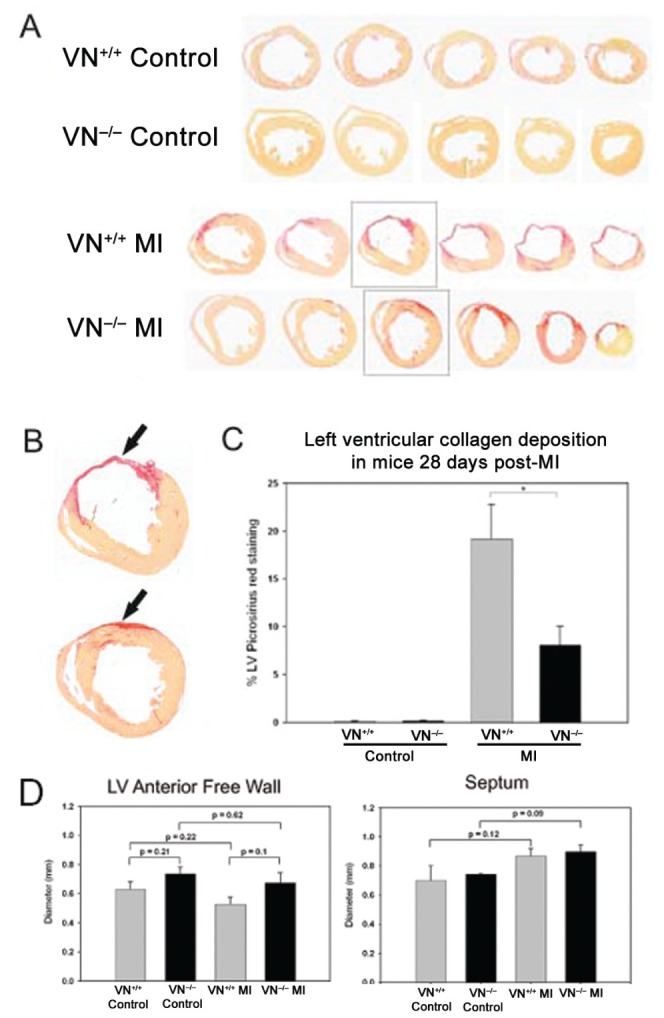
Morphometric examination of control hearts (noninfarcted) and hearts subjected to myocardial infarction (MI) (day 28) in wild-type (VN+/+) and vitronectin knockout (VN−/−) mice. Examples of sequential picrosirius red-stained cardiac sections ( A ) showing collagen deposition ( B , arrows). Analysis of the percentage of the left ventricle stained with picrosirius red showed significantly greater staining in VN+/+ infarcts than VN−/−( C, P=0.01). Analysis of the thickness of the left ventricular (LV) free wall and septum showed decreases in free wall thickness following infarction, as expected, with compensatory hypertrophic thickening of the noninfarcted septum ( D). There were no statistically significant differences between mouse genotypes (VN+/+, n=8; VN−/−, n=8)
DISCUSSION
These are the first data regarding the use of VN−/− mice as a model for MI. Noninfarcted controls exhibited no significant phenotypic differences between VN+/+ and VN−/− mice in coronary artery anatomy or echocardiographic parameters, suggesting that the absence of VN does not effect normal cardiac development and physiology, as observed by other investigators (30). Following MI, mice lacking VN exhibited a smaller reduction in echocardiographically-assessed ejection fraction compared with VN+/+ mice. Quantitative morphometry revealed significantly less collagen deposition, and although there were no other statistically significant differences in individual parameters of post-MI cardiac architecture, there were consistent trends toward improvements in VN−/− mice. Together, these findings suggest that coronary arterial ligation in the VN−/− mouse is a useful model for investigating the role of VN in MI, and that there may be improved post-MI remodelling in the absence of VN. Our findings are consistent with other studies on VN performed using mouse models that suggest VN regulates repair and fibrosis.
VN modulates the activation of proteinases and inflammatory cytokines, as well as ECM synthesis and degradation, through interactions with PAI-1. Other investigators have found that, although excessive PA or MMP activity is associated with a detrimental outcome post-MI (11,22), modest expression/activity is essential for appropriate wound healing. Complete absence of PAI-1 in a knockout mouse model was associated with unopposed fibrinolysis and an acute increase in cardiac rupture. In VN−/− mice, PAI-1 activity would be expected to be reduced rather than completely absent, which may explain why no increase in cardiac rupture was observed. This model may, therefore, be more useful than the PAI-1 knockout model to study remodelling.
Study limitations
The present study evaluated the functional effects of infarction in VN+/+ and VN−/− mice in combination with simple morphometric analysis. Now that the validity of this model has been demonstrated, further work needs to be performed to confirm that these findings were not due to chance and to evaluate the effects of the absence of VN on PAI-1 and other elements of the plasminogen activator system. More detailed analyses of cell migration and healing also need to be performed.
Our echocardiographical data were derived from images obtained in the supine position in mice anesthetized using ketamine/xylazine. This anesthetic is known to affect output parameters, particularly by causing bradycardia (31). However, the anesthetic appears to have affected both groups equally; therefore, the differences noted are likely valid. Echocardiography can also be performed on prone, conscious mice, which avoids the negative inotropic and chronotropic effects of anesthesia, offers the opportunity for assessment at multiple time-points and appears to be a more preferable means of assessment of function following MI (32).
Functional evaluation of infarcted mice using echocardiography was performed only at 28 days rather than serially at multiple time-points due to local restrictions regarding animal husbandry. Development of heart failure traditionally begins to manifest in murine models at six to eight weeks post-MI, and generally results in a rapid increase in weight caused by excessive fluid retention and development of dyspnea caused by LV dysfunction. The duration of these experiments was relatively short; therefore, we were unable to comment on the development of heart failure. These experiments would need to be performed over longer intervals to confirm whether the observed improvements in LV function in the absence of VN translate into reductions in heart failure.
CONCLUSIONS
Here, for the first time, we have used VN−/− mice in a model of MI. Our data suggest that this appears to be a valid model to study the role of VN following MI. The absence of VN appears to be associated with improved wound healing following MI, with consistent results observed using both echocardiographic and morphometric analysis. Additional work needs to be performed to confirm these findings, to understand the mechanisms by which VN regulates wound healing and to determine whether strategies aimed at preventing VN deposition at sites of myocardial ischemia may offer a therapeutic option to improve post-MI remodelling.
REFERENCES
- 1.Reilly JT, Nash JR. Vitronectin (serum spreading factor): Its localisation in normal and fibrotic tissue. J Clin Pathol. 1988;41:1269–72. doi: 10.1136/jcp.41.12.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Podor TJ, Joshua P, Butcher M, Seiffert D, Loskutoff D, Gauldie J. Accumulation of type 1 plasminogen activator inhibitor and vitronectin at sites of cellular necrosis and inflammation. Ann N Y Acad Sci. 1992;667:173–7. doi: 10.1111/j.1749-6632.1992.tb51609.x. [DOI] [PubMed] [Google Scholar]
- 3.Kostin S, Hein S, Arnon E, Scholz D, Schaper J. The cytoskeleton and related proteins in the human failing heart. Heart Fail Rev. 2000;5:271–80. doi: 10.1023/A:1009813621103. [DOI] [PubMed] [Google Scholar]
- 4.Vakeva A, Laurila P, Meri S. Regulation of complement membrane attack complex formation in myocardial infarction. Am J Pathol. 1993;141:65–75. [PMC free article] [PubMed] [Google Scholar]
- 5.Seiffert D, Loskutoff DJ. Type 1 plasminogen activator inhibitor induces multimerization of plasma vitronectin. A suggested mechanism for the generation of the tissue form of vitronectin in vivo. J Biol Chem. 1996;271:29644–51. doi: 10.1074/jbc.271.47.29644. [DOI] [PubMed] [Google Scholar]
- 6.Gibson A, Baburaj K, Day DE, Verhamme I, Shore JD, Peterson CB. The use of fluorescent probes to characterize conformational changes in the interaction between vitronectin and plasminogen activator inhibitor-1. J Biol Chem. 1997;272:5112–21. doi: 10.1074/jbc.272.8.5112. [DOI] [PubMed] [Google Scholar]
- 7.Cleutjens JP, Creemers EE. Integration of concepts: Cardiac extracellular matrix remodeling after myocardial infarction. J Card Fail. 2002;8(Suppl 6):S344–8. doi: 10.1054/jcaf.2002.129261. [DOI] [PubMed] [Google Scholar]
- 8.Creemers E, Cleutjens J, Smits J, et al. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am J Pathol. 2000;156:1865–73. doi: 10.1016/S0002-9440(10)65060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werb Z, Mainardi CL, Vater CA, Harris ED., Jr Endogenous activiation of latent collagenase by rheumatoid synovial cells. Evidence for a role of plasminogen activator. N Eng J Med. 1977;296:1017–23. doi: 10.1056/NEJM197705052961801. [DOI] [PubMed] [Google Scholar]
- 10.Chapman HA, Jr, Stone OL. Co-operation between plasmin and elastase in elastin degradation by intact murine macrophages. Biochem J. 1984;222:721–8. doi: 10.1042/bj2220721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–42. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 12.Gaertner R, Jacob MP, Prunier F, Angles-Cano E, Mercadier JJ, Michel JB. The plasminogen-MMP system is more activated in the scar than in viable myocardium 3 months post-MI in the rat. J Mol Cell Cardiol. 2005;31:193–204. doi: 10.1016/j.yjmcc.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Takeshita K, Hayashi M, Iino S, et al. Increased expression of plasminogen activator inhibitor-1 in cardiomyocytes contributes to cardiac fibrosis after myocardial infarction. Am J Pathol. 2004;164:449–56. doi: 10.1016/S0002-9440(10)63135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Askari AT, Brennan ML, Zhou X, et al. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J Exp Med. 2003;197:615–24. doi: 10.1084/jem.20021426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling – concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 16.Hekman CM, Loskutoff DJ. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J Biol Chem. 1985;260:11581–7. [PubMed] [Google Scholar]
- 17.Sobel BE, Lee YH, Pratley RE, Schneider DJ. Increased plasminogen activator inhibitor type-1 (PAI-1) in the heart as a function of age. Life Sci. 2006;79:1600–5. doi: 10.1016/j.lfs.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 18.Wun TC, Palmier MO, Siegel NR, Smith CE. Affinity purification of active plasminogen activator inhibitor-1 (PAI-1) using immobilized anhydrourokinase. Demonstration of the binding, stabilization, and activation of PAI-1 by vitronectin. J Biol Chem. 1989;264:7862–8. [PubMed] [Google Scholar]
- 19.Sobel RA, Chen M, Maeda A, Hinojoza JR. Vitronectin and integrin vitronectin receptor localization in multiple sclerosis lesions. J Neuropathol Exp Neurol. 1995;54:202–13. doi: 10.1097/00005072-199503000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Koukoulis GK, Shen J, Virtanen I, Gould VE. Vitronectin in the cirrhotic liver: An immunomarker of mature fibrosis. Hum Pathol. 2001;32:1356–62. doi: 10.1053/hupa.2001.29675. [DOI] [PubMed] [Google Scholar]
- 21.Lazar MH, Christensen PJ, Du M, et al. Plasminogen activator inhibitor-1 impairs alveolar epithelial repair by binding to vitronectin. Am J Respir Cell Mol Biol. 2004;31:672–8. doi: 10.1165/rcmb.2004-0025OC. [DOI] [PubMed] [Google Scholar]
- 22.Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res. 2000;46:214–24. doi: 10.1016/s0008-6363(00)00003-1. [DOI] [PubMed] [Google Scholar]
- 23.Lee RT. Matrix metalloproteinase inhibition and the prevention of heart failure. Trends Cardiovasc Med. 2001;11:202–5. doi: 10.1016/s1050-1738(01)00113-x. [DOI] [PubMed] [Google Scholar]
- 24.Wang AG, Yen MY, Hsu WM, Fann MJ. Induction of vitronectin and integrin alphav in the retina after optic nerve injury. Mol Vis. 2006;12:76–84. [PubMed] [Google Scholar]
- 25.Jang YC, Tsou R, Gibran NS, Isik FF. Vitronectin deficiency is associated with increased wound fibrinolysis and decreased microvascular angiogenesis in mice. Surgery. 2000;127:696–704. doi: 10.1067/msy.2000.105858. [DOI] [PubMed] [Google Scholar]
- 26.Rezai N, Walinski H, Kerjner A, et al. Methods for examining stem cells in post-ischemic and transplanted hearts. Methods Mol Med. 2005;112:223–38. doi: 10.1385/1-59259-879-x:223. [DOI] [PubMed] [Google Scholar]
- 27.Michael LH, Entman ML, Hartley CJ, et al. Myocardial ischemia and reperfusion: A murine model. Am J Physiol. 1995;269:H2147–54. doi: 10.1152/ajpheart.1995.269.6.H2147. [DOI] [PubMed] [Google Scholar]
- 28.Weisman HF, Bush DE, Mannisi JA, Bulkley BH. Global cardiac remodeling after acute myocardial infarction: A study in the rat model. J Am Coll Cardiol. 1985;5:1355–62. doi: 10.1016/s0735-1097(85)80348-x. [DOI] [PubMed] [Google Scholar]
- 29.Podor TJ, Singh D, Chindemi P, et al. Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J Biol Chem. 2002;277:7529–39. doi: 10.1074/jbc.M109675200. [DOI] [PubMed] [Google Scholar]
- 30.Zheng X, Saunders TL, Camper SA, Samuelson LC, Ginsburg D. Vitronectin is not essential for normal mammalian development and fertility. Proc Natl Acad Sci USA. 1995;92:12426–30. doi: 10.1073/pnas.92.26.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaves AA, Weinstein DM, Bauer JA. Non-invasive echocardiographic studies in mice: Influence of anesthetic regimen. Life Sci. 2001;69:213–22. doi: 10.1016/s0024-3205(01)01123-7. [DOI] [PubMed] [Google Scholar]
- 32.Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am J Physiol. 1999;277:H1967–74. doi: 10.1152/ajpheart.1999.277.5.H1967. [DOI] [PubMed] [Google Scholar]