

Abstract
Study Objectives:
In obstructive sleep apnea (OSA), arousals generally occur at apnea termination and help restore airflow. However, timing of arousals in central sleep apnea (CSA) has not been objectively quantified, and since arousals can persist even when CSA is alleviated, may not play the same defensive role as they do in OSA. We hypothesized that arousals following central events would occur longer after event termination than following obstructive events and would be related to circulation time.
Methods:
We examined polysomnograms from 20 patients with heart failure (HF) (left ventricular ejection fraction ≤ 45%): 10 with OSA and 10 with CSA (apneahypopnea index ≥ 15). Twenty central or obstructive apneas or hypopneas were analyzed in each patient.
Results:
Compared to the OSA group in whom arousals generally occurred at obstructive event termination, in the CSA group they occurred longer after central event termination (0.9 ± 1.1 versus 8.0 ± 4.1 s, p < 0.0001), but before peak hyperpnea. Time from arousal to peak hyperpnea did not differ between groups (4.3 ± 1.1 vs 4.8 ± 1.6 s, p = 0.416). Unlike the OSA group, latency from apnea termination to arousal correlated with circulation time in the CSA group (r = 0.793, p = 0.006).
Conclusions:
In HF patients with CSA, apnea-to-arousal latency is longer than in those with OSA, and arousals usually follow resumption of airflow. These observations provide evidence that arousals are less likely to act as a protective mechanism to facilitate resumption of airflow following apneas in CSA than in OSA.
Citation:
Simms T; Brijbassi M; Montemurro LT; Bradley TD. Differential timing of arousals in obstructive and central sleep apnea in patients with heart failure. J Clin Sleep Med 2013;9(8):773-779.
Keywords: Arousal, obstructive sleep apnea, central sleep apnea, heart failure
In obstructive sleep apnea (OSA), arousals occur at the end of most obstructive events,1,2 and help restore upper airway patency by activating pharyngeal dilator muscles.3 Thus, arousals generally act as a defense mechanism to reestablish airflow and prevent asphyxia in OSA. The role of arousals in central sleep apnea (CSA), however, is less clear. For example, it has been reported that arousals can occur several breaths after apnea termination in heart failure (HF) patients with CSA.4 In such cases, reinstitution of airflow prior to the occurrence of arousal suggests that such arousals may not have the same protective role as they do in OSA. In fact, arousals have been implicated in the pathogenesis of CSA by contributing to respiratory control system instability. Arousals frequently cause an abrupt increase in ventilation, which, in susceptible subjects, drives the PaCO2 below the apnea threshold, triggering a central apnea.5–7 They may also perpetuate further events by provoking ventilatory overshoot during hyperpnea by abruptly increasing chemical ventilatory responsiveness and activating the neurogenic wakefulness drive to breathe.6,7 However, the precise timing of arousals in relation to CSA has not been quantified or compared to timing of arousals in relation to OSA in patients with HF.
Many studies of patients with OSA, both with and without HF, have shown that alleviation of OSA with continuous positive airway pressure (CPAP) dramatically lowers the frequency of arousals.8–14 This observation provides further evidence that OSA causes arousals that are reversible through treatment of OSA. However, in a large, randomized controlled trial involving 258 patients with HF and CSA, alleviation of CSA with CPAP did not reduce arousal frequency.15 These findings suggest that arousals in HF patients with CSA may not be acting as a defense mechanism to the same extent as they do in OSA, and indeed, in many instances may not be a consequence of central events. These observations have given rise to the concept that arousals may play a causative role5–7 or may be an incidental finding,15 rather than serving as a protective mechanism in CSA.
BRIEF SUMMARY
Current knowledge/Study Rationale: Whereas in obstructive sleep apnea (OSA), arousals generally occur at apnea termination and act as a defense mechanism to reestablish airflow, in central sleep apnea (CSA), timing of arousals postapnea termination has not been objectively assessed, and factors related to arousal timing have not been identified. We therefore tested the hypotheses that arousals would coincide closely with termination of obstructive events, in keeping with their protective role, while they would be delayed following reestablishment of airflow after central events in proportion to circulation time, suggesting a non-protective role.
Study Impact: Among patients with heart failure, whereas in those with OSA, arousals were closely linked to apnea termination and were unrelated to circulation time, in those with CSA arousals occurred at longer and more highly variable latencies following apnea termination that were proportional to circulation time. These findings suggest (1) unlike OSA, arousals in CSA do not serve the same protective role in facilitating airflow reestablishment following apneas, and (2) in contrast to OSA, their timing is influenced by cardiac function and circulation time.
Although suppression of CSA by CPAP may not reduce total arousal frequency, the assumption that arousals are therefore not involved in facilitating resumption of respiration at the end of central apneas remains speculative. While it has been reported anecdotally that arousals usually occur several breaths after termination of central events in HF patients,4 the actual timing of arousals was not quantified. We therefore compared the timing of arousals with respect to apnea and hypopnea termination between HF patients with OSA and those with CSA. We therefore tested two hypotheses: (1) that arousals would coincide more closely with termination of obstructive events (in keeping with their protective role), while they would be more delayed until after reestablishment of airflow following central events; and (2) that owing to the longer delay of transmission of blood gas tensions from the lungs to the chemoreceptors in HF patients with CSA than in those with OSA,16 arousals following central events, but not obstructive events, would be related to circulation time.
METHODS
Subjects
Consecutive patients with HF were recruited from Mount Sinai Hospital Heart Failure Clinic in Toronto as part of an ongoing prospective epidemiological study irrespective of signs or symptoms of sleep apnea if they met the following criteria: (1) men and women ≥ 18 years of age; (2) HF with left ventricular systolic dysfunction (left ventricular ejection fraction [LVEF] ≤ 45% by radionuclide angiography or echocardiography) secondary to ischemic or nonischemic dilated cardiomyopathy for ≥ 6 months; (3) New York Heart Association (NYHA) class I to III; (4) stable clinical status on optimal medical therapy for ≥ 1 month before entry; and (5) moderatetosevere sleep apnea, defined as ≥ 15 apneas and hypopneas per hour of sleep (apneahypopnea index [AHI]). Patients were further classified as having predominantly OSA if ≥ 50% of the events were obstructive, or predominantly CSA if > 50% of the events were central. Exclusion criteria were unstable angina, myocardial infarction, cardiac surgery within the previous 3 months, and pregnancy. The protocol was approved by the local research ethics board, and all subjects provided written informed consent before entry.
Polysomnography
Diagnostic overnight polysomnography was performed in all subjects using standard techniques and scoring criteria for sleep stages.17,18 Wakefulness and sleep stages were identified by central (C3/A2; C4/A1), occipital (O1/A2; O2/A1), and frontal (F3/A2; F4/A1) electroencephalogram (EEG), bilateral electrooculogram (EOG), and submental electromyogram (EMGsm) recordings from surface electrodes. Arousals were defined by American Academy of Sleep Medicine 2007 criteria,18 and arousal index (ArI) was calculated as the total number of arousals per hour of sleep.
Thoracoabdominal movements were measured by spirometry-calibrated respiratory inductance plethysmography (Respitrace; Ambulatory Monitoring, White Plains, NY) and airflow by nasal pressure cannulae (BiNAPS model 550; Salter Labs, Arvin, CA). Central apneas were defined as > 90% reduction in tidal volume (VT) ≥ 10 sec. Central hypopneas were defined as a 50% to 90% reduction in VT from baseline ≥ 10 sec either without thoracoabdominal motion, or with inphase thoracoabdominal motion and without airflow limitation on the nasal pressure tracing. Apneas and hypopneas were classified as obstructive if outofphase thoracoabdominal motion or airflow limitation was present.15 The amplitude criterion for hypopnea termination was ≥ 50% of the baseline tidal volume. The AHI was calculated. Arterial oxygen saturation (SaO2) was measured continuously with a pulse oximeter (Nellcor N200; Tyco International Healthcare, Pleasanton, CA) placed on the ear. Mean SaO2 was derived as previously described.5
Data Analysis
Ten consecutive patients with predominantly OSA and 10 with predominantly CSA from the aforementioned epidemiological study were included in the analysis. Twenty events (apneas and hypopneas) were evaluated in each patient during stage 2 NREM sleep—10 from the beginning of the night, and 10 from the end of the night. Stage 2 sleep was analyzed, as it was the dominant sleep stage in which most events occurred and in which ventilation is predominantly under metabolic control.16 In addition, we confined data analysis to a single sleep stage to control for the potential confounding influences of different sleep stages on arousals and respiratory events. Events from both early and late in the night were analyzed in consideration of influences of possible overnight deterioration of cardiac function.19 Time from the end of an event to the beginning of an arousal (apnea-to-arousal time) and time from the end of an event to the peak of hyperpnea (apneatopeak time) were measured. Time from the beginning of an arousal to the peak of hyperpnea (arousaltopeak time) was calculated by subtracting apnea-to-arousal time from apneatopeak time. Hyperpnea time (HT) was defined as the time between the onset of inspiration of the breath terminating the apnea or hypopnea and the end of inspiration of the breath preceding the next apnea or hypopnea. Lungtoear circulation time (LECT), an estimate of lungtocarotid chemoreceptor circulation time, was measured from the end of an apnea to the subsequent nadir of SpO2 as previously described.5 This technique has been previously validated against echocardiographic Doppler as a measure of circulation time and a reflection of cardiac output.20
Data are expressed as mean ± SD unless stated otherwise. Analyses were performed using SPSS 13.0 (SPSS Inc., Chicago, IL). Data from OSA and CSA patients were compared with 2-tailed unpaired ttests for continuous variables. Nominal and ordinal variables were compared by χ2 or Fisher exact test as appropriate. Correlations among variables were performed by leastsquares linear regression analysis. P-values < 0.05 were considered statistically significant.
RESULTS
Characteristics of the Subjects
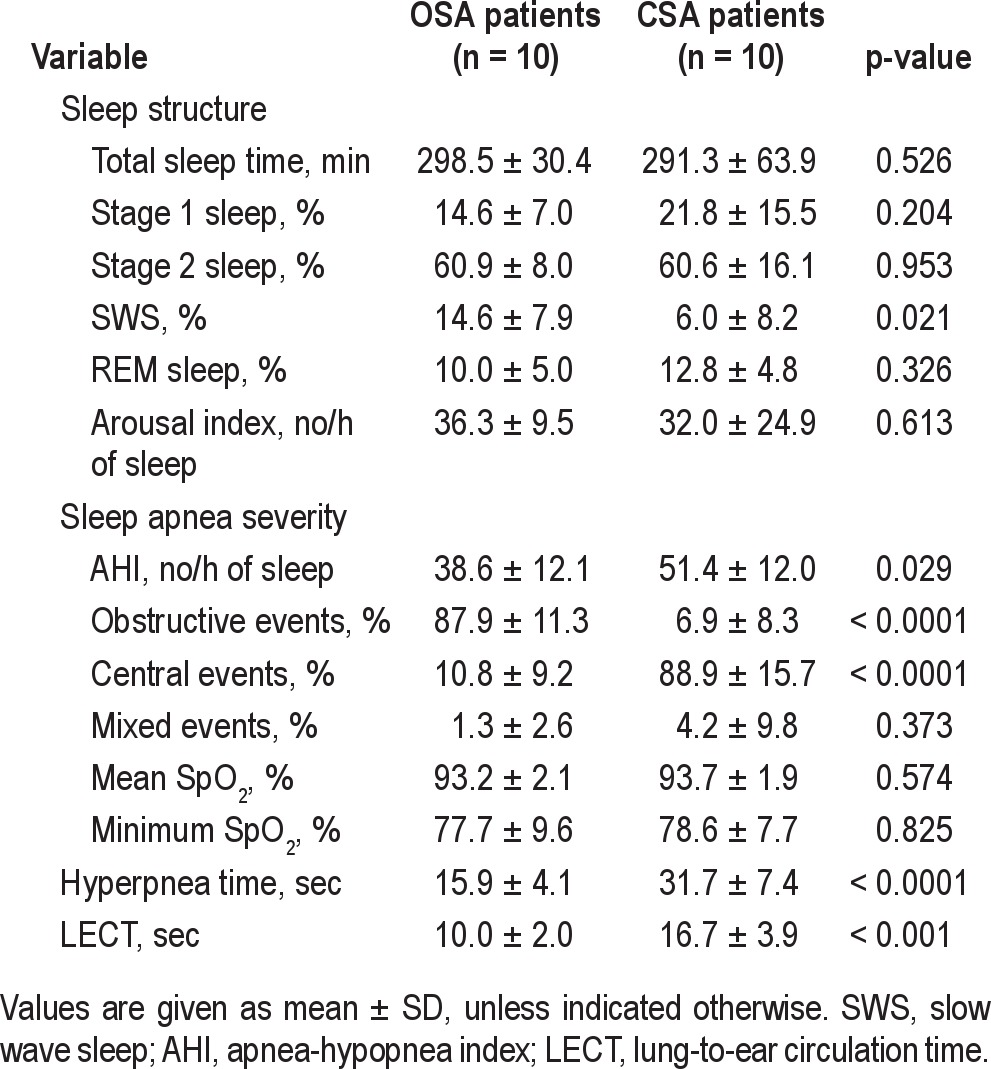
Characteristics of the 10 OSA and 10 CSA patients are shown in Table 1. Patients with CSA were older and had lower body mass index (BMI) than those with OSA. There were no significant differences in LVEF or medical therapy between the 2 groups. Sleep data are shown in Table 2. Total sleep time and the proportion of stages 1, 2, and REM sleep were similar between the 2 groups, but patients with CSA had a lower proportion of slow wave sleep (p = 0.021). The frequency of arousals was also similar in the 2 groups. However, compared to the OSA group, the ratio of arousals to apneas and hypopneas was lower in the CSA group (94% vs 62%, p = 0.042). With respect to sleep apnea severity, the AHI was higher in the CSA group, but there was no significant difference in the mean or minimum SpO2 between the 2 groups. The CSA patients had a significantly longer hyperpnea time and LECT than those with OSA.
Table 1.
Characteristics of the subjects
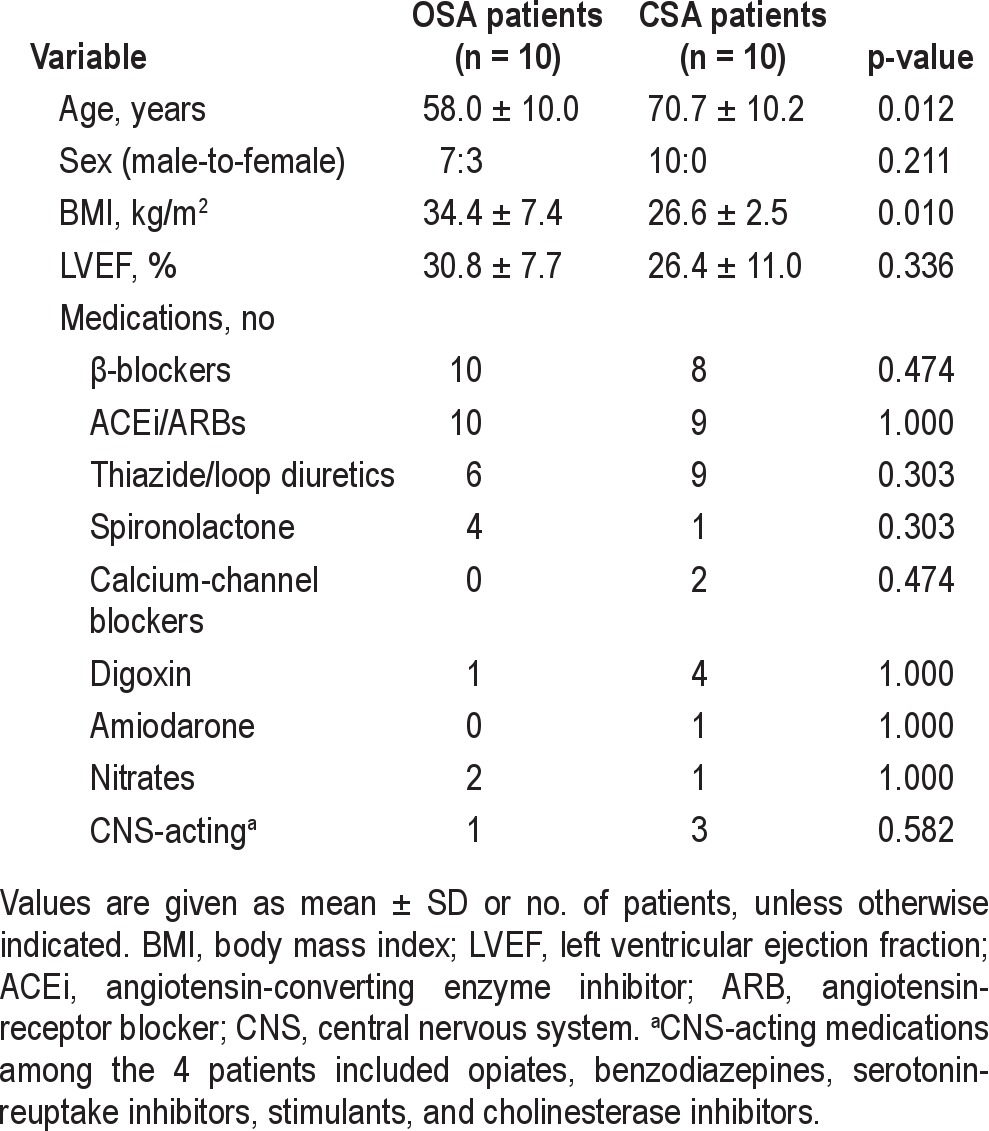
Table 2.
Polysomnographic data
Timing of Arousals
Representative polysomnographic tracings showing arousal timing with respect to the termination of apneas are shown in Figure 1. Note that in the patient with OSA, the arousal occurred coincident with the end of the event (Figure 1A). In contrast, in the patient with CSA, the arousal occurred much later after the end of the apnea, several breaths after resumption of respiration (Figure 1B). Grouped data in Figure 2 show that apnea-to-arousal time was highly variable and much longer in the CSA group than the OSA group (p < 0.0001), but occurred prior to peak hyperpnea so that arousaltopeak time did not differ between the OSA and CSA groups (p = 0.416; Figure 3). Similar to the group variability, the intraindividual variability in apnea-to-arousal time was higher in the CSA patients than in the OSA patients (mean standard deviation of 4.97 ± 2.54 vs. 1.95 ± 0.68, p = 0.002).
Figure 1. Representative polysomnographic recordings.
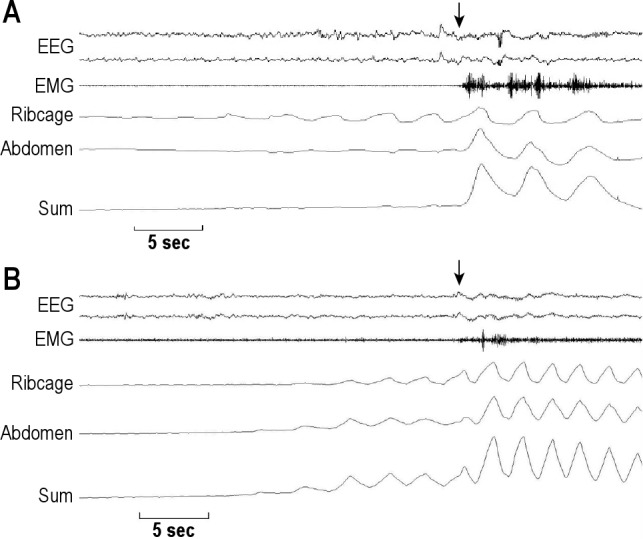
Representative polysomnographic recordings from one patient with OSA (A), and one with CSA (B), during stage 2 sleep. Arrows indicate the onset of arousals. Note outofphase thoracoabdominal motion during obstructive apnea that becomes inphase immediately following the arousal indicating termination of the obstructive event (A). In contrast, the arousal occurred several seconds after the end of the central event (B).
Figure 2. Apnea-to-arousal time.
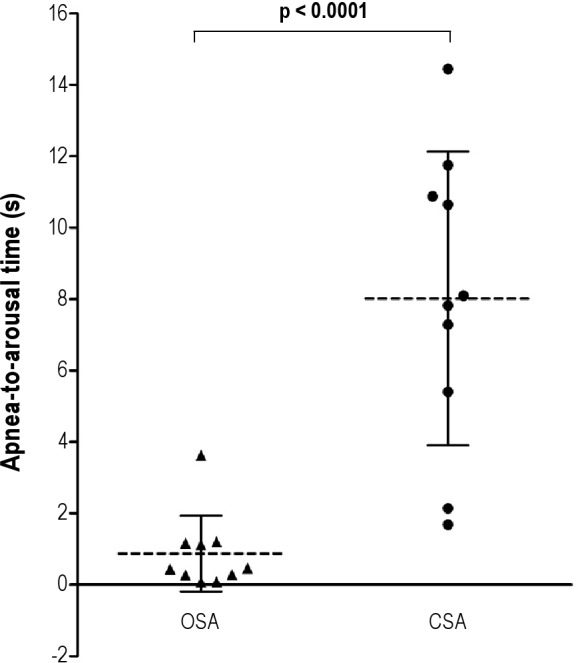
Time from the end of an apnea or hypopnea to the beginning of an arousal (apnea-to-arousal time) was significantly longer and more variable in the 10 subjects in the CSA group compared to the 10 subjects in the OSA group (8.0 ± 4.1 vs 0.9 ± 1.1 s, p < 0.0001). Data are expressed as means and standard deviations.
Figure 3. Arousal-to-peak time.
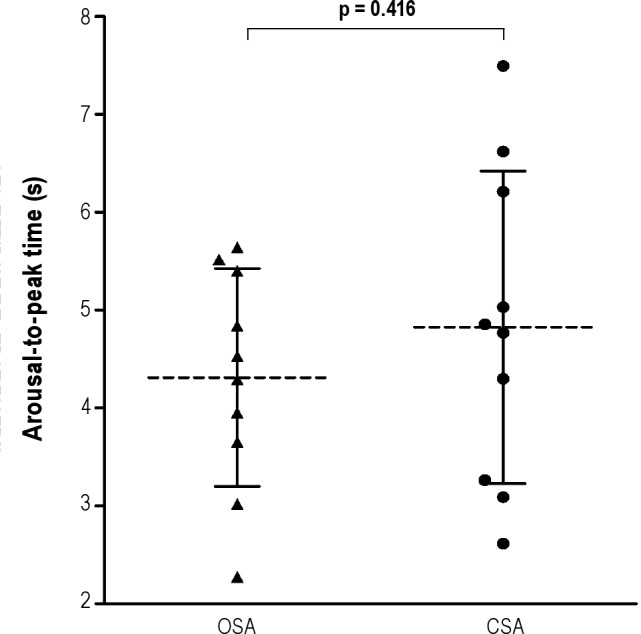
Time from the beginning of an arousal to the peak of hyperpnea (arousaltopeak time) did not differ between the 10 subjects with OSA and the 10 subjects with CSA (4.3 ± 1.1 vs 4.8 ± 1.6 s, p = 0.416).
Apnea-to-arousal time was strongly related to LECT in patients with CSA (p = 0.006, Figure 4A), but not in those with OSA (p = 0.107, Figure 4B). There was no significant relationship between arousaltopeak time and LECT in either the CSA or OSA group (p = 0.887 and p = 0.444, respectively). Length of hyperpnea was related significantly to LECT (r = 0.771, p = 0.009) in the CSA patients, as shown in previous studies,5,20 but not in the OSA patients (p = 0.366).
Figure 4. Apnea-to-arousal time and lung-to-ear circulation time.
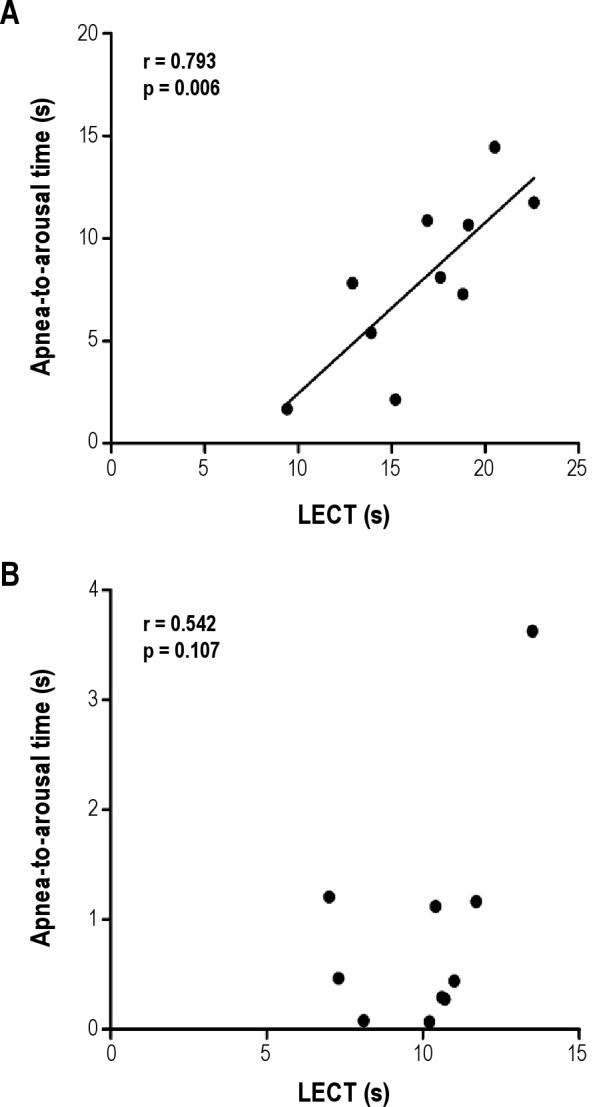
A significant correlation between apnea-to-arousal time and lungtoear circulation time (LECT) was found in the 10 subjects with CSA (A) but not the 10 subjects with OSA (B).
DISCUSSION
The results of the present study indicate that in HF patients there are marked differences in the timing of arousals following termination of apneas and hypopneas between those with OSA and those with CSA. Arousals were generally simultaneous with apnea termination in the OSA group, with a mean apnea-to-arousal time of 0.9 seconds. This is consistent with a defensive role in augmenting upper airway dilator muscle activity and reestablishing airway patency and airflow.3,15 In contrast, in the CSA group, apnea-to-arousal time was much longer, with a mean latency of 8.0 seconds. Moreover, the ratio of arousals to apneas and hypopneas was significantly lower in the CSA (62%) than in the OSA group (94%), indicating that in the former group, far fewer respiratory events were terminated by an arousal. These observations are not consistent with a similar defensive function in central than in obstructive events, since arousals were not necessary to reestablish airflow. However, the time from the onset of arousal to the peak of hyperpnea (arousaltopeak time) was similar in the two groups, suggesting that arousals may augment hyperpnea and influence the timing of peak ventilation in both OSA and CSA.
Our data are consistent with previous studies showing that arousals occur in close proximity to termination of the great majority of obstructive events,1,2 and thus play a key role in reestablishing airflow.3 In contrast, reports of the timing of arousals following termination of central events are inconsistent, with some indicating that they occur at the end of central events6,21,22 and others, several breaths later at the peak of hyperventilation.23 This discrepancy may be related to the inherent variability in arousal timing that we describe herein, or may be a result of the heterogeneity of the CSA population. Indeed, in non-HF patients with idiopathic CSA, most arousals were reported to occur at the resumption of ventilation following apneas.6 In contrast, in CSA accompanied by a Cheyne-Stokes respiratory pattern in the HF population, such as those herein, arousals have been reported to occur later during the hyperpneic phase several breaths after apnea termination.4 However, quantification of the actual time from the end of a central event until the onset of arousal has not been previously reported. Our data indicate that in HF patients with CSA, the timing of arousals is highly variable and most often occurs several seconds after termination of central events, but before the peak of hyperpnea.
The highly variable timing of arousals following central apneas and hypopneas, and their predominant occurrence several breaths after resumption of respiration are not in favor of them being an important mechanism in terminating such events or in reestablishing normal airflow. In this regard, arousals in HF patients with CSA appear to have a substantially different pathophysiological significance than in those with OSA. Indeed, our previous work suggests that arousals play a provocative role in triggering and perpetuating central apneas in both HF and non-HF patients with CSA.5–7 By causing an abrupt increase in ventilation, arousals can provoke a fall in PaCO2 below the apnea threshold that can result in central apnea. Previous studies have shown that over 90% of episodes of Cheyne-Stokes respiration in HF patients with CSA, and almost 80% of episodes of periodic breathing in non-HF patients with idiopathic CSA are precipitated by arousals that trigger an increase in VT.5,6 Arousals facilitate ventilatory overshoot and ongoing respiratory cycling by increasing statedependent chemical ventilatory responsiveness and by engaging the neurogenic wakefulness drive to breathe.6,7 Both the magnitude of hyperventilation and length of the subsequent apnea increase progressively with increasing intensity of associated arousals.6 Taken together, these data suggest a largely pathological effect of arousals in CSA.
If arousals were a result of CSA, then attenuating CSA should reduce arousal frequency, and vice versa. In this regard, the evidence is inconsistent. On the one hand, several small studies involving 11 to 16 HF patients with CSA showed a decrease in ArI by 52% to 57% in association with a 40% to 71% reduction in AHI with CPAP therapy.22,24,25 On the other hand, several studies involving 17 to 26 HF patients with CSA in which CPAP lowered the AHI by 62% to 89% showed no effect on the ArI.8,26–28 In the largest multicenter trial to date, in which 258 HF patients with CSA were randomized to CPAP treatment or control, CPAP lowered the AHI by 55% after 3 months but did not lower the ArI or improve sleep structure.15 Furthermore, there was no significant change in arousal frequency in the subgroup of 58 patients in whom CPAP reduced the AHI to < 15, nor in the 38 CPAP patients who underwent sleep studies 2 years after randomization, compared to the control group. In contrast, studies of HF patients with OSA have shown that the ArI decreases consistently and significantly in association with a reduction in the AHI in response to CPAP.8–11 CPAP has also been shown to decrease arousal frequency significantly in the non-HF OSA population.12–14 These studies therefore support the notion that arousals are less likely to be a consequence of (or defense mechanism in) HF patients with CSA than in OSA.
In OSA, arousals are triggered by inspiratory effort against the occluded airway,29 hypoxia,30 and hypercapnia.31 The stimuli for arousals following central apneas are not as well understood. The possibility of chemoreceptor-mediated stimuli is compatible with our finding that apnea-to-arousal time is strongly related to LECT (i.e., lung to chemoreceptor circulation time) in patients with CSA, such that the longer the LECT, the longer the apnea-to-arousal time. This suggests that the latency from blood gas changes in the lung to their perception at the chemoreceptors influences the timing of arousals that may be chemoreceptor mediated. Thus, while such stimuli might cause arousal at apnea termination and contribute to reinstitution of airflow in individuals with normal heart function and circulation time, if there is an increase in circulation time due to lower cardiac output in the setting of HF, arousals are delayed such that they occur after the onset of airflow. Accordingly, while the physiological purpose of arousals after central apneas may be to act as a defense mechanism to prevent asphyxia similar to that in obstructive events, prolonged circulation time related to HF defeats this purpose and delays arousal until after airflow has resumed. Further support for this hypothesis is provided by the discrepancy in arousal timing between CSA patients with HF and idiopathic CSA patients without HF and with normal circulation time. In the latter, arousals are coincident with apnea termination,6 as in OSA, while in the former, arousals are delayed until several breaths after apnea termination. This difference may well be due to lower cardiac output and longer LECT in patients with HF-related CSA than in those with idiopathic CSA and normal heart function.20
If arousals following central events arise mainly due to chemostimulation, but fail to subserve a defensive role in the setting of HF due to prolonged circulation time, alleviation of CSA by CPAP should still lower the ArI. However, as described above, most studies in HF patients with CSA showed no significant reduction in the frequency of arousals in response to CPAP, despite a decrease in AHI.8,26–28 There are two possible reasons for this discrepancy. First, arousals may be classified as spontaneous or respiratory related.32 While the stimuli for arousals related to central apneas are likely chemoreceptor mediated, the stimuli for respiratoryrelated arousals in OSA include, in addition, inspiratory efforts against an occluded airway and generation of negative intrathoracic pressure.29,32 These additional mechanical stimuli likely underlie the shorter apnea-to-arousal time found in the OSA group, as well as the lack of relationship between apnea-to-arousal time and LECT in OSA. Moreover, the stimuli for respiratory-related arousals in CSA are thus less potent than those for OSA, and arousals are not required for apnea termination.4 Accordingly, respiratory-related arousals likely comprise a smaller fraction of total arousals in CSA than in OSA. Therefore, while respiratory-related arousals may be reduced by attenuation of CSA with CPAP, the larger proportion of spontaneous arousals are unaffected, ultimately yielding nonsignificant reductions in total arousal frequency.
Second, although the relationship between apnea-to-arousal time and LECT in the CSA group suggests a chemical stimulus for arousals in CSA, it is possible that these “respiratory-relat-ed” arousals are in fact, spontaneous arousals that are entrained by the Cheyne-Stokes respiratory cycle. This may be analogous to the entrainment of oscillations in heart rate and blood pressure by periodic breathing. Experimental periodic breathing and Cheyne-Stokes respiration in HF patients can amplify oscillations in blood pressure and heart rate and entrain them at the frequency of the periodic breathing, independent of hypoxia or arousals from sleep.33–35 It is possible, therefore, that spontaneously occurring arousals may be similarly entrained at the frequency of the Cheyne-Stokes respiratory cycle, whose duration is influenced by circulation time.5 Attenuation of central apneas by CPAP may uncouple this relationship and unmask the spontaneous nature of the arousals, with little or no effect on the total arousal frequency.15 If indeed HF patients with CSA have a high frequency of spontaneous arousals, the reason is not clear. One possibility is that higher left ventricular filling pressures and greater rostral fluid shift from the legs into the thorax in HF patients with CSA than in those with OSA cause a greater degree of pulmonary congestion.36,37 Discomfort arising from such congestion might provoke arousals.
In our study, arousals following central events usually preceded the peak of postapnea hyperpnea. Arousals abruptly augment ventilation by increasing chemical ventilatory responsiveness and activating the nonchemical wakefulness drive to breathe.6,7 Both of these are neurogenic stimuli, which should not be influenced by circulation time. Indeed, arousaltopeak time did not differ between the CSA and OSA groups and bore no relationship to LECT. It is also interesting that the arousal to peak ventilation latency was approximately 5 seconds, which is very similar to the neurogenic latency between sympathetic vasoconstrictor discharge and the peak blood pressure response.38 These data also suggest that arousals may play a role in determining the timing of peak ventilation during hyperpnea in both CSA and OSA.
There are several potential limitations of this study. The detection of the start of an arousal by visual inspection has a subjective component, especially in patients who do not produce prominent alpha waves.32 However, our use of occipital32 and frontal39 EEG electrodes should have optimized arousal detection by the standard criteria. It is also possible that the standard AASM arousal definition18 we used may lack sensitivity, leading to under-appreciation of the full spectrum of arousals related to respiratory events.40 In addition, our analyses were confined to stage 2 NREM sleep to control for potential confounding effects of sleep stage on arousal and respiratory events. Therefore, the influence of sleep state on arousal timing was not examined. Finally, it may not be possible to extrapolate our results to all patients with sleep apnea, as only subjects with HF were studied.
In summary, in HF patients the latency from central events to arousals is highly variable and significantly longer than those following obstructive events. Nevertheless, arousals following both central and obstructive events occur prior to peak hyperpnea. The latency between apnea termination and arousal onset is strongly related to LECT in CSA. This observation suggests involvement of chemical respiratory arousal stimuli, with a postapneic latency to the detection of such stimuli at the chemoreceptors inversely proportional to cardiac output.20 The similarity in time from arousal to peak of hyperpnea between the OSA and CSA groups probably relates to neurogenic arousalmediated augmentation of ventilation that is not related to circulation time. These observations provide further evidence that arousals are less likely to act as a protective mechanism that facilitates resumption of airflow in HF patients with CSA than in those with OSA.
DISCLOSURE STATEMENT
This was not an industry supported study. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
This study was supported by operating grant MOP-82731 from the Canadian Institutes of Health Research. Dr. Montemurro was supported by postdoctoral research fellowships from the University of Brescia, Brescia, Italy, and the University Health Network Toronto Rehabilitation Institute.
REFERENCES
- 1.Martin SE, Engleman HM, Kingshott RN, Douglas NJ. Microarousals in patients with sleep apnoea/hypopnoea syndrome. J Sleep Res. 1997;6:276–80. doi: 10.1111/j.1365-2869.1997.00276.x. [DOI] [PubMed] [Google Scholar]
- 2.Dingli K, Fietze I, Assimakopoulos T, Quispe-Bravo S, Witt C, Douglas NJ. Arousability in sleep apnoea/hypopnoea syndrome patients. Eur Respir J. 2002;20:733–40. doi: 10.1183/09031936.02.00262002. [DOI] [PubMed] [Google Scholar]
- 3.Ryan CM, Bradley TD. Pathogenesis of obstructive sleep apnea. J Appl Physiol. 2005;99:2440–50. doi: 10.1152/japplphysiol.00772.2005. [DOI] [PubMed] [Google Scholar]
- 4.Hanly PJ, Millar TW, Steljes DG, Baert R, Frais MA, Kryger MH. Respiration and abnormal sleep in patients with congestive heart failure. Chest. 1989;96:480–8. doi: 10.1378/chest.96.3.480. [DOI] [PubMed] [Google Scholar]
- 5.Naughton M, Benard D, Tam A, Rutherford R, Bradley TD. Role of hyperventilation in the pathogenesis of central sleep apneas in patients with congestive heart failure. Am Rev Respir Dis. 1993;148:330–8. doi: 10.1164/ajrccm/148.2.330. [DOI] [PubMed] [Google Scholar]
- 6.Xie A, Wong B, Phillipson EA, Slutsky AS, Bradley TD. Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med. 1994;150:489–95. doi: 10.1164/ajrccm.150.2.8049835. [DOI] [PubMed] [Google Scholar]
- 7.Yumino D, Bradley TD. Central sleep apnea and Cheyne-Stokes respiration. Proc Am Thorac Soc. 2008;5:226–36. doi: 10.1513/pats.200708-129MG. [DOI] [PubMed] [Google Scholar]
- 8.Javaheri S. Effects of continuous positive airway pressure on sleep apnea and ventricular irritability in patients with heart failure. Circulation. 2000;101:392–7. doi: 10.1161/01.cir.101.4.392. [DOI] [PubMed] [Google Scholar]
- 9.Kaneko Y, Floras JS, Usui K, et al. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med. 2003;348:1233–41. doi: 10.1056/NEJMoa022479. [DOI] [PubMed] [Google Scholar]
- 10.Ryan CM, Usui K, Floras JS, Bradley TD. Effect of continuous positive airway pressure on ventricular ectopy in heart failure patients with obstructive sleep apnoea. Thorax. 2005;60:781–5. doi: 10.1136/thx.2005.040972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruttanaumpawan P, Gilman MP, Usui K, Floras JS, Bradley TD. Sustained effect of continuous positive airway pressure on baroreflex sensitivity in congestive heart failure patients with obstructive sleep apnea. J Hypertens. 2008;26:1163–8. doi: 10.1097/HJH.0b013e3282fb81ed. [DOI] [PubMed] [Google Scholar]
- 12.Loredo JS, Ancoli-Israel S, Dimsdale JE. Effect of continuous positive airway pressure vs placebo continuous positive airway pressure on sleep quality in obstructive sleep apnea. Chest. 1999;116:1545–9. doi: 10.1378/chest.116.6.1545. [DOI] [PubMed] [Google Scholar]
- 13.McArdle N, Douglas NJ. Effect of continuous positive airway pressure on sleep architecture in the sleep apnea-hypopnea syndrome: a randomized controlled trial. Am J Respir Crit Care Med. 2001;164:1459–63. doi: 10.1164/ajrccm.164.8.2008146. [DOI] [PubMed] [Google Scholar]
- 14.Lam B, Sam K, Mok WY, et al. Randomised study of three non-surgical treatments in mild to moderate obstructive sleep apnoea. Thorax. 2007;62:354–9. doi: 10.1136/thx.2006.063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruttanaumpawan P, Logan AG, Floras JS, Bradley TD. Effect of continuous positive airway pressure on sleep structure in heart failure patients with central sleep apnea. Sleep. 2009;32:91–8. [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan CM, Juvet S, Leung R, Bradley TD. Timing of nocturnal ventricular ectopy in heart failure patients with sleep apnea. Chest. 2008;133:934–40. doi: 10.1378/chest.07-2595. [DOI] [PubMed] [Google Scholar]
- 17.Rechtschaffen A, Kales A. A manual of standardized terminology, techniques and scoring for sleep stages of human subjects. Los Angeles: UCLA Brain Information Service/Brain Research Institute; 1968. [Google Scholar]
- 18.Iber C, Ancoli-Israel S, Chesson A, Quan S for the American Academy of Sleep Medicine. 1st ed. Westchester, IL: American Academy of Sleep Medicine; 2007. The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. [Google Scholar]
- 19.Tkacova R, Niroumand M, Lorenzi-Filho G, Bradley TD. Overnight shift from obstructive to central apneas in patients with heart failure: role of PCO2 and circulatory delay. Circulation. 2001;103:238–43. doi: 10.1161/01.cir.103.2.238. [DOI] [PubMed] [Google Scholar]
- 20.Hall MJ, Xie A, Rutherford R, Ando S, Floras JS, Bradley TD. Cycle length of periodic breathing in patients with and without heart failure. Am J Respir Crit Care Med. 1996;154:376–81. doi: 10.1164/ajrccm.154.2.8756809. [DOI] [PubMed] [Google Scholar]
- 21.Kohnlein T, Welte T, Tan LB, Elliot MW. Central sleep apnoea syndrome in patients with chronic heart disease: a critical review of the current literature. Thorax. 2002;57:547–54. doi: 10.1136/thorax.57.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohnlein T, Welte T, Tan LB, Elliott MW. Assisted ventilation for heart failure patients with Cheyne-Stokes respiration. Eur Respir J. 2002;20:934–41. doi: 10.1183/09031936.00.02622001. [DOI] [PubMed] [Google Scholar]
- 23.Mansfield DR, Solin P, Roebuck T, Bergin P, Kaye DM, Naughton MT. The effect of successful heart transplant treatment of heart failure on central sleep apnea. Chest. 2003;124:1675–81. doi: 10.1378/chest.124.5.1675. [DOI] [PubMed] [Google Scholar]
- 24.Teschler H, Dohring J, Wang YM, Berthon-Jones M. Adaptive pressure support servo-ventilation: a novel treatment for Cheyne-Stokes respiration in heart failure. Am J Respir Crit Care Med. 2001;164:614–9. doi: 10.1164/ajrccm.164.4.9908114. [DOI] [PubMed] [Google Scholar]
- 25.Hu K, Li QQ, Yang J, Chen XQ, Hu SP, Wu XJ. The role of high-frequency jet ventilation in the treatment of Cheyne-Stokes respiration in patients with chronic heart failure. Int J Cardiol. 2006;106:224–31. doi: 10.1016/j.ijcard.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Granton JT, Naughton MT, Benard DC, Liu PP, Goldstein RS, Bradley TD. CPAP improves inspiratory muscle strength in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1996;153:277–82. doi: 10.1164/ajrccm.153.1.8542129. [DOI] [PubMed] [Google Scholar]
- 27.Krachman SL, D'Alonzo GE, Berger TJ, Eisen HJ. Comparison of oxygen therapy with nasal continuous positive airway pressure on Cheyne-Stokes respiration during sleep in congestive heart failure. Chest. 1999;116:1550–7. doi: 10.1378/chest.116.6.1550. [DOI] [PubMed] [Google Scholar]
- 28.Arzt M, Schulz M, Wensel R, et al. Nocturnal continuous positive airway pressure improves ventilatory efficiency during exercise in patients with chronic heart failure. Chest. 2005;127:794–802. doi: 10.1378/chest.127.3.794. [DOI] [PubMed] [Google Scholar]
- 29.Issa FG, Sullivan CE. Arousal and breathing responses to airway occlusion in healthy sleeping adults. J Appl Physiol. 1983;55:1113–9. doi: 10.1152/jappl.1983.55.4.1113. [DOI] [PubMed] [Google Scholar]
- 30.Bowes G, Townsend ER, Kozar LF, Bromley SM, Phillipson EA. Effect of carotid body denervation on arousal response to hypoxia in sleeping dogs. J Appl Physiol. 1981;51:40–5. doi: 10.1152/jappl.1981.51.1.40. [DOI] [PubMed] [Google Scholar]
- 31.Berthon-Jones M, Sullivan CE. Ventilation and arousal responses to hypercapnia in normal sleeping humans. J Appl Physiol. 1984;57:59–67. doi: 10.1152/jappl.1984.57.1.59. [DOI] [PubMed] [Google Scholar]
- 32.Berry RB, Gleeson K. Respiratory arousal from sleep: mechanisms and significance. Sleep. 1997;20:654–75. doi: 10.1093/sleep/20.8.654. [DOI] [PubMed] [Google Scholar]
- 33.Lorenzi-Fihlo G, Dajani HR, Leung R, Floras JS, Bradley TD. Entrainment of blood pressure and heart rate oscillations by periodic breathing. Am J Respir Crit Care Med. 1999;159:1147–54. doi: 10.1164/ajrccm.159.4.9806081. [DOI] [PubMed] [Google Scholar]
- 34.Trinder J, Merson R, Rosenberg JI, Fitzgerald F, Kleiman J, Bradley TD. Patho-physiological interactions of ventilation, arousals, and blood pressure oscillations during Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med. 2000;162:808–13. doi: 10.1164/ajrccm.162.3.9806080. [DOI] [PubMed] [Google Scholar]
- 35.Leung R, Floras JS, Lorenzi-Filho G, Rankin F, Picton P, Bradley TD. Influence of Cheyne-Stokes respiration on cardiovascular oscillations in heart failure. Am J Respir Crit Care Med. 2003;167:1534–9. doi: 10.1164/rccm.200208-793OC. [DOI] [PubMed] [Google Scholar]
- 36.Solin P, Bergin P, Richardson M, Kaye DM, Walters EH, Naughton MT. Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation. 1999;99:1574–79. doi: 10.1161/01.cir.99.12.1574. [DOI] [PubMed] [Google Scholar]
- 37.Yumino D, Redolfi S, Ruttanaumpawan P, et al. Nocturnal rostral fluid shift: a unifying concept for the pathogenesis of obstructive and central sleep apnea in men with heart failure. Circulation. 2010;121:1598–605. doi: 10.1161/CIRCULATIONAHA.109.902452. [DOI] [PubMed] [Google Scholar]
- 38.Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–904. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Malley EB, Norman RG, Farkas D, Rapoport DM, Walsleben JA. The addition of frontal EEG leads improves detection of cortical arousal following obstructive respiratory events. Sleep. 2003;26:435–9. doi: 10.1093/sleep/26.4.435. [DOI] [PubMed] [Google Scholar]
- 40.Thomas RJ. Arousals in sleep-disordered breathing: patterns and implications. Sleep. 2003;26:1042–7. doi: 10.1093/sleep/26.8.1042. [DOI] [PubMed] [Google Scholar]